Abstract
The toxicity of expanded transcripts in myotonic dystrophy type 1 (DM1) is mainly mediated by the disruption of alternative splicing. However, the detailed disease mechanisms in the central nervous system (CNS) have not been fully elucidated. In our recent study, we demonstrated that the accumulation of mutant transcripts in the CNS of a mouse model of DM1 disturbs splicing in a region-specific manner. We now discuss that the spatial- and temporal-regulated expression of splicing factors may contribute to the region-specific spliceopathy in DM1 brains. In the search for disease mechanisms operating in the CNS, we found that the expression of expanded CUG-containing RNA affects the expression and phosphorylation of synaptic vesicle proteins, possibly contributing to DM1 neurological phenotypes. Although mediated by splicing regulators with a described role in DM1, the misregulation of synaptic proteins was not associated with missplicing of their coding transcripts, supporting the view that DM1 mechanisms in the CNS have also far-reaching implications beyond the disruption of a splicing program.
Myotonic Dystrophy and the Central Nervous System
Myotonic dystrophy type 1 (DM1) is the most common form of inherited muscular dystrophy in adults, with a worldwide incidence of 1 in 8,000 individuals. DM1 is a typical multisystemic disease, affecting a large number of tissues and organs in the human body.Citation1 The central nervous system (CNS) is compromised to different extents in adult, juvenile and congenital forms of the disease. Several neuropsychological symptoms have been reported in adult-onset DM1 patients, such as excessive daytime sleepiness and fatigue, visuoconstructive impairment, attention deficits, reduced initiative and apathy, increased anxiety and anhedonia, as well as reduced intelligence quotients. Marked mental retardation and delayed psychomotor development are found in the congenital cases.Citation2 In addition to the clinical evidence, imaging and histopathological techniques have also illustrated brain dysfunction in DM1. MRI scans have revealed that white and gray matter are affected in DM1 brains, while PET-SPECT imaging techniques revealed deficits in brain glucose metabolism and hypoperfusion.Citation3,Citation4 The histopathological distribution of tau protein in the brain revealed the accumulation of pathogenic protein isoforms in DM1 individuals,Citation5 in association with changes in the alternative splicing of tau transcripts,Citation6 resulting in the classification of DM1 as a tauopathy.
The neurological manifestations of the disease are highly debilitating and have a tremendous impact on the quality of life of DM1 patients and their families. As a result of their intellectual impairment and behavioral deficits, DM1 patients experience low education achievements, low employment, poor familial environment, as well as social, economic and material deprivation.Citation7
Unraveling the Molecular Mechanisms of DM1 in the CNS
DM1 is caused by the abnormal expansion of a non-coding CTG trinucleotide repeat in the 3′UTR of the DMPK gene.Citation8 Experimental evidence supports a prevailing model of disease pathogenesis, in which the DM1 phenotype is mainly mediated by a deleterious gain-of-function of expanded DMPK transcripts.Citation9,Citation10 CUG repeat-containing expanded transcripts form secondary RNA structures that bind to and sequester muscleblind-like proteins (MBNL) into ribonuclear inclusions or nuclear RNA foci,Citation11 and upregulate the CUG/Elav-like family (CELF) proteins.Citation12 Given the antagonistic role of MBNL and CELF proteins in the control of a developmentally regulated splicing program, CUG-associated RNA toxicity results in the aberrant expression of embryonic isoforms in adult skeletal muscle and heart.Citation12,Citation13 Typical DM1 symptoms, such as myotonia, muscle weakness and insulin resistance, are explained by abnormal splicing of the CLCN1 chloride channel,Citation14,Citation15 BIN1 bridging integrator proteinCitation16 and the insulin receptor,Citation17 respectively. Although splicing defects have been described in human DM1 brains,Citation18 we do not understand the functional impact of MAPT/TAU, GRIN/NMDAR1 and APP RNA missplicing in DM1 neuropathophysiology. Nor do we know the cell populations, neuronal circuits, molecular pathways and neurological functions that are primarily disturbed in DM1 brains.
In addition to spliceopathy, evidence has shown that additional elements may contribute to (or at least modify) disease pathogenesis, such as chromatin rearrangements within the DM1 locus, leaching of transcription factors away from active chromatin, dysregulated miRNA metabolism, altered protein translation, and accumulation of toxic peptides resulting from non-conventional repeat-associated RNA translation.Citation9,Citation10
Recreating RNA Toxicity in the CNS of DM1 Transgenic Mice
Given the compromised function of the CNS in DM1 patients and the impact of the neurological symptoms on their daily life, a growing effort has been made to unravel the mechanisms of DM1 neuropathogenesis over the last few years. We have been tackling this question by using DMSXL transgenic mice, previously generated in our laboratory.Citation19,Citation20 These animals carry a large fragment of the human DM1 locus containing more than 1000 CTG repeats in the 3′UTR of the DMPK gene. Homozygous DMSXL mice produce sufficient toxic RNA transcripts to reproduce some critical and highly relevant molecular features of DM1, such as RNA foci accumulation and missplicing, as well as muscle phenotypes.Citation21 In order to investigate whether RNA toxicity extended to the CNS, we have studied nuclear RNA foci accumulation and found CUG-containing ribonuclear inclusions in DMSXL mice, not only in the brain, but also in the spinal cord.Citation22 RNA foci were particularly abundant in the frontal cortex and in some neuronal nuclei of the brainstem—two brain regions that are considered to play critical roles in the development of some of the most characteristic neurological symptoms of DM1. The nuclear accumulation of toxic RNA foci was associated with sequestration of MBNL proteins, upregulation of CELF proteins and resulted in the disruption of alternative splicing in DMSXL frontal cortex and brainstem.Citation22
While most of the exons misspliced in DMSXL brains were previously reported as misregulated in post-mortem DM1 brains,Citation6,Citation18 the missplicing of GRIN1/NMDAR1 exon 21, MBNL1 and MBNL2 exon 7 in human patients was poorly documented. To confirm that the splicing changes of these exons in DMSXL mice recreate relevant molecular events characteristic of DM1, we have now validated these abnormalities in human DM1 frontal cortex and brainstem. The RT-PCR analysis confirmed increased inclusion of GRIN1/NMDAR1 exon 21, MBNL1 exon 7 and MBNL2 exon 7 in human DM1 frontal cortex and brainstem, relative to non-DM1 control individuals (), suggesting that the DMSXL splicing changes detected mimic true DM1 molecular features. Our RT-PCR analysis also revealed that missplicing was more pronounced in human frontal cortex than in brainstem, particularly for MBNL1 and MBNL2 transcripts. Coincidentally, the missplicing of Mbnl1 and Mbnl2 are among the most noticeable and reproducible defects detected in the brain of DMSXL mice, and may provide useful molecular biomarkers for the pre-clinical assessment of therapies aiming to correct DM1-spliceopathy in the CNS.
Figure 1. DM1-associated RNA spliceopathy and the expression of splicing regulators in the CNS. (A) Representative splicing analysis of candidate genes in the frontal cortex and brainstem of DM1 patients (n = 4–5) and non-DM controls (n = 2) by RT-PCR (Table S1). The alternative exons are indicated on the right of each panel. The higher molecular product results from the amplification of the transcript that includes the alternative exon (+); the lower band does not include the alternative exon (-). (B) Western blot analysis of MBNL1 and MBNL2 in the frontal cortex and brainstem of wild-type animals (Table S1), following long migration times to increase resolution and separation of protein isoforms. β-Actin was used as loading control. (C) Representative splicing analysis of alternative exons of Grin1/Nmdar1, Mapt/Tau, Mbnl1 and Mbnl2 by RT-PCR (Table S1) in the spinal cord of one-month-old DMSXL homozygous mice and wild-type controls (n = 3, per genotype). Newborn splicing profiles (P1) were determined in a cDNA pool prepared from three wild-type animals. Notably, DMSXL spinal cord exhibited increased exclusion of both Grin1/Nmdar1 exon 5 and Mapt/Tau exon 10, as well as a mild increase in the inclusion of both Mbnl1 exon 7 and Mbnl2 exon 7. (D) Western blot analysis of splicing regulators throughout wild-type embryonic development (E12.5, E14.5, E18.5), in newborn (P1), postnatal day 8 (P8), and adult mice aged one (M1), four (M4) and 10 (M10) months (Table S2). Protein extracts from three individual animals were pooled for each developmental stage, electrophoresed and analyzed in three independent assays. Representative western blots are shown for frontal cortex and brainstem. Over-exposed of MBNL1 and MBNL2 western blots are shown to confirm low protein expression during embryonic stages and at P1. (E) Western blot analysis of hnRNP H in the frontal cortex and brainstem of DMSXL and wild-type animals at one month of age (Table S2), when missplicing dysregulation is more pronounced. Protein extracts from three individual animals of each genotype were pooled. β-Tubulin was used as loading control.
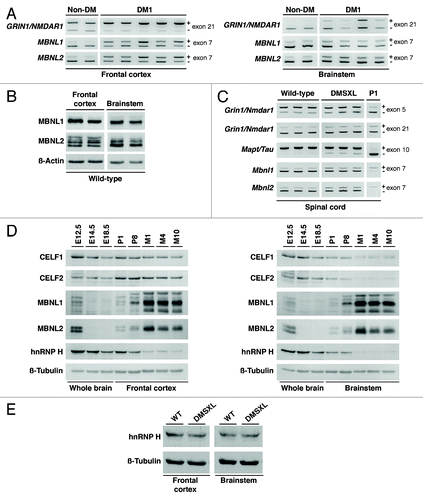
The extent and nature of the spliceopathy in DMSXL brains is region-specific, as illustrated by Grin1/Nmdar1 transcripts. Grin1/Nmdar1 exon 5 is abnormally excluded in DMSXL brainstem (but not in frontal cortex), while exon 21 is abnormally excluded in DMSXL frontal cortex (while it remains unaffected in brainstem).Citation22 The regional splicing defects of this gene (and others) might be the consequence of a spatially regulated splicing program. In support of physiological region-specific mechanisms of splicing regulation, we have found that the distribution of Grin1/Nmdar1 isoforms varied between frontal cortex and brainstem in wild-type animals: while exon 5 is preferentially included in brainstem, exon 21 shows a much higher inclusion ratio in frontal cortex.Citation22 These differences suggest that the levels and/or activity of the key splicing regulators that control these events vary between different brain territories. We have addressed this hypothesis and found higher levels of CELF1 and CELF2 proteins in adult frontal cortex than in brainstem of wild-type mice. In contrast, the levels of MBNL proteins were higher in brainstem than in frontal cortex.Citation22 Additionally, the distribution of MBNL2 protein isoforms differed significantly between frontal cortex and brainstem: western blot immunodetection following long electrophoresis migration revealed high molecular weight isoforms that were specifically present in frontal cortex, as well as a higher expression of low molecular weight isoforms in brainstem (). Given the role of CELF and MBNL proteins in the control of alternative splicing in the CNS,Citation23-Citation25 these differences may contribute to the regional splicing profiles in wild-type brains, and to the region-specific susceptibility of alternative exons to the accumulation of toxic RNA transcripts in DMSXL mice. Consistent with this view, a role of CELF2 in the regional regulation of alternative splicing in the mouse brain has been previously reported.Citation26 Our data suggest that the regional distribution of different splicing isoforms in the brain is regulated by the interplay between multiple RNA-binding proteins, which include not only CELF family members, but also MBNL proteins.
Missplicing in DMSXL CNS was not confined to frontal cortex and brainstem areas of the brain, and it was found throughout the CNS to a variable extent, including the hippocampusCitation22 and spinal cord (), among other regions. These results demonstrate that CUG toxicity operates throughout the entire CNS. In agreement with this view, signs of neuronopathy, the expression of pathological forms of MAPT/tau protein and a reduced number of lumbar motor neurons were reported in the spinal cord of DMSXL mice.Citation27 However, the correlation between histopathology and splicing dysregulation in spinal cord remains to be further explored.
Expression of Splicing Regulators is Regulated during Mouse CNS Development
The missplicing events detected in DMSXL brains increased the expression of embryonic splicing profiles in adult animals, to a limited extent,Citation22 as previously reported in skeletal muscle and heart.Citation12,Citation13 To gain insight into the mechanisms underlying the developmental splicing program in the CNS, we have studied the steady-state levels of MBNL and CELF proteins throughout brain development and aging in wild-type mice. We found that CELF1 and CELF2 protein levels experienced a pronounced decrease in brainstem. In the frontal cortex, however, only CELF1 displayed a very mild decrease from embryonic day 12.5 (E12.5) onwards. In contrast, MBNL1 and MBNL2 levels increased significantly in adult frontal cortex and brainstem, particularly between post-natal day eight (P8) and the first month of age (M1). MBNL1 and MBNL2 were hardly detected in embryonic stages and at post-natal day 1 (P1) even following long exposures (). The dramatic change in MBNL1 levels at one month of age coincided with a striking switch in the splicing patterns of Mbnl1 and Ldb3/Cypher transcripts previously detected.Citation22 This is not surprising, since the alternative splicing of the alternative exons in these genes is specifically regulated by MBNL1.Citation28 In contrast, the splicing profile of Fxr1 (a gene specifically regulated by CELF1) showed a less pronounced developmental splicing transition,Citation22 possibly due to the mild changes in the levels of CELF1 throughout brain development.
More importantly, the analysis of CELF and MBNL protein levels throughout wild-type brain development and aging revealed that DM1-associated CELF upregulation (particularly in the brainstem), and the functional inactivation of MBNL proteins by sequestration into ribonuclear foci recreated an embryonic scenario, thereby contributing to the abnormal expression of embryonic isoforms in adult DMSXL brain tissues.
We have extended our analysis to the hnRNP H ribonucleoprotein, an alternative splicing regulator with a described role in DM1 spliceopathy.Citation29 hnRNP H steady-state levels exhibited a marked decrease from wild-type embryonic stages to adult ages, in both frontal cortex and brainstem to similar extent (). In contrast to the upregulation of hnRNP H reported in DM1 myoblasts,Citation29 the steady-state levels of hnRNP H did not differ significantly between DMSXL and wild-type brain regions at one month of age ().
Changes in Synaptic Vesicle Proteins are Not Associated with Missplicing
To identify dysfunctional disease intermediates and pathways behind CUG-associated brain dysfunction, we investigated the proteomic profile of DM1 transgenic mice and found abnormal RAB3A upregulation and synapsin I (SYN1) hyperphosphorylation relative to control animals. We extended our findings from mouse to human brains, and confirmed a statistically significant RAB3A upregulation and SYN1 hyperphosphorylation in post-mortem DM1 frontal cortex.Citation22 Furthermore, higher RAB3A levels and SYN1 phosphorylation were also detected in neuronal-like PC12 cells expressing expanded CUG-containing transcripts. In these cells, the abnormal metabolism of synaptic vesicle proteins was associated with aberrant exocytosis, indicating a physiologically relevant consequence of the expression of toxic DMPK transcripts in neuronal cell lineages. Overall, we gathered electrophysiological, neurochemical, cellular and molecular data suggesting that the DM1 neuropsychological manifestations are mediated by the dysregulation of synaptic vesicle proteins, which likely affects neuronal vesicle release and exocytosis, and disrupts synaptic function in the CNS.
Since it has been suggested that DM1 molecular features, particularly splicing dysregulation, recreate embryonic events,Citation13 we asked whether synaptic protein expression and/or phosphorylation were also developmentally regulated. More importantly, we were interested in investigating whether RAB3A upregulation and SYN1 hyperphosphorylation in adult DMSXL mice recreated embryonic events, and supported a contribution of neurodevelopmental deficits behind DM1 neuropathology. To answer these questions, we studied protein expression and/or phosphorylation levels throughout wild-type brain development and aging (). The analysis revealed that RAB3A levels increased at one month of age (M1), and indicated that RAB3A upregulation detected at four months of age did not mimic an embryonic expression profile, and it was unlikely to be a direct consequence of defective developmental regulation of this protein. Similarly, the total levels of SYN1 also showed a marked post-natal increase, particularly after one month of age. However, SYN1 phosphorylation at residues serine-9 and serine-553 preceded the pronounced increase in the steady-state levels of this protein. As a result, the phosphorylation of SYN1 appears to peak at post-natal day eight (P8), but mainly as a result of the low steady-state of SYN1 at this stage (). This situation differs from the SYN1 profiles in adult DMSXL mice, which show increased SYN1 phosphorylation but unchanged steady-state levels relative to wild-type control mice.Citation22 Therefore, we conclude that the hyperphosphorylation of SYN1 in adult DMSXL brains does not truly mimic the early post-natal metabolism of this protein. In summary, our data are consistent with the view that DM1-associated RAB3A upregulation and SYN1 phosphorylation is more likely mediated by neurofunctional abnormalities, rather than CUG-associated neurodevelopmental deficits.
Figure 2. Expression, phosphorylation, and alternative splicing of RAB3A and SYN1. (A) Quantification of RAB3A steady-state levels and SYN1 phosphorylation throughout wild-type brain embryonic development (E10.4, E12.5, E14.5, E18.5), in newborn (P1), postnatal day 8 (P8), and adult mice aged one (M1), four (M4) and 10 (M10) months (Table S2). Frontal cortex protein extracts from three individual animals were pooled for each developmental stage and analyzed in two independent western blot assays (only one is shown). The graphs on the right represent the mean RAB3A and SYN1 steady-state levels (± SD) throughout the development and aging of mouse frontal cortex, as well as mean SYN1 phosphorylation on amino acid residues serine-9 (S9) and serine-553 (S553). (B) Representative RT-PCR splicing analysis of RAB3A exon 2 and the 5′ end of SYN1 exon 13 in the frontal cortex of DM1 (n = 9) and non-DM individuals (n = 4), as well as in DMSXL and wild-type mice at four months of age (n = 6, each genotype) (Table S1). The 5′ end of exon 2 of Rab3A is alternatively spliced only in mouse, and included in the analysis. The graphs show the mean inclusion ratio (± SEM) of each alternative exon or alternative 5′ end.
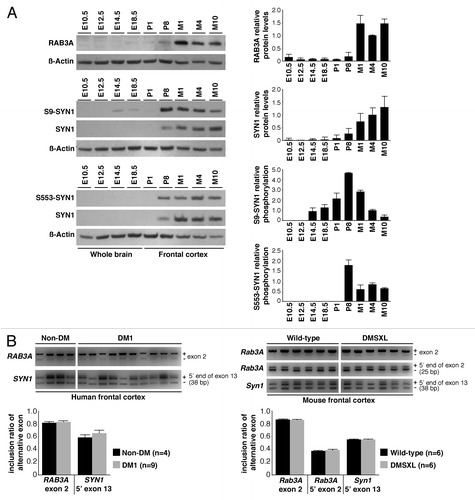
To gain insight into the mechanisms of synaptic protein dysregulation, we studied additional mouse and cell models of DM1. We found that RAB3A was upregulated in mouse brain in response to the inactivation of MBNL1, while SYN1 was hyperphosphorylated in PC12 cells overexpressing CELF1 or CELF2.Citation22 Given the role of MBNL and CELF protein families in the regulation of alternative splicing, and the prevalent role of RNA spliceopathy in the etiology of DM1, we sought to investigate whether RAB3A and SYN1 protein abnormalities were associated with missplicing of the corresponding transcripts. We initially studied the splicing profiles of the main alternative exons previously described in the literature and found no differences between DMSXL and wild-type mice at four months, an age when protein abnormalities were noticeable.Citation22 We have now extended this analysis to the remaining internal exons of the mouse and human genes. Our RT-PCR analysis confirmed additional splicing events in the RAB3A and SYN1 genes previously described in the literature, such as the alternative inclusion of exon 2 in both human and mouse RAB3A transcripts; the inclusion of an additional 25-bp sequence through the use of an alternative 5′ splice site in mouse Rab3A exon 2; and the inclusion of a short 38-bp sequence due to the use of an alternative 5′ splice site of exon 13 in both human and mouse SYN1 transcripts (). Nevertheless, we did not detect significant splicing changes induced by the expression of toxic CUG-containing transcripts in mouse or human brains (). Our data demonstrate that, although mediated by the inactivation of MBNL1 splicing regulator, RAB3A upregulation appears to be independent of missplicing events. Similarly, even if SYN1 is hyperphosphorylated as a result of the upregulation of CELF1 and/or CELF2 splicing regulators, the splicing of SYN1 transcripts remains unaltered. In summary, although involving the dysfunction of known splicing factors (such as MBNL and CELF family members), DM1 neuropathogenesis goes beyond the abnormal expression of embryonic splicing isoforms, and implicates other molecular mechanisms.
Conclusion
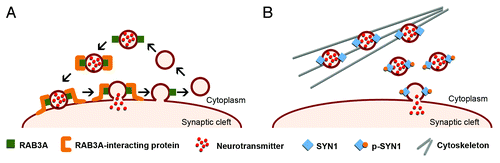
DMSXL mice recreate relevant signs of RNA toxicity in the CNS, associated with behavioral, electrophysiological and neurochemical changes. In the search for the mechanisms and dysfunctional pathways behind these phenotypes, we found altered expression and phosphorylation of synaptic proteins, which are independent of splicing dysregulation. RAB3A is an abundant synaptic vesicle protein that regulates neurotransmission, through the interaction with other synaptic proteins that control vesicle fusion to the cell membrane.Citation30 SYN1 regulates neuronal vesicle release in a phosphorylation-dependent mannerCitation31 (). As a result, RAB3A upregulation and SYN1 hyperphosphorylation disrupts synaptic function and neurotransmitter release, likely contributing to the cognitive and behavioral deficits of DMSXL mice and the neuropsychological manifestations of DM1 patients.
Figure 3. Schematic representation of the role of RAB3A and SYN1 in the dynamics of synaptic vesicles. (A) RAB3A is an abundant vesicle-associated protein that regulates synaptic efficiency. The reported functions of RAB3A in exocytosis range from docking and fusion of the synaptic vesicle to its subsequent recycling. RAB3A functions through a variety of interactions with multiple effector proteins. The upregulation of RAB3A likely perturbs the highly regulated vesicle dynamics and release, affecting neurotransmission and synaptic function in DM1. (B) SYN1 is expressed in mature neurons, where it associates with the cytoplasmic surface of synaptic vesicles. SYN1 regulates the supply of synaptic vesicles available for exocytosis by binding to both vesicles and actin cytoskeleton in a phosphorylation-dependent manner. Under resting conditions, non-phosphorylated SYN1 attaches synaptic vesicles to the actin cytoskeleton. Synaptic stimulation induces SYN1 phosphorylation that facilitates vesicle dissociation from cytoskeleton and potentiates exocytosis. Abnormal SYN1 hyperphosphorylation in DM1 likely dysregulates neuronal exocytosis and vesicle release.
| Abbreviations: | ||
| CELF | = | CUGBP/Elav-like factor |
| CUGBP | = | CUG RNA binding protein |
| DM | = | myotonic dystrophy |
| DM1 | = | myotonic dystrophy type 1 |
| DMPK | = | DM protein kinase |
| MBNL | = | muscleblind-like |
| RAB | = | RAS oncogene family |
| UTR | = | untranslated region |
Additional material
Download Zip (178 KB)Acknowledgments
We are grateful to Amine Bouallague, the personnel of CERFE (Centre d’Exploration et de Recherche Fonctionelle Expérimentale, Genopole, Evry, France) and LEAT (Laboratoire d’Expérimentation Animale et de Transgénèse, Faculté de Médecine Paris Descartes) for attentively caring for the mice. We thank our colleagues at Inserm U781 and the DM1 French Splicing Network for helpful discussions. This work was supported by ANR (Agence Nationale de Recherche, France; project grant “DM1MICE”), AFM (Association Française contre les Myopathies, France; project grant number 14687), INSERM (Institute National de la Santé et Recherche Médicale, France) and Université Paris Descartes (Paris, France). O.H.H. was partially funded by a post-doctoral fellowship and research grant from CONACyT (Consejo Nacional de Ciencia y Tecnología, Mexico; grant number 183697). G.S. was awarded a PhD student fellowship from Ministère Français de la Recherche et Technologie and AFM.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Supplemental Material
Supplemental materials may be found here: www.landesbioscience.com/journals/rarediseases/article/25553
Notes
† These authors contributed equally to this work.
References
- Harper PS. Myotonic Dystrophy. WB Saunders, 2001.
- Meola G, Sansone V. Cerebral involvement in myotonic dystrophies. Muscle Nerve 2007; 36:294 - 306; http://dx.doi.org/10.1002/mus.20800; PMID: 17486579
- Minnerop M, Weber B, Schoene-Bake JC, Roeske S, Mirbach S, Anspach C, et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain 2011; 134:3530 - 46; http://dx.doi.org/10.1093/brain/awr299; PMID: 22131273
- Romeo V, Pegoraro E, Squarzanti F, Sorarù G, Ferrati C, Ermani M, et al. Retrospective study on PET-SPECT imaging in a large cohort of myotonic dystrophy type 1 patients. Neurol Sci 2010; 31:757 - 63; http://dx.doi.org/10.1007/s10072-010-0406-2; PMID: 20842397
- Vermersch P, Sergeant N, Ruchoux MM, Hofmann-Radvanyi H, Wattez A, Petit H, et al. Specific tau variants in the brains of patients with myotonic dystrophy. Neurology 1996; 47:711 - 7; http://dx.doi.org/10.1212/WNL.47.3.711; PMID: 8797469
- Sergeant N, Sablonnière B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A, et al. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum Mol Genet 2001; 10:2143 - 55; http://dx.doi.org/10.1093/hmg/10.19.2143; PMID: 11590131
- Laberge L, Veillette S, Mathieu J, Auclair J, Perron M. The correlation of CTG repeat length with material and social deprivation in myotonic dystrophy. Clin Genet 2007; 71:59 - 66; http://dx.doi.org/10.1111/j.1399-0004.2007.00732.x; PMID: 17204048
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992; 68:799 - 808; http://dx.doi.org/10.1016/0092-8674(92)90154-5; PMID: 1310900
- Sicot G, Gomes-Pereira M. RNA toxicity in human disease and animal models: From the uncovering of a new mechanism to the development of promising therapies. Biochim Biophys Acta 2013; 1832:1390 - 409; http://dx.doi.org/10.1016/j.bbadis.2013.03.002; PMID: 23500957
- Sicot G, Gourdon G, Gomes-Pereira M. Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: new findings and future challenges. Hum Mol Genet 2011; 20:R2 R116 - 23; http://dx.doi.org/10.1093/hmg/ddr343; PMID: 21821673
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 2000; 19:4439 - 48; http://dx.doi.org/10.1093/emboj/19.17.4439; PMID: 10970838
- Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest 2007; 117:2802 - 11; http://dx.doi.org/10.1172/JCI32308; PMID: 17823658
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 2006; 15:2087 - 97; http://dx.doi.org/10.1093/hmg/ddl132; PMID: 16717059
- Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell 2002; 10:45 - 53; http://dx.doi.org/10.1016/S1097-2765(02)00572-5; PMID: 12150906
- Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 2002; 10:35 - 44; http://dx.doi.org/10.1016/S1097-2765(02)00563-4; PMID: 12150905
- Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 2011; 17:720 - 5; http://dx.doi.org/10.1038/nm.2374; PMID: 21623381
- Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 2001; 29:40 - 7; http://dx.doi.org/10.1038/ng704; PMID: 11528389
- Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet 2004; 13:3079 - 88; http://dx.doi.org/10.1093/hmg/ddh327; PMID: 15496431
- Seznec H, Lia-Baldini AS, Duros C, Fouquet C, Lacroix C, Hofmann-Radvanyi H, et al. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum Mol Genet 2000; 9:1185 - 94; http://dx.doi.org/10.1093/hmg/9.8.1185; PMID: 10767343
- Gomes-Pereira M, Foiry L, Nicole A, Huguet A, Junien C, Munnich A, et al. CTG trinucleotide repeat “big jumps”: large expansions, small mice. PLoS Genet 2007; 3:e52; http://dx.doi.org/10.1371/journal.pgen.0030052; PMID: 17411343
- Huguet A, Medja F, Nicole A, Vignaud A, Guiraud-Dogan C, Ferry A, et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1,000 CTG repeats from the human DM1 locus. PLoS Genet 2012; 8:e1003043; http://dx.doi.org/10.1371/journal.pgen.1003043; PMID: 23209425
- Hernández-Hernández O, Guiraud-Dogan C, Sicot G, Huguet A, Luilier S, Steidl E, et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain 2013; 136:957 - 70; http://dx.doi.org/10.1093/brain/aws367; PMID: 23404338
- Ladd AN. CUG-BP, Elav-like family (CELF)-mediated alternative splicing regulation in the brain during health and disease. Mol Cell Neurosci 2012; In press http://dx.doi.org/10.1016/j.mcn.2012.12.003; PMID: 23247071
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 2012; 75:437 - 50; http://dx.doi.org/10.1016/j.neuron.2012.05.029; PMID: 22884328
- Suenaga K, Lee KY, Nakamori M, Tatsumi Y, Takahashi MP, Fujimura H, et al. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PLoS One 2012; 7:e33218; http://dx.doi.org/10.1371/journal.pone.0033218; PMID: 22427994
- Zhang W, Liu H, Han K, Grabowski PJ. Region-specific alternative splicing in the nervous system: implications for regulation by the RNA-binding protein NAPOR. RNA 2002; 8:671 - 85; http://dx.doi.org/10.1017/S1355838202027036; PMID: 12022233
- Panaite PA, Kielar M, Kraftsik R, Gourdon G, Kuntzer T, Barakat-Walter I. Peripheral neuropathy is linked to a severe form of myotonic dystrophy in transgenic mice. J Neuropathol Exp Neurol 2011; 70:678 - 85; http://dx.doi.org/10.1097/NEN.0b013e3182260939; PMID: 21760538
- Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci U S A 2008; 105:20333 - 8; http://dx.doi.org/10.1073/pnas.0809045105; PMID: 19075228
- Paul S, Dansithong W, Kim D, Rossi J, Webster NJ, Comai L, et al. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J 2006; 25:4271 - 83; http://dx.doi.org/10.1038/sj.emboj.7601296; PMID: 16946708
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci 2004; 27:509 - 47; http://dx.doi.org/10.1146/annurev.neuro.26.041002.131412; PMID: 15217342
- Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, et al. Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell 1993; 75:661 - 70; http://dx.doi.org/10.1016/0092-8674(93)90487-B; PMID: 7902212