Abstract
Ataxia oculomotor apraxia type 2 (AOA2) is a rare autosomal recessive disorder characterized by cerebellar atrophy, peripheral neuropathy, loss of Purkinje cells and elevated α-fetoprotein. AOA2 is caused by mutations in the SETX gene that codes for the high molecular weight protein senataxin. Mutations in this gene also cause dominant neurodegenerative disorders. Similar to that observed for other autosomal recessive ataxias, this protein protects the integrity of the genome against oxidative and other forms of DNA damage to reduce the risk of neurodegeneration. Senataxin functions in transcription termination and RNA splicing and it has been shown to resolve RNA/DNA hybrids (R-loops) that arise at transcription pause sites or when transcription is blocked. Recent data suggest that this protein functions at the interface between transcription and DNA replication to minimise the risk of collision and maintain genome stability. Our recent data using SETX gene-disrupted mice revealed that male mice were defective in spermatogenesis and were infertile. DNA double strand-breaks persisted throughout meiosis and crossing-over failed in SETX mutant mice. These changes can be explained by the accumulation of R-loops, which interfere with Holiday junctions and crossing-over. We also showed that senataxin was localized to the XY body in pachytene cells and was involved in transcriptional silencing of these chromosomes. While the defect in meiotic recombination was striking in these animals, there was no evidence of neurodegeneration as observed in AOA2 patients. We discuss here potentially different roles for senataxin in proliferating and post-mitotic cells.
Autosomal recessive cerebellar ataxias are a class of progressive neurodegenerative disorders that result from cerebellar atrophy and spinal tract dysfunction.Citation1 One of these, ataxia oculomotor apraxia type 2 (AOA2) is characterized by progressive cerebellar atrophy and peripheral neuropathy, oculomotor apraxia and elevated α-fetoprotein serum levels, with an onset between 10–20 y of age.Citation2-Citation4 Brain MRI reveals diffuse cerebellar atrophy and electroneuromyography confirms the peripheral neuropathy.Citation4 The major clinical features of this disorder are shown in . In a post-mortem AOA2 case, Criscuolo et. al.Citation5 observed reduced brain size and cerebellar atrophy which was most evident at the level of the vermis and anterior lobe; the cerebellar cortex had marked loss of Purkinje cells and brainstem and spinal cord were slightly reduced. Thus, as with other autosomal recessive ataxias, pathology in the cerebellum features strongly. However, unlike that for the related disorder ataxia telangiectasia (A-T), there is no evidence of increased cancer susceptibility in AOA2.
Table 1. Ataxia Oculomotor Apraxia Type 2 (AOA2): Clinical Features
The gene mutated in AOA2 was initially mapped to chromosome 9q34 and subsequently identified as SETX Citation6. SETX is predicted to code for a 2,667 amino acid protein (senataxin) that contains a highly conserved C-terminal seven-motif domain found in the superfamily 1 of DNA/RNA helicases and an N-terminal domain important for protein-protein interaction.Citation7,Citation8 Generally speaking, mutations in a single gene, such as SETX, gives rise to one syndrome, which, of course, may show heterogeneity depending on the nature and localization of the mutations. In the case of SETX, up to 4 different syndromes are associated with mutations in this gene (). Juvenile amyotrophic lateral sclerosis (ALS4) is a form of juvenile ALS characterized by distal muscle weakness and atrophy, normal sensation and pyramidal tract signs. The ALS4 locus maps to chromosome 9q34. Chen et. al.Citation9 detected missense mutations in a single allele in the SETX gene (which maps to this locus) which segregated with the disease. Subsequent studies have detected SETX mutations in additional ALS4 patients. Heterozygous SETX gene mutations were also detected in patients with autosomal dominant proximal spinal muscular atrophy.Citation10 These patients showed proximal and distal muscular atrophy and pareses. While there was overlap with ALS4, this appeared to be a discrete entity. A dominant SETX mutation, causing a cerebellar phenotype termed tremor-ataxia syndrome, has been described for a mother and daughter. These patients showed cerebellar atrophy, oculomotor defects and tremor but no evidence of peripheral neuropathy or pyramidal signs.Citation11 In short, mutations in SETX can give rise to both dominant and recessive disorders with some overlap in features. A greater insight into the function of senataxin and the proteins it interacts with will help to resolve the quandary of several distinct disorders from mutations in a single gene.
Table 2. Mutations in SETX give rise to different neurodegenerative disorders
Senataxin shares extensive homology with the yeast Saccharomyces cerevisiae splicing endonuclease 1 protein (Sen1p), which possesses helicase activity, and is involved in the processing of tRNA, rRNA, small nuclear and small nucleolar RNA.Citation12 Sen1p also interacts with Rad2, which is required for DNA repair, suggesting that the protein may be involved in protecting the genome.Citation13 We demonstrated that this might also be the case for senataxin by showing that AOA2 patient cells display sensitivity to DNA damaging agents such as H2O2, camptothecin and mitomycin C and the cells had elevated levels of oxidative DNA damage.Citation8 In support of a role for senataxin in the DNA damage response, it has also been demonstrated that telomere length is constitutively reduced in AOA2 lymphocytes and the rate of telomere shortening by DNA damage is increased in these cells.Citation14 Interaction of Sen1p with Rnt1p (an endoribonuclease required for RNA maturation) suggested that Sen1p is also involved in RNA processing and transcription.Citation13 We provided evidence for a similar role in human cells by identifying novel senataxin-interacting proteins, the majority of which are involved in transcription and RNA processing, including RNA polymerase II.Citation15 Binding of RNA polymerase II to candidate genes was significantly reduced in senataxin deficient cells and this was accompanied by decreased transcription of these genes, suggesting a role for senataxin in the regulation/modulation of transcription. RNA polymerase II-dependent transcription termination was defective in cells depleted of senataxin in keeping with the observed interaction of senataxin with poly(A) binding proteins 1 and 2. Splicing efficiency of specific mRNAs and alternate splice-site selection of both endogenous genes and artificial minigenes were altered in senataxin-depleted cells.Citation15 A role for senataxin in transcription elongation and termination is further supported by a report showing that cells with senataxin knockdown displayed an increase in RNA read-through and Pol II density downstream of the Poly (A) site and also exhibited increased levels of R-loop formation.Citation16 R-loops are RNA/DNA hybrids that form over transcription pause sites by interaction with a ssDNA template behind an elongating Pol II complex (). These structures are potentially harmful and can cause genomic instability if left unresolved.Citation16-Citation18 The yeast ortholog of senataxin Sen1p, has also been shown to protect its heavily transcribed genome from R-loop-mediated DNA damage.Citation19 More recently, Hazelbaker et. al.Citation20 showed that kinetic competition between elongating RNA pol II and Sen1p helicase likely explains the temporal and spatial window for early Pol II termination. Loss of Sen1p results in transient R-loop accumulation, giving rise to transcription-associated recombination and genome instability. More insight into this was provided recently by Alzu et. al.Citation21 who showed that Sen1p associates with DNA replication forks to protect their integrity across RNA Pol II – transcribed genes. Thus, Sen1p plays an important role in coordinating replication with transcription to protect the genome. Support for a similar role for the human ortholog, senataxin, was provided recently by Yuce-Petronczki and WestCitation22 who showed that senataxin localized to distinct nuclear foci in S/G2 phase cells and that the number of these foci increased in response to impaired replication. These data suggest that senataxin localizes to collision sites between the transcription apparatus and components of the replisome.
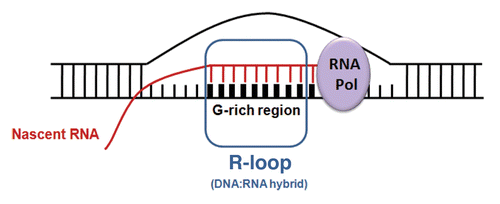
Figure 1. R-loop structure. R-loops form at sites when the RNA polymerase complex encounters G-rich sequences such as those found at transcription pause sites, CpG islands in promoter regions, repeat sequences and telomeric regions.Citation31,Citation32 Pairing of the nascent RNA remains with the ssDNA region behind the elongating RNA polymerase complex leads to R-loop (DNA/RNA hybrid) formation.
All of the investigations above have described a role for senataxin in preventing collision between DNA replication forks and ongoing transcription to preserve genome integrity in proliferating cells. However, the major phenotype in AOA2 patients is progressive neurodegeneration in post-mitotic tissue. Under those conditions, ongoing transcription will not encounter DNA replication forks. What then is the role of senataxin in the brain? Vantaggiato et. al.Citation23 provided evidence that senataxin plays a role in neuritogenesis and cytoprotection during neuronal differentiation which is mediated by fibroblast growth factor 8 . However, since this is a progressive disease, it is unlikely that the role of senataxin is restricted to development. To address this further, we disrupted the SETX gene in a mouse model for AOA2, which was the subject matter of our recent publication.Citation24 Unfortunately, we did not observe a neurodegenerative phenotype in the Setx−/− mice and there was no evidence of more subtle behavioral differences in these mice, which limited investigation into the nature of the defect in the brains of these mice. This was not altogether surprising since knockout of several of the genes causing autosomal recessive ataxias in humans fails to re-capitulate the phenotype in mouse models.Citation25 However, we observed that Setx−/− male mice were infertile and fertility was reduced in females. While there is no information on male fertility in AOA2, there are a few reports of hypogonadism in females. Criscuolo et. al.Citation4 reported two patients who entered menopause in early adulthood which was also observed in a separate study.Citation5 Ovarian failure has also been observed in a patient with AOA2Citation26 and another patient had a diagnosis of polycystic ovarian syndrome.Citation27 We subsequently showed that senataxin had an essential role in spermatogenesis in mice and in its absence these cells failed to progress past the pachytene stage of prophase 1 of meiosis.Citation24 The DNA double-strand breaks (DSB) introduced by Spo11 in readiness for meiotic recombination were inefficiently repaired on autosomes, resulting in a failure to complete crossing-over. During the process of crossing-over, autosomes remain transcriptionally active, so it was possible that in the absence of senataxin, R-loops would accumulate in the vicinity of unrepaired DNA DSB leading to collapse of Holiday junctions and inhibition of the crossing-over step. Indeed this was the case since we detected elevated levels of R-loops in both spermatocyte spreads and testes sections (). Wild-type mice showed a very much reduced level of signal. So in the case of spermatocyte differentiation, the R-loops that accumulate in the absence of senataxin appear to collide with Holiday junctions rather than with advancing replication forks. We also screened for the presence of R-loops in the brains of Setx mutant mice but failed to detect these structures by immunofluorescence (unpublished data). This was not altogether surprising since neither DNA replication nor DNA recombination is taking place in this tissue. It is possible that persistence of DNA damage in post-mitotic cells might lead to the accumulation of these structures, which in turn could contribute to the neurodegenerative changes in AOA2 patients. However, we and others have provided evidence for a broader role for senataxin in transcription and other cellular processes. Senataxin plays an important role in transcription termination to prevent RNA readthrough, which may or may not be related to R-loop resolution.Citation15,Citation16 The presence of significant readthrough of mRNA may lead to inefficient or aberrant protein synthesis and consequently cell toxicity. Senataxin has also been shown to play a role in the regulation of splicingCitation15 and deficiency of the SR splicing factor ASF/SF2 leads to R-loop accumulation and genome instability.Citation18 This in turn may interfere with the fidelity of the transcriptome in AOA2 cerebellum.
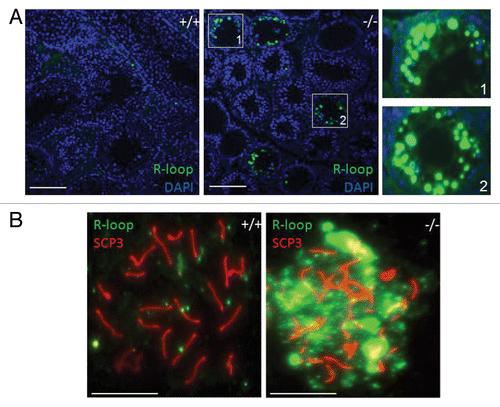
Figure 2. Accumulation of R-loops in Setx−/− germ cells. (A) Immunostaining of testes cross-sections from wildtype (+/+) and Setx knockout (−/−) mice with the S9.6 DNA/RNA (R-loop) antibody. Nuclei were stained with Hoechst 33342. Scale bar, 100µm. Regions 1 and 2 show magnifications. (B) Immunostaining of pachytene spermatocytes from wildtype (+/+) and Setx knockout (−/−) with S9.6 antibody shows a dramatic accumulation of R-loops in senataxin deficient germ cells. SCP3 was used to stain for the synaptonemal complex and identify pachytene stage cells. Scale bar, 20 µm.
AOA2 is just one of several neurodegenerative disorders characterized by defects in RNA metabolism that impact either gene transcription, pre-mRNA splicing, ribonucleoprotein complex formation, mRNA transport, RNA translation or RNA degradation.Citation28 One form of the motor neuron disease, amyotrophic lateral sclerosis (ALS) is caused by defects in TDP-43 and FUS/TLS, both of which contain RNA-binding motifsCitation29 and spinal muscular atrophy (SMA) is caused by deletion or mutation in survival of motor neuron 1 (SMN1). Profound loss of splicesome integrity is a critical mechanism common to ALS and SMA.Citation30 It is also of interest that we previously identified SMN as one of the proteins that interact with senataxin, pointing to an overlapping role in RNA processing.Citation15
In summary, much progress has been made recently on the function of senataxin and its yeast ortholog Sen1p in resolving potential conflict between transcription and DNA replication in proliferating cells. Resolution of R-loops is prominent in that role but it is evident that senataxin has a broader involvement in RNA processing (). Its role in post-mitotic cells is not clear, but it is likely that this will be on some aspect of RNA metabolism to protect the integrity of the genome/transcriptome. What remains intriguing is how mutations in a single gene SETX, often in close proximity to one another, can cause both autosomal dominant and recessive disorders. However, while mutations in SETX give rise to what appears to be 4 different syndromes, it is also evident that there is some overlap across these disorders. To date, several senataxin-interacting proteins have been identified, all of which are involved in some aspect of RNA processing.Citation15 Mutations in some of these also give rise to other neurodegenerative disorders.Citation28 It seems likely that these proteins function in complexes to control RNA metabolism and different mutations in the various subunits may impact differently on the function of the complex(es), giving rise to the heterogeneity of neurodegenerative disorders observed. The challenge that lies ahead is to understand the relationships of these proteins to one another in the complexes and how they function to control processes in neurons and other cell types in the brain to minimise the risk of neurodegeneration.
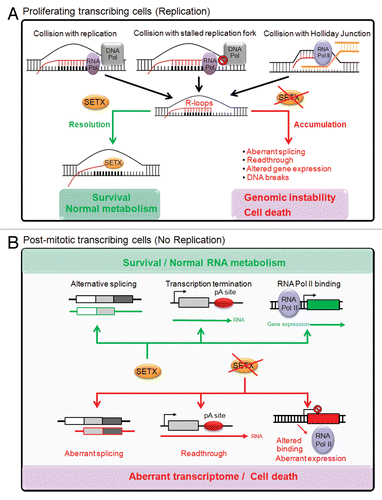
Figure 3. Protection of the genome by senataxin in proliferating vs. post-mitotic cells. (A) In proliferating cells, collision of the transcription apparatus (RNA Pol) with replication forks (DNA Pol), stalled replication forks following DNA damage exposure, or Holiday junctions during homologous recombination, lead to the formation and accumulation of R-loop structures. In the presence of senataxin, R-loops are effectively resolved by its putative DNA/RNA helicase activity, thus leading to normal cellular metabolism and cell survival. In the absence of senataxin, R-loops accumulate and impact on RNA metabolism through the alteration of mRNA splicing, the inhibition of transcription termination, the promotion of readthrough, the alteration of gene expression and the formation of DNA breaks. The accumulation of these defects drives genomic instability and ultimately cell death. (B) In contrast, in post-mitotic cells, such as neurons, the absence of DNA replication and homologous recombination, and the lack of R-loops accumulation suggest senataxin’s role in protecting the genome may be directly due to its effect on mRNA splicing, transcription termination and the modulation of gene expression through its interaction with RNA binding proteins.
Acknowledgments
We thank the Australian National Health and Medical Research Council for funding support and Tracey Laing for assistance in preparation of this manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Palau F, Espinós C. Autosomal recessive cerebellar ataxias. Orphanet J Rare Dis 2006; 1:47; http://dx.doi.org/10.1186/1750-1172-1-47; PMID: 17112370
- Le Ber I, Bouslam N, Rivaud-Péchoux S, Guimarães J, Benomar A, Chamayou C, et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain 2004; 127:759 - 67; http://dx.doi.org/10.1093/brain/awh080; PMID: 14736755
- Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol 2007; 6:245 - 57; http://dx.doi.org/10.1016/S1474-4422(07)70054-6; PMID: 17303531
- Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, et al. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain 2009; 132:2688 - 98; http://dx.doi.org/10.1093/brain/awp211; PMID: 19696032
- Criscuolo C, Chessa L, Di Giandomenico S, Mancini P, Saccà F, Grieco GS, et al. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology 2006; 66:1207 - 10; http://dx.doi.org/10.1212/01.wnl.0000208402.10512.4a; PMID: 16636238
- Moreira MC, Klur S, Watanabe M, Németh AH, Le Ber I, Moniz JC, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 2004; 36:225 - 7; http://dx.doi.org/10.1038/ng1303; PMID: 14770181
- Chen YZ, Hashemi SH, Anderson SK, Huang Y, Moreira MC, Lynch DR, et al. Senataxin, the yeast Sen1p orthologue: characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease. Neurobiol Dis 2006; 23:97 - 108; http://dx.doi.org/10.1016/j.nbd.2006.02.007; PMID: 16644229
- Suraweera A, Becherel OJ, Chen P, Rundle N, Woods R, Nakamura J, et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol 2007; 177:969 - 79; http://dx.doi.org/10.1083/jcb.200701042; PMID: 17562789
- Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 2004; 74:1128 - 35; http://dx.doi.org/10.1086/421054; PMID: 15106121
- Rudnik-Schöneborn S, Eggermann T, Kress W, Lemmink HH, Cobben JM, Zerres K. Clinical utility gene card for: proximal spinal muscular atrophy. Eur J Hum Genet 2012; 20; http://dx.doi.org/10.1038/ejhg.2012.62; PMID: 22510849
- Bassuk AG, Chen YZ, Batish SD, Nagan N, Opal P, Chance PF, et al. In cis autosomal dominant mutation of Senataxin associated with tremor/ataxia syndrome. Neurogenetics 2007; 8:45 - 9; http://dx.doi.org/10.1007/s10048-006-0067-8; PMID: 17096168
- Ursic D, Himmel KL, Gurley KA, Webb F, Culbertson MR. The yeast SEN1 gene is required for the processing of diverse RNA classes. Nucleic Acids Res 1997; 25:4778 - 85; http://dx.doi.org/10.1093/nar/25.23.4778; PMID: 9365256
- Ursic D, Chinchilla K, Finkel JS, Culbertson MR. Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res 2004; 32:2441 - 52; http://dx.doi.org/10.1093/nar/gkh561; PMID: 15121901
- De Amicis A, Piane M, Ferrari F, Fanciulli M, Delia D, Chessa L. Role of senataxin in DNA damage and telomeric stability. DNA Repair (Amst) 2011; 10:199 - 209; http;//dx.doi.org/10.1016/j.dnarep/2010.10.012 http://dx.doi.org/10.1016/j.dnarep.2010.10.012; PMID: 21112256
- Suraweera A, Lim Y, Woods R, Birrell GW, Nasim T, Becherel OJ, et al. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet 2009; 18:3384 - 96; http://dx.doi.org/10.1093/hmg/ddp278; PMID: 19515850
- Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 2011; 42:794 - 805; http://dx.doi.org/10.1016/j.molcel.2011.04.026; PMID: 21700224
- Aguilera A, Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet 2008; 9:204 - 17; http://dx.doi.org/10.1038/nrg2268; PMID: 18227811
- Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005; 122:365 - 78; http://10.1016/j.cell.2005.06.008 http://dx.doi.org/10.1016/j.cell.2005.06.008; PMID: 16096057
- Mischo HE, Gómez-González B, Grzechnik P, Rondón AG, Wei W, Steinmetz L, et al. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol Cell 2011; 41:21 - 32; http://dx.doi.org/10.1016/j.molcel.2010.12.007; PMID: 21211720
- Hazelbaker DZ, Marquardt S, Wlotzka W, Buratowski S. Kinetic competition between RNA Polymerase II and Sen1-dependent transcription termination. Mol Cell 2013; 49:55 - 66; PMID: 23177741
- Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 2012; 151:835 - 46; http://dx.doi.org/10.1016/j.cell.2012.09.041; PMID: 23141540
- Yüce O, West SC. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol 2013; 33:406 - 17; http://dx.doi.org/10.1128/MCB.01195-12; PMID: 23149945
- Vantaggiato C, Bondioni S, Airoldi G, Bozzato A, Borsani G, Rugarli EI, et al. Senataxin modulates neurite growth through fibroblast growth factor 8 signalling. Brain 2011; 134:1808 - 28; http://dx.doi.org/10.1093/brain/awr084; PMID: 21576111
- Becherel OJ, Yeo AJ, Stellati A, Heng EY, Luff J, Suraweera AM, et al. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet 2013; 9:e1003435; http://dx.doi.org/10.1371/journal.pgen.1003435; PMID: 23593030
- Lavin MF. The appropriateness of the mouse model for ataxia-telangiectasia: Neurological defects but no neurodegeneration. DNA Repair (Amst) 2013; In press http://dx.doi.org/10.1016/j.dnarep.2013.04.014; PMID: 23731731
- Lynch DR, Braastad CD, Nagan N. Ovarian failure in ataxia with oculomotor apraxia type 2. Am J Med Genet A 2007; 143A:1775 - 7; http://dx.doi.org/10.1002/ajmg.a.31816; PMID: 17593543
- Fogel BL, Lee JY, Perlman S. Aberrant splicing of the senataxin gene in a patient with ataxia with oculomotor apraxia type 2. Cerebellum 2009; 8:448 - 53; http://dx.doi.org/10.1007/s12311-009-0130-8; PMID: 19727998
- Strong MJ, Volkening K. TDP-43 and FUS/TLS: sending a complex message about messenger RNA in amyotrophic lateral sclerosis?. FEBS J 2011; 278:3569 - 77; http://dx.doi.org/10.1111/j.1742-4658.2011.08277.x; PMID: 21810174
- Fiesel FC, Kahle PJ. TDP-43 and FUS/TLS: cellular functions and implications for neurodegeneration. FEBS J 2011; 278:3550 - 68; http://dx.doi.org/10.1111/j.1742-4658.2011.08258.x; PMID: 21777389
- Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med 2013; 5:221 - 34; http://dx.doi.org/10.1002/emmm.201202303; PMID: 23255347
- Wongsurawat T, Jenjaroenpun P, Kwoh CK, Kuznetsov V. Quantitative model of R-loop forming structures reveals a novel level of RNA-DNA interactome complexity. Nucleic Acids Res 2012; 40:e16; http://dx.doi.org/10.1093/nar/gkr1075; PMID: 22121227
- Ginno PA, Lott PL, Christensen HC, Korf I, Chédin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 2012; 45:814 - 25; http://dx.doi.org/10.1016/j.molcel.2012.01.017; PMID: 22387027