Abstract
We used whole exome sequence analysis to investigate a possible genetic etiology for a patient with the phenotype of intrauterine growth restriction, microcephaly, developmental delay, failure to thrive, congenital bilateral hip dysplasia, cerebral and cerebellar atrophy, hydrocephalus, and congenital diaphragmatic hernia (CDH).
Whole exome sequencing identified a novel de novo c.2722G > T (p.E908X) mutation in the Myosin Heavy Chain 10 gene (MYH10) which encodes for non-muscle heavy chain II B (NMHC IIB). Mutations in MYH10 have not been previously described in association with human disease. The E908X mutation is located in the coiled-coil region of the protein and is expected to delete the tail domain and disrupt filament assembly.
Nonmuscle myosin IIs (NM IIs) are a group of ubiquitously expressed proteins, and NM II B is specifically enriched in neuronal tissue and is thought to be important in neuronal migration. It is also expressed in cardiac myocytes along with NM IIC.
Homozygous NMHC II B-/B- mouse knockouts die by embryonic day (E)14.5 with severe cardiac defects (membranous ventricular septal defect and cardiac outflow tract abnormalities) and neurodevelopmental disorders (progressive hydrocephalus and neuronal migrational abnormalities).
A heterozygous MYH10 loss of function mutation produces a severe neurologic phenotype and CDH but no apparent cardiac phenotype and suggests that MYH10 may represent a novel gene for brain malformations and/or CDH.
Introduction
Many rare genetic conditions with low reproductive fitness have been difficult to genetically define using methods of linkage and association. Genetic conditions with strong negative selection and reduced reproductive fitness can in part be explained by genes with a high new mutation rate.Citation1 Whole exome sequencing (WES) offers the opportunity to define many of these rare genetic conditions, even within single families. Common, serious neuropsychiatric disorders like intellectual disability, schizophrenia, and autism are genetically heterogeneous and frequently caused by rare de novo mutations.Citation1-Citation4 Global developmental delay and intellectual disability affects 3% of the general population. Genetic evaluation of such individuals is challenging in the absence of the recognizable genetic syndrome or structural chromosomal abnormalities. Mutations in more than 400 different genes have been associated with intellectual disability.Citation5 In clinical situations in which extensive conventional genetic evaluation (chromosome microarray and single-gene testing) has failed to identify the genetic etiology, whole exome sequencing with a trio design to compare proband and both parents has been shown to be particularly successful in rare genetic diseases.Citation2,Citation6 Vissers et al. have shown that de novo mutations discovered by WES are the major cause of the unexplained severe developmental disability in a cohort of 10 patients with a moderate to severe mental retardation.Citation3 In a larger cohort of 100 patients with moderate to severe unexplained intellectual disability, WES has been shown to have a diagnostic yield of 16% supporting the use of whole exome sequencing as a diagnostic tool for patients with unexplained severe intellectual disability.Citation7
Results
Case presentation
The proband is an 8 y old male from non-consanguineous family of mixed Ashkenazi Jewish and Caucasian ethnicity. He was born at 38 weeks of gestation via induced vaginal delivery. The prenatal history was significant for intrauterine growth restriction and unilateral multicystic dysplastic kidney. Decreased fetal movement was noted initially at the beginning of the third trimester. His birth weight was 5 pounds 12 ounces (< 5%), and his head circumference was 31 cm (< 5%). After birth he was noted to have bilateral dislocated hips due to an underlying developmental hip dysplasia. He was also noted to have foot abnormalities consistent with talipes cavus and bilateral undescended testicles. The immediate neonatal period was characterized by a prolonged hospitalization, hypotonia and feeding difficulties. MRI of the brain in the newborn period was abnormal demonstrating microcephaly and small thalami with increased intensities in bilateral basal ganglia and thalami. His growth continued to be poor, and he was severely hypotonic and microcephalic. MRI of the brain at the age of 4 y showed evidence of diffuse volume loss with decreased thickness of the white matter and increased ventricular size. The atrophy was also evident in the inferior cerebellar hemispheres and inferior cerebellar vermis. MRI demonstrated a left sided diaphragmatic hernia which has not been repaired with partial herniation of the stomach into the chest. At the age of 8 y he is severely developmentally delayed and microcephalic. He is non-ambulatory and non-verbal. He uses an assistive communication device. He is able to recognize objects and faces. All his feeds are via gastrostomy tube.
Whole exome sequencing analysis
Exome sequencing of the family trio (proband, mother and father) resulted in an average of ~19 Gb of sequence per sample. Mean coverage of captured regions was 154 × per sample, with > 90% covered with at least 10 × coverage, an average of ~90% base call quality of Q30 or greater, and an overall average mean quality score of < Q35. Stepwise filtering removal of common SNPs, intergenic and 3′/5′ UTR variants, non-splice-related intronic variants, and synonymous variants resulted in ~12,000 variants per sample (Table S1). Family history inheritance model filtering based on autosomal and X-linked dominant and recessive and Y-linked inheritance models of the proband, mother and father revealed 15 genes with 19 alterations (Table S2). Manual review of each alteration to rule out sequencing artifacts and polymorphisms along with medical interpretation to rule out genes lacking clinical overlap with the patient's evaluated phenotype resulted in 11 genes with 14 unique alterations (Table S3). Among these, two notable genes with a single alteration each with potential clinical relevance underwent confirmation and co-segregation analysis using automated fluorescence dideoxy sequencing: c.626C > T (p.S209L) X-linked recessive alteration in BCOR and a novel variant c.2722G > T (p.E908X) in MYH10. Both were confirmed with dideoxy sequencing. Co-segregation analysis revealed that the mother is a carrier of the BCOR alteration which was inherited from the unaffected maternal grandfather; and the MYH10 alteration was absent in the mother, father, and brother, establishing a de novo origin of the alteration.
The novel, de novo, nonsense variant p.E908X (c.2722G > T) in MYH10, results in premature termination of the protein truncating the protein with loss of the myosin tail (). The truncating loss of function mutation, the de novo nature of the mutation and the overlapping phenotype of our patient and mice deficient in MYH10 () led us to believe this was likely a pathogenic mutation in our patient.
Figure 1. Schematic of the de novo nonsense heterozygous mutation (c.2722G>T, p.E908X) in exon 22 of MY10 (uc002gll.3). Top: 41 exons of MY10 gene with black bars represent coding exons. The c.2722G>T mutation is indicated Middle: MY10 protein showing the N terminal SH3-like domain, motor domain, and myosin tail. Bottom: Cross species amino acid alignment of a portion of MY10 indicating the glutamate 908 mutated position.
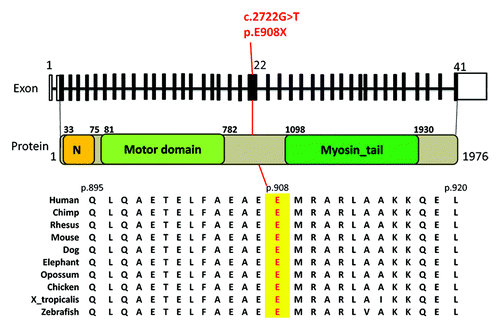
Table 1. Comparison of human phenotype with different mice genotype for NMHC IIB
Discussion
We describe the application of WES to identify a novel de novo heterozygous p.E908X variant in MYH10 that is predicted to be deleterious. MYH10 (OMIM 160776) encodes the heavy chain of one of the three isoforms of non-muscle myosin II (NM II) namely NM IIB. Unlike the findings for MYH9 and MYH14, mutations in MYH10 have not previously been implicated in human diseases. NM IIs are a group of ubiquitously expressed proteins that bind actin and are involved in cytokinesis, migration and cell-cell adhesion.Citation8-Citation10 In humans the heavy chains are encoded by three different genes (MYH9, MYH10 and MYH14) encoding NMHC IIA, NMHC IIB and NMHC IIC, respectively.Citation11,Citation12 The three NM II isoforms have considerable homology in primary sequence (60–80%) and have similar molecular structure consisting of 2 structurally defined regions12. The NM II is a hexamer composed of 2 heavy chains and 2 pairs of light chains. Each non-muscle heavy chain isoform has an N-terminal globular head or motor domain and a long α-helix coiled coil tail domain. The motor domain reversibly binds actin filaments and contains the site for actin activated MgATP-ase activity.Citation13 Both NM IIA and IIB are critical for early mouse embryonic development and show overlapping localization in most of the embryonic tissue with some distinct tissue specific distributions.Citation14,Citation15 NM IIB is specifically enriched in neuronal tissue during mouse embryonic development and has been shown to be critical for neuronal cell migration in both cerebral cortex, cerebellum and pontine and facial neurons16,17. Furthermore, NM IIB plays a critical role in mouse cardiac development and is expressed along with NM IIC in the cardiac myocytes during embryonic development.Citation18 NM IIB is also expressed in a variety of other tissues including murine diaphragm. Germline homozygous but not heterozygous loss of function mutations in NM IIB are embryonic lethal by E14.5 in mice and are associated with development of severe progressive hydrocephalus that destroys brain architecture ().Citation19
The mice develop severe abnormalities in brain and heart, including severe progressive hydrocephalus, starting at the embryonic day 12.5 (E12.5) that destroys the brain architecture and prevents further investigation of the role of NM IIB in the brain development.Citation19 In order to enable study of NM IIB in brain development, a mouse model with a hypomorphic, homozygous R709C missense mutation in the motor domain of NM IIB was generated, and homozygous mutant mice die in the early postnatal period, and those that survive longer showed severe growth retardation and progressive hydrocephalus and ataxia.Citation16 Heterozygous mutants survive to adulthood without any obvious abnormalities. The homozygous mutants show 73% reduction in the expression of the NMHC-IIB. Brain tissue obtained from homozygous mutant mice show distorted cerebral cortex due to hydrocephalus and small and underdeveloped cerebellum. In addition to small cerebellar size, the mice also showed marked abnormalities in cerebellar foliation pattern with shallow and missing fissures primarily in anterior and midline part of the cerebellum. The mutant mice also show abnormal cerebellar layer formation with decreased thickness of the internal germinal layer and abnormalities in migratory pattern of certain major groups of neurons including cerebellar granule cells, pontine neurons, reticular neurons of the pons, and facial neurons.Citation17
Further studies on germline ablated NMHC IIB mice and R709C mutant mice show that both develop progressive hydrocephalus starting from E11.5, although the R709C mice develop it at a slower rate. It appears that the hydrocephalus is communicating with no obstruction of aqueduct of Sylvius. The spinal canal of these mice show invasion of neuroepithelial cells which are thought to be responsible for development of hydrocephalus. It has been shown that neuroepithelial cells surrounding the spinal canal are specifically enriched with NM II B compared with other areas of spinal cord.Citation20
In addition to being critical for mouse brain development, NM IIB appears to be critical for normal development of the mouse heart. It has been shown that NM IIB is the predominant isoform present in the developing cardiac myocytes. Homozygous NMHC IIB B-/B- mice show abnormalities in cardiac development including membranous ventricular septal defect, overriding aorta, narrowing of RVOT due to right ventricular hypertrophy and marked cardiac myocyte hypertrophy.Citation18 In addition to major abnormalities in brain and heart, homozygous (NMHC IIB R709C) mutants also show other defects in ventral body wall closure including ectopia cordis, omphalocele and anterior diaphragmatic hernia resembling Pentlogy of Cantrell (unpublished observation by X. Ma and R. S. Adelstein).Citation8
Although no human mutations have been previously reported in MYH10, human mutations have been identified in MYH9 which encodes for NMHC IIA. NM IIA is the predominant isoform of NM II in blood cells including platelets, where myosin accounts for a 2% of total protein.Citation20 MYH-9 related disorders are inherited in an autosomal-dominant manner and are primarily characterized by hematological abnormalities including thrombocytopenia with distinct giant platelets and neutrophils with inclusion bodiesCitation21-Citation24 and occasionally sensorineural hearing loss, presenile cataracts and severe proteinuric nephropathy that can lead to end-stage renal diseaseCitation25
Heterozygous missense and nonsense mutations in MYH14 encoding the NMHC IIC isoform have been implicated in non-syndromic autosomal-dominant hearing loss.Citation26 Mutations in MYH14 have also been described in a family with peripheral neuropathy, myopathy, hoarseness and hearing loss.Citation27 MYH14 is highly expressed in cochlea, peripheral nerves, brain, skeletal muscle and small and large intestine.Citation28
Mutations in MYH10 have not yet been described in any known human genetic disorder. Our patient shows predominately a severe central nervous system phenotype characterized by microcephaly and severe intellectual disability with marked cerebral and cerebellar atrophy involving both cerebellar hemispheres and vermis with development of ex-vacuo hydrocephalus. NM IIB is specifically enriched in neuronal cells and appears to be critical for a process of neuronal migration. The C-terminal tail domain is important for bipolar filament formation which is important for cell-cell adhesion and function. The E908X mutation in our patient is located in the coiled-coil domain and is predicted to cause the loss of essentially the whole downstream tail domain. There are striking similarities in the phenotype between our patient and the murine models of MYH10 mutations. The murine Myh10 knockout and missense mutations (R709C) are autosomal recessively inherited, and it is unclear whether the E908X dominant mutation in our patient acts through loss of function and haploinsufficiency or a dominant negative effect. The closely related MYH9 gene that encodes for NMHC-IIA, one of the other NM IIs, has been extensively studied and suggests that the human mutation may act by a dominant negative effect. Human MYH9-related disorders are due to heterozygous missense and premature termination mutations that are dominantly inherited. In the mouse, heterozygous Myh9 knockouts have no phenotype while homozygous knockouts are embryonic lethal at day 7.5 because of the failure of visceral endoderm formation.Citation29 This parallels MYH10.
Franke et al., studied the 4 most common MYH9 human mutants (R1165C, D1424N, E1841K and R1933X) in vitro using electron microscopy to analyze paracrystal morphology to visualize assembly of individual myosin-II molecules. To participate in cellular contractility, NM IIs must first form functional bipolar filaments after dimerization. All 4 mutants failed to assemble normally when visualized by electron microscopy, and none of the four mutations formed large, well ordered paracrystals but formed only aberrant chain-like structures. To study the potential dominant negative effect, the R1933X mutant was mixed with the wild type allele, and aberrant chain-like structures similar to the mutant alone were predominately observed, supporting a dominant negative mechanism. With insufficient wild-type filament, there is loss of contractility and impaired cellular dynamics of the affected cells.Citation30 We propose that our patient’s E908X MYH10 mutation may act through a similar dominant negative mechanism. Further functional studies to analyze whether mutant protein is expressed are needed to address these questions.
In summary, we describe for the first time an association of a novel nonsense heterozygous de novo mutation in MYH10 gene with a human phenotype characterized by severe intellectual disability, microcephaly and congenital diaphragmatic hernia. The disease-association of this novel gene is supported by its de novo occurrence, the deleterious nature of nonsense alterations, the gene function, and the phenotype observed in mice with loss of function mutations in the same gene. A major limitation of this study is that there is only a single patient identified with a de novo novel variant in MYH10. Functional in vitro analysis to further define the impact of the altered gene would also substantiate these findings. We encourage others engaged in the genetic study of children with intellectual disabilities and/or CDH to include MYH10 as a candidate gene in genetic studies to find additional similarly affected patients in order to better define this novel clinical disorder.
Material and Methods
DNA from the parents and proband were prepared using the SureSelect Target Enrichment System (Agilent Technologies). The exome libraries were sequenced using paired-end, 100-cycle chemistry on the Illumina HiSeq 2000 (Illumina). Sequence quality filtering was executed with the Illumina CASAVA software (ver 1.8.2, Illumina). Data yield (Mbases), %PF (pass-filter), # of reads, % of raw clusters per lane, and quality scores were examined in Demultiplex_Stats.htm file. The sequence data were aligned to the reference human genome (GRCh37), variant calls generated using CASAVA and Pindel, and alterations annotated with the Ambry Variant Analyzer tool (AVA) including sequence changes on cDNA and protein, nucleotide and amino acid conservation, population frequency, predicted functional impact (including PolyPhen and SIFT in silico prediction tools), etc. Exons plus at least 2 bases into the 5′ and 3′ ends of all the introns were analyzed. Multiple sequence alignments were viewed using IGV (integrative genomics Viewer software). Data analysis focused on nonsense variants, small insertions and deletions, canonical splice site alterations or non-synonymous missense alterations. The Human Gene Mutation Database (HGMD), the Single Nucleotide Polymorphism database (dbSNP), 1000 genomes, HapMap data and online search engines (e.g., PubMed) were used to search for previously described gene mutations and polymorphisms. The filtering pipeline protects all variants annotated within the Human Gene Mutation Database (HGMD) and/or the Online Mendelian Inheritance in Man (OMIM) databases. Stepwise filtering included the removal of common SNPs, intergenic and 3′/5′ UTR variants, non-splice-related intronic variants, and lastly synonymous variants. Variants were then filtered further based on family history and possible inheritance models. Identified candidate alterations were confirmed using automated fluorescence dideoxy sequencing. Co-segregation analysis was performed using each available family member. Amplification primers were designed using PrimerZ PCR primers were tagged with established sequencing primers on the 5′ end. Sequencing was performed on an ABI3730 (Life Technologies) using standard procedures.
Additional material
Download Zip (144.8 KB)Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/rarediseases/article/26144.
Acknowledgments
We thank the family for their generous contribution. We thank Mary Anne Conti, Xuefei Ma and Robert Adelstein for helpful discussions and review of the manuscript. This work was supported by NIH grant R01 HD057036.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Julie G, Hamdan FF, Rouleau GA. A strategy to identify de novo mutations in common disorders such as autism and schizophrenia. J Vis Exp 2011; 52:2534; PMID: 21712793
- Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol 2011; 12:228; http://dx.doi.org/10.1186/gb-2011-12-9-228; PMID: 21920049
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, et al. A de novo paradigm for mental retardation. Nat Genet 2010; 42:1109 - 12; http://dx.doi.org/10.1038/ng.712; PMID: 21076407
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011; 43:585 - 9; http://dx.doi.org/10.1038/ng.835; PMID: 21572417
- van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet 2011; 45:81 - 104; http://dx.doi.org/10.1146/annurev-genet-110410-132512; PMID: 21910631
- Willemsen MH, Vissers LE, Willemsen MA, van Bon BW, Kroes T, de Ligt J, de Vries BB, Schoots J, Lugtenberg D, Hamel BC, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet 2012; 49:179 - 83; http://dx.doi.org/10.1136/jmedgenet-2011-100542; PMID: 22368300
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med 2012; 367:1921 - 9; http://dx.doi.org/10.1056/NEJMoa1206524; PMID: 23033978
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 2009; 10:778 - 90; http://dx.doi.org/10.1038/nrm2786; PMID: 19851336
- Takeda K, Kishi H, Ma X, Yu ZX, Adelstein RS. Ablation and mutation of nonmuscle myosin heavy chain II-B results in a defect in cardiac myocyte cytokinesis. Circ Res 2003; 93:330 - 7; http://dx.doi.org/10.1161/01.RES.0000089256.00309.CB; PMID: 12893741
- Lo CM, Buxton DB, Chua GC, Dembo M, Adelstein RS, Wang YL. Nonmuscle myosin IIb is involved in the guidance of fibroblast migration. Mol Biol Cell 2004; 15:982 - 9; http://dx.doi.org/10.1091/mbc.E03-06-0359; PMID: 14699073
- Simons M, Wang M, McBride OW, Kawamoto S, Yamakawa K, Gdula D, Adelstein RS, Weir L. Human nonmuscle myosin heavy chains are encoded by two genes located on different chromosomes. Circ Res 1991; 69:530 - 9; http://dx.doi.org/10.1161/01.RES.69.2.530; PMID: 1860190
- Leal A, Endele S, Stengel C, Huehne K, Loetterle J, Barrantes R, Winterpacht A, Rautenstrauss B. A novel myosin heavy chain gene in human chromosome 19q13.3. Gene 2003; 312:165 - 71; http://dx.doi.org/10.1016/S0378-1119(03)00613-9; PMID: 12909352
- Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions. J Cell Sci 2008; 121:11 - 8; http://dx.doi.org/10.1242/jcs.007112; PMID: 18096687
- Wang A, Ma X, Conti MA, Liu C, Kawamoto S, Adelstein RS. Nonmuscle myosin II isoform and domain specificity during early mouse development. Proc Natl Acad Sci U S A 2010; 107:14645 - 50; http://dx.doi.org/10.1073/pnas.1004023107; PMID: 20679233
- Bhatia-Dey N, Taira M, Conti MA, Nooruddin H, Adelstein RS. Differential expression of non-muscle myosin heavy chain genes during Xenopus embryogenesis. Mech Dev 1998; 78:33 - 6; http://dx.doi.org/10.1016/S0925-4773(98)00136-1; PMID: 9858676
- Ma X, Kawamoto S, Hara Y, Adelstein RS. A point mutation in the motor domain of nonmuscle myosin II-B impairs migration of distinct groups of neurons. Mol Biol Cell 2004; 15:2568 - 79; http://dx.doi.org/10.1091/mbc.E03-11-0836; PMID: 15034141
- Ma X, Kawamoto S, Uribe J, Adelstein RS. Function of the neuron-specific alternatively spliced isoforms of nonmuscle myosin II-B during mouse brain development. Mol Biol Cell 2006; 17:2138 - 49; http://dx.doi.org/10.1091/mbc.E05-10-0997; PMID: 16481398
- Tullio AN, Accili D, Ferrans VJ, Yu ZX, Takeda K, Grinberg A, Westphal H, Preston YA, Adelstein RS. Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc Natl Acad Sci U S A 1997; 94:12407 - 12; http://dx.doi.org/10.1073/pnas.94.23.12407; PMID: 9356462
- Tullio AN, Bridgman PC, Tresser NJ, Chan CC, Conti MA, Adelstein RS, Hara Y. Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II-B heavy chain. J Comp Neurol 2001; 433:62 - 74; http://dx.doi.org/10.1002/cne.1125; PMID: 11283949
- Ma X, Bao J, Adelstein RS. Loss of cell adhesion causes hydrocephalus in nonmuscle myosin II-B-ablated and mutated mice. Mol Biol Cell 2007; 18:2305 - 12; http://dx.doi.org/10.1091/mbc.E07-01-0073; PMID: 17429076
- Kelley MJ, Jawien W, Ortel TL, Korczak JF. Mutation of MYH9, encoding non-muscle myosin heavy chain A, in May-Hegglin anomaly. Nat Genet 2000; 26:106 - 8; http://dx.doi.org/10.1038/79069; PMID: 10973260
- Seri M, Pecci A, Di Bari F, Cusano R, Savino M, Panza E, Nigro A, Noris P, Gangarossa S, Rocca B, et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine (Baltimore) 2003; 82:203 - 15; http://dx.doi.org/10.1097/01.md.0000076006.64510.5c; PMID: 12792306
- Heath KE, Campos-Barros A, Toren A, Rozenfeld-Granot G, Carlsson LE, Savige J, Denison JC, Gregory MC, White JG, Barker DF, et al. Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am J Hum Genet 2001; 69:1033 - 45; http://dx.doi.org/10.1086/324267; PMID: 11590545
- Zhang Y, Conti MA, Malide D, Dong F, Wang A, Shmist YA, Liu C, Zerfas P, Daniels MP, Chan CC, et al. Mouse models of MYH9-related disease: mutations in nonmuscle myosin II-A. Blood 2012; 119:238 - 50; http://dx.doi.org/10.1182/blood-2011-06-358853; PMID: 21908426
- De Rocco D, Zieger B, Platokouki H, Heller PG, Pastore A, Bottega R, Noris P, Barozzi S, Glembotsky AC, Pergantou H, et al. MYH9-related disease: five novel mutations expanding the spectrum of causative mutations and confirming genotype/phenotype correlations. Eur J Med Genet 2013; 56:7 - 12; http://dx.doi.org/10.1016/j.ejmg.2012.10.009; PMID: 23123319
- Donaudy F, Snoeckx R, Pfister M, Zenner HP, Blin N, Di Stazio M, Ferrara A, Lanzara C, Ficarella R, Declau F, et al. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am J Hum Genet 2004; 74:770 - 6; http://dx.doi.org/10.1086/383285; PMID: 15015131
- Choi BO, Kang SH, Hyun YS, Kanwal S, Park SW, Koo H, Kim SB, Choi YC, Yoo JH, Kim JW, et al. A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss is linked to an autosomal dominant mutation in MYH14. Hum Mutat 2011; 32:669 - 77; http://dx.doi.org/10.1002/humu.21488; PMID: 21480433
- Golomb E, Ma X, Jana SS, Preston YA, Kawamoto S, Shoham NG, Goldin E, Conti MA, Sellers JR, Adelstein RS. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J Biol Chem 2004; 279:2800 - 8; http://dx.doi.org/10.1074/jbc.M309981200; PMID: 14594953
- Conti MA, Even-Ram S, Liu C, Yamada KM, Adelstein RS. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J Biol Chem 2004; 279:41263 - 6; http://dx.doi.org/10.1074/jbc.C400352200; PMID: 15292239
- Franke JD, Dong F, Rickoll WL, Kelley MJ, Kiehart DP. Rod mutations associated with MYH9-related disorders disrupt nonmuscle myosin-IIA assembly. Blood 2005; 105:161 - 9; http://dx.doi.org/10.1182/blood-2004-06-2067; PMID: 15339844