Abstract
Prader-Willi syndrome (PWS) is a rare (~1 in 12,000) genetic disorder that involves at least six genes on chromosome 15q11–q13. Children with PWS not only rapidly gain weight and become severely obese because of reduced voluntary activity and increased food intake, but also exhibit growth hormone deficiency, excessive daytime sleepiness, endocrine dysregulation and infertility. These phenotypes suggest dysfunction of the hypothalamus, the brain region that regulates short- and long-term energy balance and other body functions. The physiological basis for obesity in children with PWS has eluded researchers for decades. Mercer et al. now demonstrate that Magel2, the murine ortholog of one of the PWS genes, is a component of the hypothalamic leptin-melanocortin pathway that is critical for energy balance. Most interestingly, disruptions of other components of this pathway cause obesity in both mice and humans, suggesting a mechanistic link between PWS and other rare genetic forms of severe childhood-onset obesity.
Obesity and obesity-associated complications are a leading cause of morbidity, mortality and excess health care costs.Citation1 While the heritability of body mass index in adults is estimated at 40–70%,Citation2 genetic factors contribute to over 80% of weight variation in children and adolescents.Citation3 Many obesity susceptibility genes act in the central nervous system, interacting with each other and with an environment that provides easy access to cheap, calorically dense and highly palatable food.Citation4-Citation6 Studies that identify and characterize novel obesity genes and pathways have already been shown to have great potential to help us understand, prevent and treat obesity.
Most childhood-onset, severe, syndromic or heritable forms of obesity are caused by rare mutations in genes important in energy balance control circuits in the hypothalamus ().Citation7-Citation12 This small region of the brain coordinates the nervous and endocrine systems to regulate energy balance and other homeostatic activities.Citation13 Both rare mutations and common variants have been found in genes encoding proteins that are involved in the neural responses to leptin, a key hormone produced by adipose tissue. Mutations in these genes profoundly affect body weight and are not compensated for by other genes or pathways, highlighting their physiological importance. Further, leptin resistance is a hallmark of diet-induced obesity.Citation14 The recent report by Mercer et al. demonstrating that loss of a protein named Magel2 impairs leptin signaling in the hypothalamus introduces a new member to the cast of characters essential for energy homeostasis.Citation15 Magel2 is the murine ortholog of MAGEL2,Citation16-Citation23 one of the genes that is inactivated in the orphan genetic obesity disorder Prader-Willi syndrome (PWS).Citation21,Citation24-Citation26 This raises a question about this rare and poorly understood disease: is PWS still an orphan genetic obesity disorder or is it too caused by a defect in hypothalamic leptin signaling?
Figure 1. Genes mutated in childhood obesity act in the hypothalamus to regulate energy balance. Mutations that cause rare monogenic or syndromic forms of childhood obesity have been identified in at least 10 different genes, listed on the right. The proteins encoded by these genes act in the hypothalamus (gray) and participate in the leptin - melanocortin pathway that regulates appetite and body weight. MAGEL2 is one of the genes inactivated in Prader-Willi syndrome, a rare genetic disorder that causes childhood-onset severe obesity. Mercer et al. showed that Magel2, the murine ortholog of MAGEL2, is essential for the leptin-mediated responses of a specific set of neurons, those that express POMC in the arcuate nucleus of the hypothalamus.
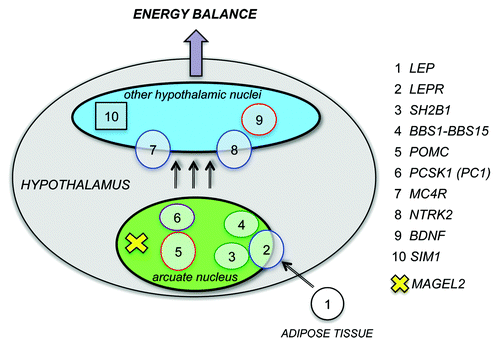
Severely obese children with mutations in the genes encoding leptin, its receptor and downstream signaling components have intractable feelings of hunger (hyperphagia) and aggressive behavior around food.Citation27 Likewise, mice lacking leptin or its receptor are hypoactive, hyperphagic, infertile and obese.Citation28 Other obese children carry mutations in genes encoding proteins in pathways that intersect leptin circuits, such as those involving the downstream melanocortin or brain-derived neurotrophic factor (BDNF) pathways.Citation29 For example, variants in the melanocortin-4 receptor (MC4R) gene are the most frequent cause of monogenic obesity, and disruptions to the SIM1 gene cause syndromic obesity by impairing the hypothalamic leptin-melanocortin pathway. Other rare childhood obesity syndromes, such as Bardet-Biedl syndrome (BBS) are caused by mutations in genes encoding components of cilia, which are highly conserved organelles that act as cellular antennae to coordinate signal transduction pathways and control cell growth or differentiation.Citation30
Of all the syndromic forms of obesity, Prader-Willi syndrome has been the most perplexing to geneticists and obesity specialists. Like children with leptin pathway mutations, children with PWS have an increased ratio of fat to muscle, relentless hunger that can lead to morbid obesity, as well as reduced voluntary activity and infertility.Citation24-Citation26 People with PWS have been known to steal, hoard and beg for food, steal money for food, eat spoiled or frozen food and binge on food to the point of gastric distension and sometimes stomach rupture. For this reason, PWS has been described as a genetic model of starvation, because parallel food foraging behavior and reduced energy expenditure are observed in food-deprived animals.Citation31 In contrast to most human genetic childhood onset obesity disorders where the causative genes disrupt the leptin, melanocortin, BDNF or related pathways, the cause of hyperphagic obesity in PWS in unknown. A priori, a genetic defect involving the leptin - melanocortin pathway is the most parsimonious explanation for obesity in PWS. However, until now, a mechanistic link between any one PWS gene and a specific pathway in the brain had not been made.
PWS is most commonly caused by a microdeletion that occurs on the paternally inherited chromosome 15 and genomic imprinting that silences the maternal allele of six key genes in this region. In this respect, the genetic odds have not been in our favor. Multiple genes, including at least five protein-coding genes and a gene that produces a long non-coding RNA are simultaneously inactivated either by this microdeletion, by uniparental disomy or by a mutation that disrupts the imprinting process. Nature has thrown almost every possible genetic twist at the PWS region: in addition to genomic imprinting, clusters of small nucleolar RNAs are encoded in the introns of the non-coding RNA that also serves as an antisense RNA for the UBE3A gene responsible for Angelman syndrome.Citation32,Citation33 Other inactivated genes encode proteins of unknown function. One protein-coding gene located in the human PWS region is not found in rodents, complicating the studies of animal models.Citation34 The PWS region is in the pericentromeric region of chromosome 15, so the genomic DNA is littered with repeated elements and sequence coverage is poor. In spite of this complexity, researchers have been systematically investigating the genes involved, one by one, in clusters or by studying deletions of the entire region of conserved synteny.Citation35
A first clue that MAGEL2 could be implicated in obesity in PWS came with the observation that Magel2 is most highly expressed in regions of the hypothalamus that control circadian rhythm and energy balance.Citation36 Gene targeted knockout mice lacking Magel2 are overweight and have twice the fat mass, proportionately higher leptin, low lean mass and low bone mass.Citation16,Citation17 Other hypothalamic deficiencies include blunted circadian rhythm, progressive infertility and neuroendocrine deficits, recapitulating phenotypes seen in people with PWS.Citation18,Citation20 As described above, excess fat, reduced energy expenditure and infertility are also characteristic of mice with leptin pathway mutations. Mercer et al. therefore tested whether Magel2 mice have reduced leptin sensitivity and indeed found that, while control mice become anorexic when injected with leptin, adult Magel2 mice do not consume less food after leptin treatment.
Many neurons in the brain respond to leptin, but the leptin-sensitive neurons considered most critical for energy homeostasis are located in the arcuate nucleus of the hypothalamus.Citation37 Two distinct neuronal subtypes respond to leptin within the arcuate nucleus: neurons that produce Agouti-related peptide (AgRP) and Neuropeptide Y (NPY) and neurons that produce pro-opiomelanocortin (POMC). Leptin hyperpolarizes (inhibits) AgRP/NPY neurons, reducing the release of orexigenic (appetite-inducing) peptides. In contrast, leptin depolarizes (excites) POMC neurons, promoting the release of anorexigenic (appetite-suppressing) peptides. One such peptide is α-MSH (melanocyte-stimulating hormone), which acts on melanocortin receptors in other brain sites to promote satiety and increase energy expenditure.Citation38,Citation39 Both the anti-orexigenic and pro-anorexigenic responses to leptin are blunted in animals with acquired (diet-induced obese) or congenital (leptin receptor mutation) leptin insensitivity. To test leptin-mediated electrical activity in hypothalamic neurons, Mercer et al. used whole-cell patch recordings in individual neurons, which surprisingly revealed a defect specific to the anorexigenic POMC neurons in Magel2 mice. That is, while POMC neurons are present in near normal numbers in the ARC of the hypothalamus, they fail to depolarize in response to leptin, while normal hyperpolarizing responses were detected in AgRP/NPY cells.
The finding that Magel2 mice have a primary defect in leptin responses in the POMC-expressing class of anorexigenic neurons was intriguing because previous studies had shown that mice with inactivation of the leptin receptor in only the POMC neurons have increased fat mass, but like Magel2 mice are not massively obese.Citation40 This is presumably because their AgRP/NPY and other neurons still respond to leptin, as is the case with Magel2 mice that retain responses to leptin in non-POMC neurons. Likewise, inactivating the signaling protein STAT3 in only POMC neurons causes mild obesity similar to that of Magel2 mice,Citation41 and inactivating ciliary function in only POMC neurons is sufficient to cause obesity.Citation42 The murine Magel2 and human MAGEL2 genes share 72% amino acid similarity,Citation22,Citation36 and likely share a conserved function in POMC neurons. A defect in the leptin-melanocortin system in children with PWS who lack MAGEL2 would explain many aspects of their disorder, including but not limited to excess fat mass, lower lean mass, reduced voluntary activity and infertility. Further studies, including clinical trials, will be needed to address whether the melanocortin system can be manipulated pharmacologically to improve appetite control in children with PWS.
We still have far to go in understanding the neural basis for PWS. Mice lacking Magel2 do not eat voraciously when freely fed, so they do not become morbidly obese like mice with leptin-pathway mutations or like children with PWS whose food environment is not strictly controlled. Their body weight regulation is abnormal though, as they return more slowly to a normal body weight after under- and over-feeding. The loss of other genes, such as the NDN gene encoding necdinCitation43-Citation45 and the clusters of small nucleolar RNAs have also been proposed to account for other endophenotypes in PWS, including neonatal respiratory insufficiencyCitation46 and failure to thrive in infancy.Citation47 Nonetheless, we now have a foothold into energy imbalance in one of the last remaining and arguably most intriguing rare genetic disorders causing obesity. The new actor on this stage—Magel2—will have to find its niche in the complex protein network in hypothalamic neurons that governs the delicate balance between food intake and energy expenditure. This discovery also adds a new pharmacological target for improving hormone sensitivity and activating pathways downstream of appetitive hormones, both promising avenues for treatment of the obesity that has become rampant in today’s society.
Acknowledgments
Studies in the authors’ laboratories involving PWS mice were funded by grants from the CIHR (MT10520 and OTG 88592 to W.F.C.), the Prader-Willi Syndrome Association (Best Ideas Grant) and the Women and Children’s Health Research Institute at the University of Alberta. W.F.C. is an AHFMR Medical Scientist.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta-analysis. JAMA 2013; 309:71 - 82; http://dx.doi.org/10.1001/jama.2012.113905; PMID: 23280227
- Rokholm B, Silventoinen K, Tynelius P, Gamborg M, Sørensen TI, Rasmussen F. Increasing genetic variance of body mass index during the Swedish obesity epidemic. PLoS One 2011; 6:e27135; http://dx.doi.org/10.1371/journal.pone.0027135; PMID: 22087252
- Dubois L, Ohm Kyvik K, Girard M, Tatone-Tokuda F, Pérusse D, Hjelmborg J, et al. Genetic and environmental contributions to weight, height, and BMI from birth to 19 years of age: an international study of over 12,000 twin pairs. PLoS One 2012; 7:e30153; http://dx.doi.org/10.1371/journal.pone.0030153; PMID: 22347368
- Bradfield JP, Taal HR, Timpson NJ, Scherag A, Lecoeur C, Warrington NM, et al, Early Growth Genetics Consortium. A genome-wide association meta-analysis identifies new childhood obesity loci. Nat Genet 2012; 44:526 - 31; http://dx.doi.org/10.1038/ng.2247; PMID: 22484627
- Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, et al, Wellcome Trust Case Control Consortium, Genetic Investigation of ANthropometric Traits Consortium. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 2009; 41:25 - 34; http://dx.doi.org/10.1038/ng.287; PMID: 19079261
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al, MAGIC, Procardis Consortium. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010; 42:937 - 48; http://dx.doi.org/10.1038/ng.686; PMID: 20935630
- Kimber W, Peelman F, Prieur X, Wangensteen T, O’Rahilly S, Tavernier J, et al. Functional characterization of naturally occurring pathogenic mutations in the human leptin receptor. Endocrinology 2008; 149:6043 - 52; http://dx.doi.org/10.1210/en.2008-0544; PMID: 18703626
- Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med 2007; 356:237 - 47; http://dx.doi.org/10.1056/NEJMoa063988; PMID: 17229951
- Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997; 387:903 - 8; http://dx.doi.org/10.1038/43185; PMID: 9202122
- Bochukova EG, Huang N, Keogh J, Henning E, Purmann C, Blaszczyk K, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature 2010; 463:666 - 70; http://dx.doi.org/10.1038/nature08689; PMID: 19966786
- Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci 2004; 7:1187 - 9; http://dx.doi.org/10.1038/nn1336; PMID: 15494731
- van Vliet-Ostaptchouk JV, Hofker MH, van der Schouw YT, Wijmenga C, Onland-Moret NC. Genetic variation in the hypothalamic pathways and its role on obesity. Obes Rev 2009; 10:593 - 609; http://dx.doi.org/10.1111/j.1467-789X.2009.00597.x; PMID: 19712437
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature 2006; 443:289 - 95; http://dx.doi.org/10.1038/nature05026; PMID: 16988703
- Könner AC, Brüning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab 2012; 16:144 - 52; http://dx.doi.org/10.1016/j.cmet.2012.07.004; PMID: 22883229
- Mercer RE, Michaelson SD, Chee MJ, Atallah TA, Wevrick R, Colmers WF. Magel2 is required for leptin-mediated depolarization of POMC neurons in the hypothalamic arcuate nucleus in mice. PLoS Genet 2013; 9:e1003207; http://dx.doi.org/10.1371/journal.pgen.1003207; PMID: 23341784
- Kozlov SV, Bogenpohl JW, Howell MP, Wevrick R, Panda S, Hogenesch JB, et al. The imprinted gene Magel2 regulates normal circadian output. Nat Genet 2007; 39:1266 - 72; http://dx.doi.org/10.1038/ng2114; PMID: 17893678
- Bischof JM, Stewart CL, Wevrick R. Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum Mol Genet 2007; 16:2713 - 9; http://dx.doi.org/10.1093/hmg/ddm225; PMID: 17728320
- Mercer RE, Kwolek EM, Bischof JM, van Eede M, Henkelman RM, Wevrick R. Regionally reduced brain volume, altered serotonin neurochemistry, and abnormal behavior in mice null for the circadian rhythm output gene Magel2. Am J Med Genet B Neuropsychiatr Genet 2009; 150B:1085 - 99; http://dx.doi.org/10.1002/ajmg.b.30934; PMID: 19199291
- Mercer RE, Wevrick R. Loss of magel2, a candidate gene for features of Prader-Willi syndrome, impairs reproductive function in mice. PLoS One 2009; 4:e4291; http://dx.doi.org/10.1371/journal.pone.0004291; PMID: 19172181
- Tennese AA, Wevrick R. Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in Magel2-null mice. Endocrinology 2011; 152:967 - 78; http://dx.doi.org/10.1210/en.2010-0709; PMID: 21248145
- Mercer RE, Wevrick R. Energy homeostasis in Prader-Willi syndrome: how clinical research informs studies of animal models of genetic obesity: comment on “Nutritional phases in Prader-Willi syndrome,” Miller et al., 2011. Am J Med Genet Part A, 155:1040-1049. Am J Med Genet A 2012; 158A:966 - 8; http://dx.doi.org/10.1002/ajmg.a.35249; PMID: 22419655
- Boccaccio I, Glatt-Deeley H, Watrin F, Roëckel N, Lalande M, Muscatelli F. The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader-Willi region. Hum Mol Genet 1999; 8:2497 - 505; http://dx.doi.org/10.1093/hmg/8.13.2497; PMID: 10556298
- Schaller F, Watrin F, Sturny R, Massacrier A, Szepetowski P, Muscatelli F. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum Mol Genet 2010; 19:4895 - 905; http://dx.doi.org/10.1093/hmg/ddq424; PMID: 20876615
- Goldstone AP. The hypothalamus, hormones, and hunger: alterations in human obesity and illness. Prog Brain Res 2006; 153:57 - 73; http://dx.doi.org/10.1016/S0079-6123(06)53003-1; PMID: 16876568
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med 2012; 14:10 - 26; http://dx.doi.org/10.1038/gim.0b013e31822bead0; PMID: 22237428
- Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A 2011; 155A:1040 - 9; http://dx.doi.org/10.1002/ajmg.a.33951; PMID: 21465655
- Ramachandrappa S, Farooqi IS. Genetic approaches to understanding human obesity. J Clin Invest 2011; 121:2080 - 6; http://dx.doi.org/10.1172/JCI46044; PMID: 21633175
- Friedman JM. Leptin at 14 y of age: an ongoing story. Am J Clin Nutr 2009; 89:973S - 9S; http://dx.doi.org/10.3945/ajcn.2008.26788B; PMID: 19190071
- Ranadive SA, Vaisse C. Lessons from extreme human obesity: monogenic disorders. Endocrinol Metab Clin North Am 2008; 37:733 - 51, x; http://dx.doi.org/10.1016/j.ecl.2008.07.003; PMID: 18775361
- Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet 2012; 21:8 - 13; http://dx.doi.org/10.1038/ejhg.2012.115; PMID: 22713813
- Holland A, Whittington J, Hinton E. The paradox of Prader-Willi syndrome: a genetic model of starvation. Lancet 2003; 362:989 - 91; http://dx.doi.org/10.1016/S0140-6736(03)14370-X; PMID: 14511934
- Ding F, Li HH, Zhang S, Solomon NM, Camper SA, Cohen P, et al. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS One 2008; 3:e1709; http://dx.doi.org/10.1371/journal.pone.0001709; PMID: 18320030
- Bortolin-Cavaillé ML, Cavaillé J. The SNORD115 (H/MBII-52) and SNORD116 (H/MBII-85) gene clusters at the imprinted Prader-Willi locus generate canonical box C/D snoRNAs. Nucleic Acids Res 2012; 40:6800 - 7; http://dx.doi.org/10.1093/nar/gks321; PMID: 22495932
- Neumann LC, Markaki Y, Mladenov E, Hoffmann D, Buiting K, Horsthemke B. The imprinted NPAP1/C15orf2 gene in the Prader-Willi syndrome region encodes a nuclear pore complex associated protein. Hum Mol Genet 2012; 21:4038 - 48; http://dx.doi.org/10.1093/hmg/dds228; PMID: 22694955
- Resnick JL, Nicholls RD, Wevrick R. Recommendations for the investigation of animal models of Prader-Willi Syndrome. Mamm Genome 2013; In press
- Lee S, Kozlov S, Hernandez L, Chamberlain SJ, Brannan CI, Stewart CL, et al. Expression and imprinting of MAGEL2 suggest a role in Prader-willi syndrome and the homologous murine imprinting phenotype. Hum Mol Genet 2000; 9:1813 - 9; http://dx.doi.org/10.1093/hmg/9.12.1813; PMID: 10915770
- Woods SC, D’Alessio DA. Central control of body weight and appetite. J Clin Endocrinol Metab 2008; 93:Suppl 1 S37 - 50; http://dx.doi.org/10.1210/jc.2008-1630; PMID: 18987269
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001; 411:480 - 4; http://dx.doi.org/10.1038/35078085; PMID: 11373681
- Qiu J, Fang Y, Rønnekleiv OK, Kelly MJ. Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J Neurosci 2010; 30:1560 - 5; http://dx.doi.org/10.1523/JNEUROSCI.4816-09.2010; PMID: 20107083
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 2004; 42:983 - 91; http://dx.doi.org/10.1016/j.neuron.2004.06.004; PMID: 15207242
- Xu AW, Ste-Marie L, Kaelin CB, Barsh GS. Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes decreased pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology 2007; 148:72 - 80; http://dx.doi.org/10.1210/en.2006-1119; PMID: 17023536
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, et al. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol 2007; 17:1586 - 94; http://dx.doi.org/10.1016/j.cub.2007.08.034; PMID: 17825558
- MacDonald HR, Wevrick R. The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum Mol Genet 1997; 6:1873 - 8; http://dx.doi.org/10.1093/hmg/6.11.1873; PMID: 9302265
- Gérard M, Hernandez L, Wevrick R, Stewart CL. Disruption of the mouse necdin gene results in early post-natal lethality. Nat Genet 1999; 23:199 - 202; http://dx.doi.org/10.1038/13828; PMID: 10508517
- Muscatelli F, Abrous DN, Massacrier A, Boccaccio I, Le Moal M, Cau P, et al. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum Mol Genet 2000; 9:3101 - 10; http://dx.doi.org/10.1093/hmg/9.20.3101; PMID: 11115855
- Ren J, Lee S, Pagliardini S, Gérard M, Stewart CL, Greer JJ, et al. Absence of Ndn, encoding the Prader-Willi syndrome-deleted gene necdin, results in congenital deficiency of central respiratory drive in neonatal mice. J Neurosci 2003; 23:1569 - 73; PMID: 12629158
- Ding F, Prints Y, Dhar MS, Johnson DK, Garnacho-Montero C, Nicholls RD, et al. Lack of Pwcr1/MBII-85 snoRNA is critical for neonatal lethality in Prader-Willi syndrome mouse models. Mamm Genome 2005; 16:424 - 31; http://dx.doi.org/10.1007/s00335-005-2460-2; PMID: 16075369