Abstract
Giant axonal neuropathy (GAN)Citation1 is a rare autosomal recessive neurological disorder caused by mutations in the GAN gene that encodes gigaxonin, a member of the BTB/Kelch family of E3 ligase adaptor proteins.Citation1 This disease is characterized by the aggregation of Intermediate Filaments (IF)—cytoskeletal elements that play important roles in cell physiology including the regulation of cell shape, motility, mechanics and intra-cellular signaling. Although a range of cell types are affected in GAN, neurons display the most severe pathology, with neuronal intermediate filament accumulation and aggregation; this in turn causes axonal swellings or “giant axons.” A mechanistic understanding of GAN IF pathology has eluded researchers for many years. In a recent studyCitation1 we demonstrate that the normal function of gigaxonin is to regulate the degradation of IF proteins via the proteasome. Our findings present the first direct link between GAN mutations and IF pathology; moreover, given the importance of IF aggregations in a wide range of disease conditions, our findings could have wider ramifications.
Giant Axonal Neuropathy (GAN) (OMIM # 256850) is a rare, autosomal recessive disorder caused by mutations in the GAN gene that encodes gigaxonin.Citation2,Citation3 More than 30 distinct GAN mutations are known to cause this devastating disease. Patients first show muscle weakness, gait disturbances and sensory complaints, typically when they are just toddlers. It progresses slowly, with patients becoming wheelchair-bound in early childhood. The range of symptoms is quite variable, with some patients displaying problems with ocular motility, seizures and cerebellar ataxia. Eventually patients lose control of vital functions like breathing and swallowing; they require ventilators and feeding tubes before finally succumbing from respiratory compromise, usually in their second decade of life.
GAN was first reported as a distinct disease in the early 1970s, based on the characteristic engorgement of nerve cells and aggregation of intermediate filaments (IF) . These pathologic findings, typically identified by sural nerve biopsy, were the only method of establishing the diagnosis until the GAN gene was discovered.Citation4,Citation5 Neurofilaments belong to the large family of IF proteins that form 10 nm cytoskeletal intermediate filaments. IF have been classified into five distinct types based on their primary sequence, their intron-exon gene structure and the specific cells in which they are expressed.Citation6 Types I and II IF constitute the epithelial keratins; type III include vimentin in fibroblasts, desmin in muscles, GFAP in astrocytes and peripherin in neurons. Neurons also express Type IV neuronal IF consisting of NF-heavy (200kDa), medium (145 kDa) and light (68 kDa) chains and α internexin. Finally, Type V IF consists of the nuclear lamins that form a network subjacent to the nuclear envelope and are also dispersed throughout the nucleoplasm.
Interestingly, GAN is not just a disease of neurofilaments; rather it affects several IF types, the most visible being abnormalities in keratin IF that lead to characteristic kinky hair—noted in the earliest descriptions of the disease.Citation7,Citation8 The other cell type that shows major pathological changes are dermal fibroblasts containing large vimentin IF aggregates.Citation9-Citation14 Because of their accessibility and availability in quantities sufficient to carry out biochemical and cell biological experiments, we focused our attention on these dermal fibroblasts obtained from GAN patients. This enabled us to concentrate on the relationships between gigaxonin and the Type III IF protein vimentin, which forms relatively simple homopolymeric IF in contrast to Type IV IF, such as neurofilaments, which are complex heteropolymers.Citation6
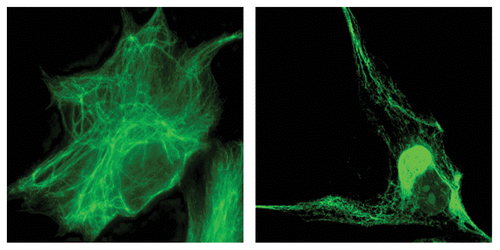
Microscopic studies of these fibroblasts confirmed the presence of abnormally bundled and aggregated IF, which in many cases, form a discrete perinuclear body (). The enormous size of some of these bodies, frequently larger than the nucleus, distorts the organization of cellular organelles such as the endoplasmic reticulum and mitochondria. Evidence linking these abnormal accumulations of IF to a loss of gigaxonin function came from recent observations that mouse embryonic fibroblasts (MEFs) from the GAN null mouse model contain similar vimentin IF aggregates.Citation1 Histological studies of these mice have also revealed neuropathological features similar to those found in GAN patients, including accumulation of neuronal IF proteins with a significant loss of peripheral neurons and muscle atrophy.Citation15,Citation16 We next speculated that if the aggregates are caused by a loss of normally functioning gigaxonin, expressing exogenous gigaxonin might rid the GAN patients’ cells of the aggregates. If so, this could then represent a valid therapeutic approach, as is being attempted.Citation17 To our surprise, we found that overexpression of gigaxonin has an even more dramatic effect: not only did it clear the cells of aggregates, but it cleared the cell not only of IF aggregates but also of all vimentin proteins. This degradation occurred coincidently with the disassembly of long, mature IF into non-filamentous particles over a three day period. Intriguingly, the IF aggregates were the last to be cleared. This was a specific effect on the IF cytoskeletal system, since other cytoskeletal networks, such as the actin and microtubule networks, appeared unperturbed.
Figure 1. Left: A control fibroblast from an unaffected individual shows the well-dispersed IF network and the absence of aggregates. Right: A GAN patient fibroblast showing perinuclear vimentin IF aggregation.
So how exactly does gigaxonin lead to IF clearance? Of course we already had an important clue. Gigaxonin belongs to the growing BTB-KELCH domain family of E3 ligase adaptor proteins.Citation3,Citation18 These proteins act by targeting substrates for ubiquitination and degradation through the proteasome. The KELCH domain interacts with the protein to be degraded, while the BTB domain interacts with Cullin-3, a component of the catalytic ubiquitin ligase complex.Citation18-Citation21 We immediately hypothesized that the role of gigaxonin might be to regulate the ubiquitination of vimentin for degradation via the ubiquitin-proteasome system (UPS) and that a lack of this function in GAN could explain the abnormal buildup of IF.
To test this hypothesis, we first demonstrated that wild-type gigaxonin, and not disease causing mutants, can clear vimentin from cells as determined by immunofluorescence and immunoblotting.Citation1 Second, we showed that the clearance of vimentin is not due to either altered levels or stability of its mRNA. Third, we demonstrated that vimentin clearance is mediated by the proteasome, since inhibition by MG-132 prevents its degradation. Using drugs to inhibit lysosomes and autophagosomes, we essentially eliminated the possibility that these two pathways for protein degradation might be involved in the clearance of vimentin. Finally, we found that vimentin physically binds gigaxonin, as would be expected for an E3 ligase adaptor. By making deletions in vimentin, we discovered that gigaxonin interacts with vimentin via its highly α-helical central rod domain. This finding is extremely interesting in light of the conserved nature of this domain across all classes of IF, and thus, might suggest how gigaxonin exerts its effects on a range of IF types. Although, we have not yet performed a systemic evaluation of other IF proteins, we have found that gigaxonin is efficient at clearing the neurofilament proteins NF-L and peripherin.
Our experiments bring up several interesting questions and point us in several directions for future studies. From a mechanistic viewpoint, we can now begin to ask how polymerized IF are degraded under normal conditions. It is possible, for instance, that gigaxonin clears a small pool of soluble IF protein and that this mechanism shifts the equilibrium toward IF depolymerization. Another possibility is that gigaxonin binds vimentin that is still in a polymer form, disassembles it through unknown biochemical mechanisms and then targets the disassembly products for degradation. Regardless, in either of these scenarios, the polymeric network is maintained by newly synthesized vimentin. It is also not clear whether IF proteins are directly targeted for ubiquitination by gigaxonin. Indeed, in our studies to date, we have found no evidence of vimentin ubiquitination. It is possible that our assays aimed at detecting ubiquitination are not sensitive enough or that there is a very active de-ubiquitinase that makes the detection of IF ubiquitination difficult. But it is also possible that gigaxonin links vimentin to an unknown protein which is ubiquitinated, hereby targeting vimentin to the proteasome for degradation. Precedents for this latter type of mechanism have been recently reported.Citation22,Citation23
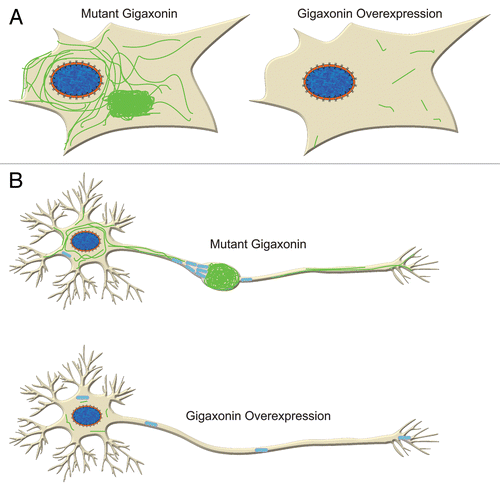
Despite numerous unanswered questions, we believe that our results convincingly demonstrate that gigaxonin is an important player in the degradation of IF. Clearly, this is an important insight for GAN. We speculate that gigaxonin might play a much broader role in a number of diseases where IF accumulate, ranging from liver diseases that display keratin accumulations (called Mallory bodies),Citation24 Alexander disease with glial fibrillary acidic protein (GFAP) accumulations (called Rosenthal fibers)Citation25 and a whole host of diseases in the nervous system including Alzheimer disease, Parkinson disease, Charcot-Marie-Tooth disease and many others that have neuro- IF accumulations.Citation26,Citation27 Understanding GAN, albeit an extremely rare disease, might well lead to the clues required to truly understand these various disorders. It should also be noted that very little is known about the normal turnover of IF proteins in general, so GAN has already spun off an exciting new insight into the turnover and degradation of several types of IF proteins long thought to assemble into one of the most stable cytoskeletal networks in vertebrate cells. Finally, our findings raise the possibility that one therapeutic avenue for GAN and related diseases might be to deliver wild-type GAN or to screen for small molecules that take apart the large aggregates of IF. This overexpression should remove aggregates of IF not only in fibroblasts, but also in neurons, allowing the free movement of organelles, such as mitochondria ().
Figure 2. Gigaxonin overexpression clears IF in fibroblasts (A) and neurons (B). In neurons this is likely to lead to improvement of axonal transport of organelles such as mitochondria as they are no longer hindered by IF aggregation.
Acknowledgments
We acknowledge the contributions of all the co-authors of our recent JCI publication. We thank Lori Sames and the Hannah’s Hope Fund for providing support to enable us to carry out this research as well as NIH 1P01GM096971–01 to R.D.G. and NIH R01 NS062051 to P.O. We thank Dale Shumaker for help with the artwork.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Mahammad S, Murthy SN, Didonna A, Grin B, Israeli E, Perrot R, et al. Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J Clin Invest 2013; 123:1964 - 75; http://dx.doi.org/10.1172/JCI66387; PMID: 23585478
- Yang Y, Allen E, Ding J, Wang W. Giant axonal neuropathy. Cell Mol Life Sci 2007; 64:601 - 9; http://dx.doi.org/10.1007/s00018-007-6396-4; PMID: 17256086
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet 2000; 26:370 - 4; http://dx.doi.org/10.1038/81701; PMID: 11062483
- Berg BO, Rosenberg SH, Asbury AK. Giant axonal neuropathy. Pediatrics 1972; 49:894 - 9; PMID: 4339350
- Carpenter S, Karpati G, Andermann F, Gold R. Giant axonal neuropathy. A clinically and morphologically distinct neurological disease. Arch Neurol 1974; 31:312 - 6; http://dx.doi.org/10.1001/archneur.1974.00490410060005; PMID: 4153361
- Eriksson JE, Dechat T, Grin B, Helfand B, Mendez M, Pallari HM, et al. Introducing intermediate filaments: from discovery to disease. J Clin Invest 2009; 119:1763 - 71; http://dx.doi.org/10.1172/JCI38339; PMID: 19587451
- Peiffer J, Schlote W, Bischoff A, Boltshauser E, Müller G. Generalized giant axonal neuropathy: a filament-forming disease of neuronal, endothelial, glial, and schwann cells in a patient without kinky hair. Acta Neuropathol 1977; 40:213 - 8; http://dx.doi.org/10.1007/BF00691956; PMID: 602684
- Treiber-Held S, Budjarjo-Welim H, Reimann D, Richter J, Kretzschmar HA, Hanefeld F. Giant axonal neuropathy: a generalized disorder of intermediate filaments with longitudinal grooves in the hair. Neuropediatrics 1994; 25:89 - 93; http://dx.doi.org/10.1055/s-2008-1071592; PMID: 8072681
- Bomont P, Koenig M. Intermediate filament aggregation in fibroblasts of giant axonal neuropathy patients is aggravated in non dividing cells and by microtubule destabilization. Hum Mol Genet 2003; 12:813 - 22; http://dx.doi.org/10.1093/hmg/ddg092; PMID: 12668605
- Cleveland DW, Yamanaka K, Bomont P. Gigaxonin controls vimentin organization through a tubulin chaperone-independent pathway. Hum Mol Genet 2009; 18:1384 - 94; http://dx.doi.org/10.1093/hmg/ddp044; PMID: 19168853
- Klymkowsky MW, Plummer DJ. Giant axonal neuropathy: a conditional mutation affecting cytoskeletal organization. J Cell Biol 1985; 100:245 - 50; http://dx.doi.org/10.1083/jcb.100.1.245; PMID: 3880753
- Leung CL, Pang Y, Shu C, Goryunov D, Liem RK. Alterations in lipid metabolism gene expression and abnormal lipid accumulation in fibroblast explants from giant axonal neuropathy patients. BMC Genet 2007; 8:6; http://dx.doi.org/10.1186/1471-2156-8-6; PMID: 17331252
- Pena SD. Giant axonal neuropathy: intermediate filament aggregates in cultured skin fibroblasts. Neurology 1981; 31:1470 - 3; http://dx.doi.org/10.1212/WNL.31.11.1470; PMID: 6273766
- Pena SD, Opas M, Turksen K, Kalnins VI, Carpenter S. Immunocytochemical studies of intermediate filament aggregates and their relationship to microtubules in cultured skin fibroblasts from patients with giant axonal neuropathy. Eur J Cell Biol 1983; 31:227 - 34; PMID: 6315439
- Dequen F, Bomont P, Gowing G, Cleveland DW, Julien JP. Modest loss of peripheral axons, muscle atrophy and formation of brain inclusions in mice with targeted deletion of gigaxonin exon 1. J Neurochem 2008; 107:253 - 64; http://dx.doi.org/10.1111/j.1471-4159.2008.05601.x; PMID: 18680552
- Ganay T, Boizot A, Burrer R, Chauvin JP, Bomont P. Sensory-motor deficits and neurofilament disorganization in gigaxonin-null mice. Mol Neurodegener 2011; 6:25; http://dx.doi.org/10.1186/1750-1326-6-25; PMID: 21486449
- Mussche S, Devreese B, Nagabhushan Kalburgi S, Bachaboina L, Fox JC, Shih HJ, et al. Restoration of cytoskeleton homeostasis after gigaxonin gene transfer for giant axonal neuropathy. Hum Gene Ther 2013; 24:209 - 19; http://dx.doi.org/10.1089/hum.2012.107; PMID: 23316953
- Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, et al. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature 2003; 425:316 - 21; http://dx.doi.org/10.1038/nature01985; PMID: 13679922
- Perez-Torrado R, Yamada D, Defossez PA. Born to bind: the BTB protein-protein interaction domain. Bioessays 2006; 28:1194 - 202; http://dx.doi.org/10.1002/bies.20500; PMID: 17120193
- Furukawa M, He YJ, Borchers C, Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol 2003; 5:1001 - 7; http://dx.doi.org/10.1038/ncb1056; PMID: 14528312
- Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J 2004; 23:1681 - 7; http://dx.doi.org/10.1038/sj.emboj.7600186; PMID: 15071497
- Gonzalez SL, Stremlau M, He X, Basile JR, Münger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol 2001; 75:7583 - 91; http://dx.doi.org/10.1128/JVI.75.16.7583-7591.2001; PMID: 11462030
- Berezutskaya E, Bagchi S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26 S proteasome. J Biol Chem 1997; 272:30135 - 40; http://dx.doi.org/10.1074/jbc.272.48.30135; PMID: 9374493
- Denk H, Stumptner C, Zatloukal K. Mallory bodies revisited. J Hepatol 2000; 32:689 - 702; http://dx.doi.org/10.1016/S0168-8278(00)80233-0; PMID: 10782920
- Li R, Messing A, Goldman JE, Brenner M. GFAP mutations in Alexander disease. Int J Dev Neurosci 2002; 20:259 - 68; http://dx.doi.org/10.1016/S0736-5748(02)00019-9; PMID: 12175861
- Rudrabhatla P, Jaffe H, Pant HC. Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimer’s NFTs. FASEB J 2011; 25:3896 - 905; http://dx.doi.org/10.1096/fj.11-181297; PMID: 21828286
- Liu Q, Xie F, Siedlak SL, Nunomura A, Honda K, Moreira PI, et al. Neurofilament proteins in neurodegenerative diseases. Cell Mol Life Sci 2004; 61:3057 - 75; http://dx.doi.org/10.1007/s00018-004-4268-8; PMID: 15583867