Abstract
Xeroderma Pigmentosum (XP), Trichothiodystrophy (TTD) and Cockayne Syndrome (CS) are rare, recessive disorders caused by mutational defects in the Nucleotide Excision Repair (NER) pathway and/or disruption of basic cellular DNA transcription. To date, a multitude of mutations in the XPD/ERCC2 gene have been described, many of which give rise to NER- and DNA transcription related diseases, which share certain diagnostic features and few overlap patients have been described. Despite increasing understanding of the roles of XPD/ERCC2 in mammalian cells, there is still weak predictability of somatic outcome from many of these mutations. We demonstrate a patient, believed to represent an overlap between XP and TTD/CS. In addition to other organ dysfunctions, the young man presented with Photosensitivity, Ichthyosis, Brittle hair, Impaired physical and mental development, Decreased fertility and Short stature (PIBIDS) suggestive of TTD, but lacking the almost patognomonic “tiger tail” banding of the hair under polarized light. Additionally, he developed basal cell carcinoma aged 28, as well as adult onset kidney failure, features normally not associated with TTD but rather XP/CS. His freckled appearance also suggested XP, but fibroblast cultures only demonstrated x2 UV-sensitivity with expected NER and TFIIH-activity decrease. Genetic sequencing of the XPD/ERCC2 gene established the patient as heterozygote compound with a novel, N-terminal Y18H mutation and a known C-terminal (TTD) mutation, A725P. The possible interplay between gene products and the patient phenotype is discussed.
Introduction
Xeroderma Pigmentosum (XP), Trichothiodystrophy (TTD) and Cockayne Syndrome (CS) are rare, recessive disorders caused by mutational defects in the Nucleotide Excision Repair (NER) pathway and/or disruption of basic cellular DNA transcription. Briefly, XP is characterized by a freckled appearance with highly increased skin UV sensitivity and proneness for skin cancer sometimes with late onset neurological features. About half the TTD patients suffer from UV sensitivity but have no apparent predisposition for skin cancer. These patients share a cardinal sign of short, brittle hair with abnormally low sulfur content. With polarizing microscopy, individual hair strands display alternating light and dark bands, thus earning the nickname “tiger-tail pattern,” which is highly suggestive of TTD.Citation1,Citation2 Other TTD features are mental and growth retardation, infection, ichthyosis, nail abnormalities, decreased fertility and various progeroid features. CS share some XP/TTD features but has a more severe phenotype with many developmental defects and a shortened life expectancy.Citation3 On rare occasions, individual patients’ symptoms overlap criteria for more than one disease as exemplified by the XP/TTD overlap patients.Citation4,Citation5
XP was the first DNA-repair disorder to be identified and continuous work throughout the past two decades has revealed intriguing findings on the close genetic relationship between XP, TTD and CS despite their phenotypic heterogeneity. Several gene defects have been identified as causative of XP, TTD and CS but mutations in ERCC2 (XPD) alone can result in all three diseases, fueling efforts into elucidating the role of the XPD protein in eukaryotic cells.Citation6 To date, a collection of different mutations throughout the XPD-gene have been described and attention toward the cause-and-effect of various mutations within the XPD protein has been increased. Consequently, it is important to thoroughly characterize the individual patients, as in this study, where we describe a patient with a novel XP/TTD pheno- and genotype.
Results
NER analysis
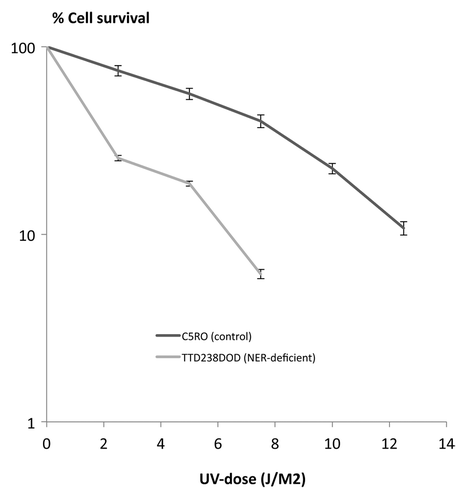
Skin fibroblast cultures (designated TTD238DOD) were initiated from the patient to test his nucleotide excision repair (NER) capabilities, using standardized methods. Global-genome NER activity, reflected by UV-induced unscheduled DNA synthesis (UDS), was decreased to about 55% of normal control cells on the same microscope slide (). Transcription-coupled NER, measured as the ability to recover from transcription inhibition after UV exposure (RRS), was reduced to background levels (12% of normal cells, ). These defects resulted in an overall UV-hypersensitivity of 2.2 × , measured in a cellular survival assay ().
Figure 1. Cell cultures and results. Patient's cells were mixed 1:1 with normal control fibroblasts preloaded with cytoplasmic beads. The mixed cultures were assayed for unscheduled DNA synthesis (A) and recovery from transcription inhibition (B). Red fluorescence shows the NER activity, and blue color reflects the DAPI signal. White arrows indicate the 238DOD cells, as recognized by not containing cytoplasmic beads. (C) Mixed cultures as described for panels A/B,immunostained for TFIIH core component XPB (green fluorescence). (D) Measured NER-activity in TTD238DOD and a control cell line. (E) Measured XPB expression in TTD238DOD and a control cell line
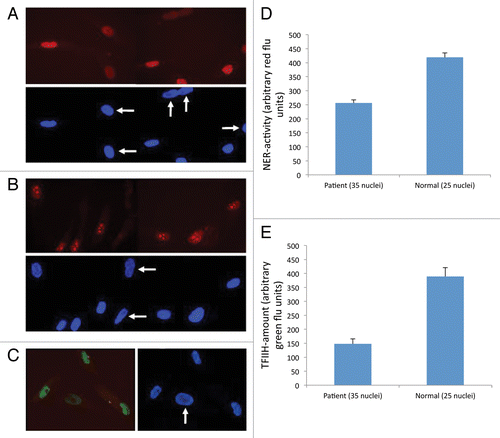
Figure 2. Cell survival assay.
TFIIH
By comparative immunofluorescence of XPB, a core component of the TFIIH complex, we found the TFIIH levels of the cells from our patient reduced to an average of 38% of normal cells ().
Mutations in ERCC2/XPD
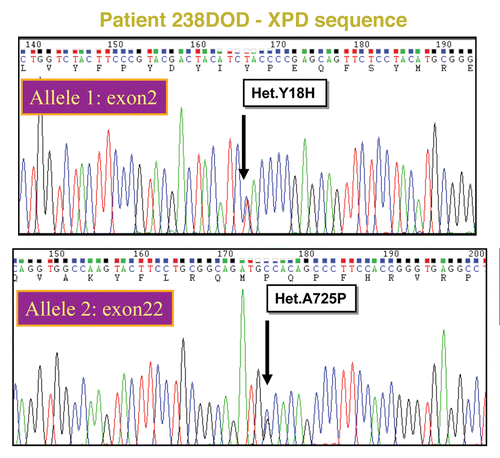
The sequence analysis of ERCC2 of fibroblastic DNA from our patient revealed two missense mutations, g.45873444t > C, p.Y18H in exon 2 and g.45855484G > C, p.A725P in exon 22. Analysis of the parents showed that they each carried one of the mutations, thereby confirming that the patient is compound heterozygous for the mutations. The paternal allele had the G to C change in exon 22, causing a change from Alanine to Proline at position 725 in the protein ( top). The maternal allele had the T to C substitution in exon 2, resulting in Histidine at position 18 instead of Tyrosine in the encoded protein ( bottom). Both mutated alleles are expressed at normal levels and were found in paraffin-embedded biopsies from the deceased brother as well.
Figure 3. ERCC2/XPD sequence analysis of TTD238DOD Relevant parts of peak plots from standard capillary sequencing analysis of TTD238DOD fibroblast DNA. The patient is a compound heterozygote for two mutations. Top: XPD exon 2, bottom XPD exon 22.
Discussion
The clinical presentation aroused suspicion of syndromic ichthyosis, possibly steroid sulfatase (STS) deficiency and contiguous gene deletion syndrome or TTD. However FISH analysis for STS deficiency and array-CGH were normal and polarizing hair microscopy was almost normal as well. Other diagnostic considerations were Refsum disease, LEOPARD syndrome, dyskeratosis congenita and mitochondrial disease, but no positive biochemical analyses or genetic findings could support these diagnoses. Since the patient displayed progressive progeroid features, fulfilling PIBIDS criteria and developed non melanoma skin cancer it was decided to perform DNA repair evaluations.Citation7,Citation8 The 55% NER reduction in fibroblasts suggested NER-defective TTDCitation9,Citation10 and doubled UV sensitivity warranted further detailed analysis of the XPD gene, consequently proving the patient to be compound heterozygous for a novel and a known mutation.Citation11
There can be significant variations on phenotypes produced by clustered, or even identical, mutations in XPD.Citation4 XP patients are distinctly skin cancer prone, whereas UV-sensitive TTD patients display UV sensitivity in vivo as well as in vitro, but paradoxically show no tendency toward skin cancer. The patient in casu demonstrates mild in vitro UV sensitivity comparable to that of the only other patient with the p.A725P mutationCitation11 but is also cancer-prone. A mouse model with an UV sensitive-specific TTD mutation (R722W) demonstrated phenotypic changes that correlate to those in humans, but the mice also developed skin cancer.Citation12
Remarkably, despite having visibly unruly and brittle hair with slight morphologic abnormalities at light microscopy as well as other TTD-specific changes, our patient TTD238DOD lacks the “tiger-tail pattern” and has near-normal cystine levels. While the relatively cystine-rich hair correlates well with a missing tiger tail pattern,Citation1 the higher-than-expected cystine levels for a TTD patient could be explained by the fact that the patient is compound heterozygote with two active alleles offers another probable explanation. It has previously been suggested that homozygous or even hemizygous patients may fare better due to the fact that phenotypic changes are determined more by gene dosage rather than specific mutations.Citation9 However, the opposite could also be true in a patient with both XP-inducing as well as TTD-inducing mutations.Citation13
The kidney problems of our patient are not common features of XP/TTD but are frequently encountered in CS patients, although they usually present at an earlier age.Citation14 We are unsure how to interpret this, as the normal mitochondria and MRI-c contradict a dominant CS phenotype. Furthermore, the ocular findings of TTD238DOD are in line with those previously reported for XP/TTD patients, although cataract was severe and he also had iridal pigmentary changes.Citation15 Overall, the phenotype in the presented case is indeed that of mild XP together with a slightly altered TTD, suggestive of a beneficial interplay between the gene-products and it should be emphasized that lack of the tell-tale tiger tail pattern does not rule out TTD.
Materials and Methods
Case presentation
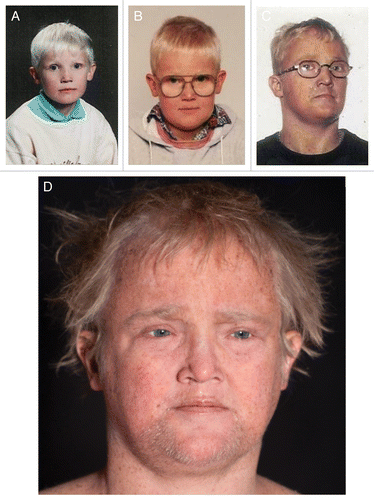
A 25-y old male () was referred with dry skin, lentigines, cutaneous warts and dysmorphic features. He was conceived by healthy, non-consanguineous Caucasian parents and born after an uneventful pregnancy. At the age of three, he was diagnosed with strabismus and hypermetropia and since 12 y of age he used hearing aids because of sensorineural hearing loss. Since early childhood the patient suffered from very dry skin and became easily sunburned despite preventive measures although he never developed blisters. From early puberty he gradually developed many freckles, sparse, thin hair, photophobia and growth retardation ().
Table 1. Clinical findings of TTD238DOD.
Figure 4.(A) TTD238DOD age 8. (B) TTD238DOD age 10. (C) TTD238DOD age 22. (D) TTD238DOD age 28.
He had an older brother who died at age 26 due to ruptured appendicitis and salmonella sepsis. The deceased brother had a similar phenotype with short stature, ichthyosis, hypogonadism, epilepsy, mild mental retardation and diabetes mellitus. A second brother and a sister displayed none of these traits and were considered healthy. The patient’s height was 165 cm (healthy siblings had a height between 180 and 200 cm). Medical history, clinical examination and laboratory and imaging test revealed diseases in many organs.
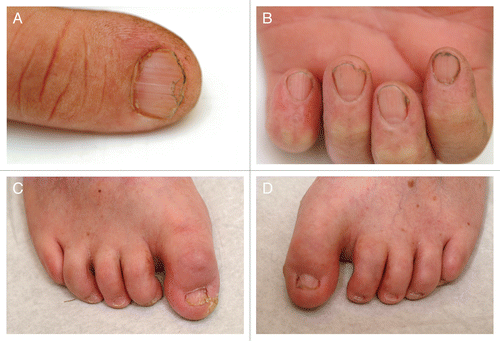
He had dry, parchment-like skin with ichthyosiform scaling mainly on the back and scattered, but pronounced, lentiginous pigmentation (). Viral warts were seen on his hands, knees and feet. He had sparse, brittle and unruly hair and koilonychia with distal nail splitting (). Hair microscopy showed some variation in the calibre of the hair shafts with few trichorrhexis nodosa-like defects. Some hairs had focal ribboning/undulation and irregular contours. No “tiger-tail pattern” was observed using polarizing light microscopy. Cystine content in hair was only slightly decreased to 8.2 mol% (normal values 10.0−10.5 mol%, analysis performed by Dr Josef Föhles, Aachen, who provided reference values). At the age of 28, basal cell carcinoma was diagnosed on his shoulder.
Figure 5. Lentiginous and dry skin. (A) Lentiginous back of TTD238DOD. (B) Close-up of (A) with ichthyosiform skin.
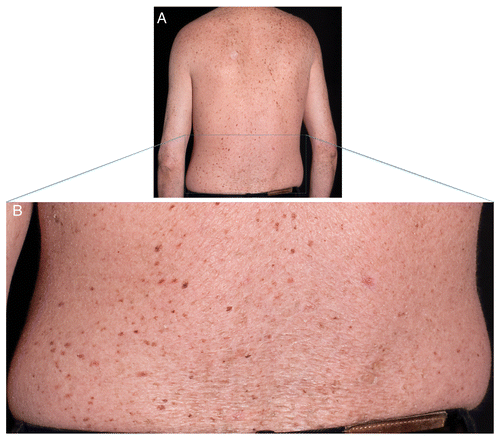
Figure 6.(A−D) Nail changes with distal nail splitting and koilonychias.
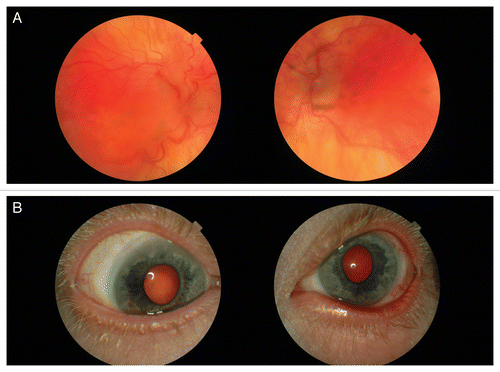
Eye examination revealed photophobia and severely decreased visual acuity (1/60 on the right eye and 12/60 on the left eye) (). He had esotropia but no nystagmus. He had bilateral microphthalmos with an axial length of 18.39 mm on the right eye and 18.53 mm on the left eye, but normal corneal size (12 mm on both eyes). He had hypopigmentation of the iris with transillumination. Corneas were normal with the exception of limbal vascularization. Before cataract surgery he had an excessive hyperopia +13 D on both eyes. He had bilateral cataract with punctate lens opacities. Ophthalmoscopy revealed hypopigmentation of the retina, tortuosity of the vessels and macular lesions with pigment epithelial derangement and atrophy, retinal fibrosis and macular folds on both eyes. Earlier performed visual evoked potential (VEP) was normal and electroretinography did not show any signs of tapetoretinal degeneration.
Figure 7. Eye changes. (A) Retinal degenerative changes. (B) Limbal corneal neovascularization.
He had a high-pitched voice, micropenis (2 cm) and cryptorchidism with high levels of plasma luteinizing- and follicle stimulating hormones and very low plasma testosterone. Pubic and axillary hairs were absent. Type 2 diabetes was diagnosed at the age of 26 and required insulin treatment. The pituitary-adrenal axis and thyroid function were normal.
Neurologic examination showed progressive ataxia and a broad-gauged gait. Varying imaging studies showed thickened theca cranii and a dysmorphic sella turcica as the only clear-cut abnormal findings and EEG was normal. Multiple CT scans as well as an MRI of the cerebrum were normal at around age 27.
Between ages 25 to 31, he developed progressive renal disease with an increase in P-creatinine from 114 to 252 µmol/l, persistent albuminuria around 1.0 g/l and intermittent hematuria. Renal biopsy revealed partial atrophy and sclerosis with scattered hyaline tubular casts but immunoflourescent evaluations were all negative. Electron microscopy showed expanded mesangium and unevenly thickened glomerular basement membranes with varying density but without deposits. The renal disease was complicated by nephrogenic anemia, renal osteodystrophy and arterial hypertension. MCV was low, but within normal range and HBa2 was not tested. Due to hypercholesterolemia treatment with Simvastatin was initiated but had to be discontinued due to acute renal failure caused by rhabdomyolysis. Kidney function was restored after three weeks on dialysis.
Mitochondrial function was normal when respiratory enzymes were measured in a liver biopsy. Plasma concentrations of amino acids were normal except for a slight elevation of P-cysteine. A biopsy from striated muscles did not reveal any primary muscular disorder.
Due to recalcitrant viral warts and frequent pneumonias, a lymphocyte-subpopulation evaluation was conducted showing normal fractions and concentrations of T-, B- and NK-cells. CD4+ count was slightly below normal levels and CD4+/CD8+ levels inversed T- and B- memory cell levels were unexpected high, mimicking levels of a much older person. It was concluded that the patient was slightly immunodeficient.
Methods
Single-cell fluorescent repair assays were performed in 1−2 d old 1:1 mixed cultures with normal control fibroblasts (C5RO) preloaded with cytoplasmic polystyrene beads. The assays were conducted essentially as described by Jaspers and coworkers.Citation16 However, radioactive precursors were replaced by ethynyl-deoxyurine and ethynyl-uridine, which can be demonstrated using a specific click-reaction with Alexa-azide [Invitrogen], as recently described.Citation17,Citation18 In short, for unscheduled DNA synthesis (UDS, a measure of global-genome NER), coverslip cultured cells were exposed to 16J/m2 of 254nm UV light, then treated with ethynylated thymidine analog for 3h and fixed for fluorescent staining. Parallel coverslip cultures also exposed to 16J/m2 of UV were cultured for another 16h to recover from transcription inhibition and then labeled for 2h with ethynyl-uridine, to measure overall rates of RNA synthesis. Immunostaining for TFIIH using XPB antibodies was performed as described by Vermeulen et al.Citation10 In this assay, unirradiated patient and normal primary fibroblasts are known to display similar levels of transcription, even with lowered TFIIH levels in patient cells (data not shown here.) Average nuclear fluorescence intensities, from 20−30 randomly selected cells, were quantified using standard image software (Adobe Photoshop).
Our standard cellular survival protocol involved measurement of incorporated tritiated thymidine in a scintillation counter, 3–5 d after exposure to graded doses of UV light. For UV, this method produces exactly the same survival curves as very tedious colony assays.Citation19 Total RNA was extracted from skin fibroblasts from the patient and mRNA was reverse-transcribed to cDNA. Genomic DNA was extracted from blood lymphocytes of the patient, his deceased brother and his parents using standard methods. The cDNA was screened for mutations in ERCC2 (NM_000400) by primers covering all 22 exons.
| Abbreviations: | ||
| XP | = | xeroderma Pigmentosum |
| TTD | = | trichothiodystrophy |
| CS | = | Cockayne syndrome |
| NER | = | nucleotide excision Repair |
| UDS | = | unscheduled DNA synthesis |
| RRS | = | recovery of mRNA synthesis |
Acknowledgments
The authors would like to thank pathologists Niels Marcussen and Ole J. Clemmensen (Dept of Pathology, Odense University Hospital) for their invaluable efforts with renal and hair microscopy and description, respectively as well as immunologist Torben Barington for fruitful discussions and immunotesting.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Liang C, Kraemer KH, Morris A, Schiffmann R, Price VH, Menefee E, et al. Characterization of tiger-tail banding and hair shaft abnormalities in trichothiodystrophy. J Am Acad Dermatol 2005; 52:224 - 32; http://dx.doi.org/10.1016/j.jaad.2004.09.013; PMID: 15692466
- Sperling LC, DiGiovanna JJ. “Curly” wood and tiger tails: an explanation for light and dark banding with polarization in trichothiodystrophy. Arch Dermatol 2003; 139:1189 - 92; http://dx.doi.org/10.1001/archderm.139.9.1189; PMID: 12975162
- Natale V. A comprehensive description of the severity groups in Cockayne syndrome. Am J Med Genet A 2011; 155A:1081 - 95; http://dx.doi.org/10.1002/ajmg.a.33933; PMID: 21480477
- Broughton BC, Berneburg M, Fawcett H, Taylor EM, Arlett CF, Nardo T, et al. Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene. Hum Mol Genet 2001; 10:2539 - 47; http://dx.doi.org/10.1093/hmg/10.22.2539; PMID: 11709541
- Boyle J, Ueda T, Oh KS, Imoto K, Tamura D, Jagdeo J, et al. Persistence of repair proteins at unrepaired DNA damage distinguishes diseases with ERCC2 (XPD) mutations: cancer-prone xeroderma pigmentosum vs. non-cancer-prone trichothiodystrophy. Hum Mutat 2008; 29:1194 - 208; http://dx.doi.org/10.1002/humu.20768; PMID: 18470933
- Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience 2007; 145:1388 - 96; http://dx.doi.org/10.1016/j.neuroscience.2006.12.020; PMID: 17276014
- Faghri S, Tamura D, Kraemer KH, Digiovanna JJ. Trichothiodystrophy: a systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J Med Genet 2008; 45:609 - 21; http://dx.doi.org/10.1136/jmg.2008.058743; PMID: 18603627
- Itin PH, Sarasin A, Pittelkow MR. Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J Am Acad Dermatol 2001; 44:891 - 920, quiz 921-4; http://dx.doi.org/10.1067/mjd.2001.114294; PMID: 11369901
- Botta E, Nardo T, Lehmann AR, Egly JM, Pedrini AM, Stefanini M. Reduced level of the repair/transcription factor TFIIH in trichothiodystrophy. Hum Mol Genet 2002; 11:2919 - 28; http://dx.doi.org/10.1093/hmg/11.23.2919; PMID: 12393803
- Vermeulen W, Rademakers S, Jaspers NG, Appeldoorn E, Raams A, Klein B, et al. A temperature-sensitive disorder in basal transcription and DNA repair in humans. Nat Genet 2001; 27:299 - 303; http://dx.doi.org/10.1038/85864; PMID: 11242112
- Takayama K, Danks DM, Salazar EP, Cleaver JE, Weber CA. DNA repair characteristics and mutations in the ERCC2 DNA repair and transcription gene in a trichothiodystrophy patient. Hum Mutat 1997; 9:519 - 25; http://dx.doi.org/10.1002/(SICI)1098-1004(1997)9:6<519::AID-HUMU4>3.0.CO;2-X; PMID: 9195225
- de Boer J, de Wit J, van Steeg H, Berg RJ, Morreau H, Visser P, et al. A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol Cell 1998; 1:981 - 90; http://dx.doi.org/10.1016/S1097-2765(00)80098-2; PMID: 9651581
- Ueda T, Compe E, Catez P, Kraemer KH, Egly JM. Both XPD alleles contribute to the phenotype of compound heterozygote xeroderma pigmentosum patients. J Exp Med 2009; 206:3031 - 46; http://dx.doi.org/10.1084/jem.20091892; PMID: 19934020
- Sato H, Saito T, Kurosawa K, Ootaka T, Furuyama T, Yoshinaga K. Renal lesions in Cockayne’s syndrome. Clin Nephrol 1988; 29:206 - 9; PMID: 3365865
- Brooks BP, Thompson AH, Clayton JA, Chan CC, Tamura D, Zein WM, et al. Ocular manifestations of trichothiodystrophy. Ophthalmology 2011; 118:2335 - 42; http://dx.doi.org/10.1016/j.ophtha.2011.05.036; PMID: 21959366
- Jaspers NG, Raams A, Silengo MC, Wijgers N, Niedernhofer LJ, Robinson AR, et al. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet 2007; 80:457 - 66; http://dx.doi.org/10.1086/512486; PMID: 17273966
- Limsirichaikul S, Niimi A, Fawcett H, Lehmann A, Yamashita S, Ogi T. A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU). Nucleic Acids Res 2009; 37:e31; http://dx.doi.org/10.1093/nar/gkp023; PMID: 19179371
- Nakazawa Y, Yamashita S, Lehmann AR, Ogi T. A semi-automated non-radioactive system for measuring recovery of RNA synthesis and unscheduled DNA synthesis using ethynyluracil derivatives. DNA Repair (Amst) 2010; 9:506 - 16; http://dx.doi.org/10.1016/j.dnarep.2010.01.015; PMID: 20171149
- Jaspers NG, Raams A, Kelner MJ, Ng JM, Yamashita YM, Takeda S, et al. Anti-tumour compounds illudin S and Irofulven induce DNA lesions ignored by global repair and exclusively processed by transcription- and replication-coupled repair pathways. DNA Repair (Amst) 2002; 1:1027 - 38; http://dx.doi.org/10.1016/S1568-7864(02)00166-0; PMID: 12531012