Abstract
Whole exome sequencing using a parent-child trio design to identify de novo mutations provides an efficient method to identify novel genes for rare diseases with low reproductive fitness that are difficult to study by more classical genetic methods of linkage analysis. We describe a 15 y old female with severe static encephalopathy, intellectual disability, and generalized epilepsy. After extensive metabolic and genetic testing, whole exome sequencing identified a novel de novo variant in Synaptosomal-associated protein-25 (SNAP25), c.142G > T p.Phe48Val alteration. This variant is predicted to be damaging by all prediction algorithms. SNAP25 is part of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein complex which is involved in exocytotic release of neurotransmitters. Genetic alterations in Snap25 in animal models can cause anxiety-related behavior, ataxia and seizures. We suggest that SNAP25 mutations in humans are a novel genetic cause of intellectual disability and epilepsy.
Introduction
Many rare genetic conditions with low reproductive fitness have been difficult to genetically define using methods of linkage and association. Genetic conditions with strong negative selection and reduced reproductive fitness can in part be explained by genes with a high new mutation rate.Citation1 Whole exome sequencing now offers the opportunity to define many of these rare genetic conditions, even within single families. Common, serious neuropsychiatric disorders like intellectual disability, schizophrenia, and autism are genetically heterogeneous and frequently caused by rare de novo mutations.Citation1-Citation4
Over 340 genes are associated with epilepsy, some of which are associated with other specific phenotypic features.Citation5 Neuronal migration disorders causing lissencephaly and heterotopias have been linked to mutations in LIS1, DCX, ARX, TUBA1A, RELN, FLNA, and ARFGEF2.Citation6 TSC1 and TSC2 mutations impair regulation of the mTOR pathway and cause tuberous sclerosis.Citation5 Rett and atypical Rett syndromes have been linked to mutations in CDKL5, FOXG1 and MECP2.Citation7,Citation8 Early onset epileptic encephalopathy has been associated with mutations in ALDH7A1, ATP6AP2, PCDH19, POLG, PNPO, SLC2A1, SLC25A22, SPTAN1, and STXBP1.Citation5 Mutations in KCNQ2 and KCNQ3, which control voltage gated potassium channels, have been identified in patients with benign familial neonatal seizures.Citation9 Myoclonic epilepsy has been linked to mutations in CACNB4, EFHC1, GABRA1, CSTB, EPM2A, EPM2B, and PRICKLE1.Citation5,Citation10 The most common familial epilepsies result from mutations in the voltage gated sodium channels genes SCN1A, SCN1B, and SCN2A.Citation6
Whole exome sequencing was initiated on a patient with intellectual disability and epilepsy who had evaded a specific diagnosis despite significant laboratory investigations into the cause of her disorder. A mutation in a gene previously not associated with a specific human disease was identified. This analysis demonstrates the strength of whole exome sequencing as a means of molecular diagnosis for patients who have non-specific neuropsychiatric presentations.
Results
Case presentation
The proband is a 15 y old female, delivered at 41 weeks of gestation. Early prenatal ultrasounds showed bilateral choroid plexus cysts in the brain which were no longer observed by 21 weeks of gestation. APGAR scores were 8 and 9 at 1 and 5 min, respectively. Birth weight was appropriate at 3360 g. The newborn physical exam identified hip dysplasia requiring a Pavlik harness for 4 mo.
Developmentally, she was always delayed in reaching milestones. At 6 weeks of age, the proband’s mother was concerned that she was not making eye contact. At 2.5 mo she was unable to lift her head when prone. Generalized hypotonia was noted at 4 mo. She first sat unsupported at 3 y of age. She can now ambulate with assistance, but does so infrequently. She is non-verbal. She gained weight well from 0–5 mo, maintaining her weight between the 50th–75th centiles but dropped to the 5th centile by 8 mo. Head circumference was also maintained at the 50th centile from 0–5 mo but dropped to the 5th centile by 9 mo. There was no history of developmental regression, but rather a severe delay in development from early life. To optimize nutrition and increase her caloric intake, nasogastric feeds were started at 1 y of age followed shortly thereafter with gastrostomy tube placement.
Seizures began at 5 mo of age with episodes of repetitive blinking, upward eye deviation and impaired awareness, and were later associated with bilateral kicking and arm abduction accompanied by eye deviation to either side. Video EEG monitoring revealed mild generalized slowing, 2–2.5 Hz generalized spike-and-slow wave complexes. Subsequent video EEG studies revealed (1) mild generalized slowing, (2) interictal partial and symptomatic generalized discharges, and (3) ictal partial and generalized seizures with left (95%) or right-sided (~5%) hemiclonic seizures and lateralized frontocentral discharges (~90% left-sided, ~10% right sided) as well as seizures with bilateral motor features at onset, including tonic-clonic seizures, with generalized discharges. She currently averages 3–10 focal clonic seizures per hour, with an average duration of 15 s. Tonic-clonic seizures occur once every two weeks. Her most recent video EEG showed frequent epileptiform discharges but did not comprise more than 30% of her waking or sleep background. Seizures remain refractory to more than eight antiepileptic drugs in up to three drug combinations, as well as a ketogenic diet and intravenous methylprednisolone trials.
At 5 y of age, she required bilateral varus rotation osteotomies of the proximal femurs. She required spinal fusion at 11 y of age for scoliosis.
Her current neurological examination reveals that she is nonverbal, with global developmental delays. She can intermittently follow simple commands and make intermittent, but fleeting eye contact. Eye tracking is impaired. She is microcephalic with diminished oralmotor tone, excessive drooling and spastic quadriparesis.
The proband is an only child. There is no family history of birth defects, seizures, intellectual disability or consanguinity.
Brain MRI at 8 mo showed delayed myelination, but no structural abnormalities. MR spectroscopy showed increased lactate, raising concern for a mitochondrial disorder but given normal cerebrospinal fluid lactate levels it was thought that the MR spectroscopy lactate peaks were secondary to frequent seizure activity.
Metabolic screening including quantitative plasma amino acids, acylcarnitine profile, carbohydrate deficient transferrin electrophoresis, urine organic acid analysis, and cerebrospinal fluid neurotransmitters, amino acids, lactate and glucose were all normal. Sulfite oxidase testing for molybdenum cofactor deficiency was normal. Muscle biopsy pathology and electron microscopy, and mitochondrial oxidative phosphorylation enzyme activity in muscle were normal. Electromyogram was normal. Karyotype, subtelomeric FISH, and chromosome microarray were normal. Molecular genetic testing for MECP2, CDKL5, Angelman syndrome methylation, ACAT1, FOXG1, Mitochondrial DNA 3243 allele was negative, MERRF testing on muscle, 101 nuclear encoded mitochondrial genes, 53 epilepsy genes, and 64 genes associated with X linked intellectual disabilities were all normal.
Whole exome sequencing
Exome sequencing of the family trio (proband, mother and father) resulted in an average of ~16 Gb of sequence per sample (). Mean coverage of captured regions was 119x per sample, with > 90% covered with at least 10x coverage, an average of >91% base call quality of Q30 or greater, and an overall average mean quality score of <Q35. Stepwise filtering removal of common SNPs, intergenic and 3′/5′ UTR variants, non-splice-related intronic variants, and synonymous variants resulted in ~12 000 variants per sample. Family history inheritance model filtering based on de novo autosomal and X-linked dominant and recessive inheritance models of the proband, mother and father revealed 37 genes (55 unique alterations) (). Manual review of each alteration to rule out sequencing artifacts and polymorphisms along with medical interpretation to rule out genes lacking clinical overlap with the patient's evaluated phenotype resulted in 7 genes (10 unique alterations) ( and ). Review of the individuals genes demonstrated that heterozygous and homozygous loss of function mutations in FAM90A1 and SGK223 were common in normal individuals in the ESP database, suggesting these are not responsible for the phenotype. PCMTD1 has pseudogenes with the same 9 bp insertion suggesting this is a mapping error. Among the three possible recessive genes ABCC10 and CAPRIN2, and IGSF3, at least one of the alleles was predicted to be benign by either PolyPhen-2 or SIFT and none of the three are highly expressed within the brain, making them less likely candidates. Among these, the most likely gene with one alteration with potential clinical relevance underwent confirmation and co-segregation analysis using automated fluorescence dideoxy sequencing: SNAP25 c.142G > T (p.V48F) ().
Figure 1. (A) Family pedigree. Shaded shapes indicate affected individuals. (B) Next-gen sequence alignment of the SNAP25: c.142G > T (p.V48F alteration in the proband, mother, and father. (C) Sequence conservation plots at the mutated site amino acid position across different species.
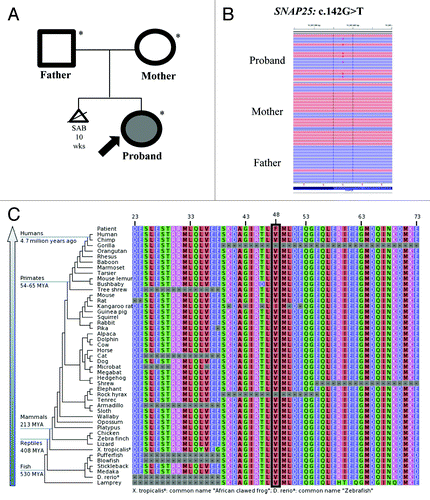
Table 1. Variant filtering based on inheritance model and interpretation
Table 2. Summary of variants remaining after manual filtering
The c.142G > T (p.V48F) alteration is located in exon 4 (3rd coding exon) of the SNAP25 gene. The V48 amino acid is highly conserved throughout vertebrates (). The amino acid substitution results in changing a non-polar, neutral amino acid to a larger, non-polar, neutral amino acid and is predicted to be probably damaging by PolyPhenCitation16 and deleterious by SIFTCitation17 in silico analysis. Based on data from the NHLBI Exome Sequencing Project (ESP),Citation14 no alterations were observed among 6,503 individuals tested (0.0%). Allele frequency data for this nucleotide position not currently available from the 1000 Genomes ProjectCitation14 and the alteration is not currently listed in the Database of Single Nucleotide Polymorphisms (dbSNP). Confirmatory sequencing and co-segregation analysis using automated fluorescence dideoxy sequencing revealed that the alteration was absent in the mother and father, establishing a de novo origin of the alteration.
Discussion
We suggest that the novel variant, c.142G > T in SNAP25, identified via whole exome sequencing is the likely cause of the proband’s epilepsy and intellectual disabilities. SNAP25 is one of the best characterized components of the SNARE protein complex which is involved in the exocytotic release of neurotransmitters during synaptic transmission by mediating synaptic vesicle fusion.Citation19 SNAP25 assembles with syntaxin-1 and synaptobrevin to form the exocytotic fusion complex in neurons.Citation20 Electroretinogram recordings of Snap25 null drosophila show a complete loss of synaptic transmission.Citation21
SNAP25 has not previously been associated with any Mendelian human disease. It was discovered that blind-drunk mice (Bdr), which display ataxia and impaired gait, harbor a heterozygous single amino acid substitution (I67T) in Snap25.Citation22 Substituting serine at position 187 with alanine in Snap25 induced anxiety related behavior in the mouse.Citation23 The Coloboma mouse, which is heterozygous for 4.6Mb deletion of chromosome 2 which encompasses 12 genes including Snap25, have shown seizure predisposition.Citation24
Snap25 exists as two isoforms.Citation25 Mice with only Snap25a develop seizures and show learning deficits and anxiety. Snap25b knockout/ Snap25a knock-in mouse mutants that only express Snap25a showed frequent episodes of tremors and seizure activity.Citation25 This experiment demonstrated that small changes in the regulation of Snap25 caused neurological problems including epilepsy.
Synapsin 1 (Syn1) and Synapsin II (SynII) knock out mice exhibit seizure activity.Citation26SYN1 mutations are associated with seizure activity in humans.Citation27 Synapsins are presynaptic proteins implicated in the regulation of neurotransmitter release.Citation6 This was an important model confirming the role of neurotransmitter release machinery in epilepsy.Citation9
Syntaxin binding protein-1 (STXBP1) is another component of the SNARE complex, and it is implicated in regulating synaptic vesicle docking and fusion similar to SNAP25.Citation22 Mutations in STXBP1 cause early infantile epilepsy.Citation28-Citation30 Paciorkowski et al. review of cases of infantile spasms associated with copy number variants and demonstrated that many components of the SNARE complex are implicated in epilepsy.Citation31
Review of the DECIPHER databaseCitation32 revealed 10 patients who had deletions of chromosome 20 including SNAP25. Four patients had no description of their phenotype. Three patients had seizures, and one had intellectual disability. This is a small number of patients but it further supports the association of SNAP25 mutations and seizures and intellectual disability.
We hypothesize that the novel p.Phe48Val de novo variant in SNAP25 disturbs neurotransmitter release at the synapse leading to seizures and generalized central nervous system dysfunction leading to intellectual disability. A major limitation of this study is that there is only a single patient identified with a de novo novel variant in SNAP25. Functional in vitro analysis to further define the impact of the altered gene would also substantiate these findings. We encourage others engaged in the genetic study of children with epilepsy and/or intellectual disabilities to include SNAP25 as a candidate gene in genetic studies to find additional similarly affected patients in order to better define this novel neurological disorder.
Methods
DNA from the parents and proband were prepared using the SureSelect Target Enrichment System (Agilent Technologies).Citation11 The exome libraries were then applied to the solid surface flow cell for clonal amplification and sequencing using paired-end, 100-cycle chemistry on the Illumina HiSeq 2000 (Illumina). Sequence quality filtering script was executed with the Illumina CASAVA software (ver 1.8.2, Illumina). Data yield (Mbases), %PF (pass-filter), # of reads, % of raw clusters per lane, and quality scores were examined in Demultiplex_Stats.htm file. The sequence data were aligned to the reference human genome (GRCh37) and variant calls were generated using CASAVA. Exons plus at least 2 bases into the 5′ and 3′ ends of all the introns were analyzed. The Human Gene Mutation Database (HGMD),Citation12 the Single Nucleotide Polymorphism database (dbSNP),Citation13 1000 genomes,Citation14 HapMap data,Citation15 and online search engines (e.g., PubMed) were used to search for previously described gene mutations and polymorphisms. Stepwise filtering removed common SNPs, intergenic and 3′/5′ UTR variants, non-splice-related intronic variants, and synonymous variants. Variants were then filtered further based on possible recessive, X linked or de novo mutations. Data were annotated with the Ambry Variant Analyzer tool (AVA), including nucleotide and amino acid conservation, biochemical nature of amino acid substitutions, population frequency (ESP and 1000 genomes,Citation14 and predicted functional impact (including PolyPhenCitation16 and SIFTCitation17 in silico prediction tools). Multiple sequence alignments were viewed using IGV (integrative genomics Viewer software).Citation18 Identified candidate alterations were confirmed using automated fluorescence dideoxy sequencing in the proband and parents on an ABI3730 (Life Technologies) using standard procedures.
| Abbreviations: | ||
| ADHD | = | attention deficit hyperactivity disorder |
| DICIPHER | = | Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources |
| EEG | = | electroencephalogram |
| FISH | = | fluorescent in Situ hybridization |
| MERRF | = | myoclonic epilepsy ragged red fibers |
| SNAP25 | = | synaptosomal associated protein-25kDa |
| SNARE | = | soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| SNP | = | single nucleotide polymorphisms |
Acknowledgments
We thank the patient and her family for their contribution.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Julie G, Hamdan FF, Rouleau GA. A strategy to identify de novo mutations in common disorders such as autism and schizophrenia. J Vis Exp 2011; http://dx.doi.org/10.3791/2534; PMID: 21712793
- Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol 2011; 12:228; http://dx.doi.org/10.1186/gb-2011-12-9-228; PMID: 21920049
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, et al. A de novo paradigm for mental retardation. Nat Genet 2010; 42:1109 - 12; http://dx.doi.org/10.1038/ng.712; PMID: 21076407
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011; 43:585 - 9; http://dx.doi.org/10.1038/ng.835; PMID: 21572417
- Garofalo S, Cornacchione M, Di Costanzo A. From genetics to genomics of epilepsy. Neurol Res Int 2012; 2012:876234; http://dx.doi.org/10.1155/2012/876234; PMID: 22645681
- Bozzi Y, Casarosa S, Caleo M. Epilepsy as a neurodevelopmental disorder. Front Psychiatry 2012; 3:19; http://dx.doi.org/10.3389/fpsyt.2012.00019; PMID: 22457654
- Ariani F, Hayek G, Rondinella D, Artuso R, Mencarelli MA, Spanhol-Rosseto A, Pollazzon M, Buoni S, Spiga O, Ricciardi S, et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet 2008; 83:89 - 93; http://dx.doi.org/10.1016/j.ajhg.2008.05.015; PMID: 18571142
- Tao J, Van Esch H, Hagedorn-Greiwe M, Hoffmann K, Moser B, Raynaud M, Sperner J, Fryns JP, Schwinger E, Gécz J, et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet 2004; 75:1149 - 54; http://dx.doi.org/10.1086/426460; PMID: 15499549
- Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci 2003; 26:599 - 625; http://dx.doi.org/10.1146/annurev.neuro.26.010302.081210; PMID: 14527270
- Bassuk AG, Wallace RH, Buhr A, Buller AR, Afawi Z, Shimojo M, Miyata S, Chen S, Gonzalez-Alegre P, Griesbach HL, et al. A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am J Hum Genet 2008; 83:572 - 81; http://dx.doi.org/10.1016/j.ajhg.2008.10.003; PMID: 18976727
- Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 2009; 27:182 - 9; http://dx.doi.org/10.1038/nbt.1523; PMID: 19182786
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med 2009; 1:13; http://dx.doi.org/10.1186/gm13; PMID: 19348700
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001; 29:308 - 11; http://dx.doi.org/10.1093/nar/29.1.308; PMID: 11125122
- Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA, 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010; 467:1061 - 73; http://dx.doi.org/10.1038/nature09534; PMID: 20981092
- International HapMap Consortium. The International HapMap Project. Nature 2003; 426:789 - 96; http://dx.doi.org/10.1038/nature02168; PMID: 14685227
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7:248 - 9; http://dx.doi.org/10.1038/nmeth0410-248; PMID: 20354512
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4:1073 - 81; http://dx.doi.org/10.1038/nprot.2009.86; PMID: 19561590
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol 2011; 29:24 - 6; http://dx.doi.org/10.1038/nbt.1754; PMID: 21221095
- Südhof TC, Rizo J. Synaptic vesicle exocytosis. Cold Spring Harb Perspect Biol 2011; 3:1 - 14; http://dx.doi.org/10.1101/cshperspect.a005637; PMID: 22026965
- Hodel A. SNAP-25. Int J Biochem Cell Biol 1998; 30:1069 - 73; http://dx.doi.org/10.1016/S1357-2725(98)00079-X; PMID: 9785471
- Vilinsky I, Stewart BA, Drummond J, Robinson I, Deitcher DL. A Drosophila SNAP-25 null mutant reveals context-dependent redundancy with SNAP-24 in neurotransmission. Genetics 2002; 162:259 - 71; PMID: 12242238
- Jeans AF, Oliver PL, Johnson R, Capogna M, Vikman J, Molnár Z, Babbs A, Partridge CJ, Salehi A, Bengtsson M, et al. A dominant mutation in Snap25 causes impaired vesicle trafficking, sensorimotor gating, and ataxia in the blind-drunk mouse. Proc Natl Acad Sci U S A 2007; 104:2431 - 6; http://dx.doi.org/10.1073/pnas.0610222104; PMID: 17283335
- Kataoka M, Yamamori S, Suzuki E, Watanabe S, Sato T, Miyaoka H, Azuma S, Ikegami S, Kuwahara R, Suzuki-Migishima R, et al. A single amino acid mutation in SNAP-25 induces anxiety-related behavior in mouse. PLoS One 2011; 6:e25158; http://dx.doi.org/10.1371/journal.pone.0025158; PMID: 21949876
- Corradini I, Donzelli A, Antonucci F, Welzl H, Loos M, Martucci R, De Astis S, Pattini L, Inverardi F, Wolfer D, et al. Epileptiform Activity and Cognitive Deficits in SNAP-25+/- Mice are Normalized by Antiepileptic Drugs. Cereb Cortex 2012; http://dx.doi.org/10.1093/cercor/bhs316; PMID: 23064108
- Johansson JU, Ericsson J, Janson J, Beraki S, Stanić D, Mandic SA, Wikström MA, Hökfelt T, Ogren SO, Rozell B, et al. An ancient duplication of exon 5 in the Snap25 gene is required for complex neuronal development/function. PLoS Genet 2008; 4:e1000278; http://dx.doi.org/10.1371/journal.pgen.1000278; PMID: 19043548
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 1995; 375:488 - 93; http://dx.doi.org/10.1038/375488a0; PMID: 7777057
- Garcia CC, Blair HJ, Seager M, Coulthard A, Tennant S, Buddles M, Curtis A, Goodship JA. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet 2004; 41:183 - 6; http://dx.doi.org/10.1136/jmg.2003.013680; PMID: 14985377
- Hamdan FF, Piton A, Gauthier J, Lortie A, Dubeau F, Dobrzeniecka S, Spiegelman D, Noreau A, Pellerin S, Côté M, et al. De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol 2009; 65:748 - 53; http://dx.doi.org/10.1002/ana.21625; PMID: 19557857
- Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, Uruno K, Kumada S, Nishiyama K, Nishimura A, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008; 40:782 - 8; http://dx.doi.org/10.1038/ng.150; PMID: 18469812
- Otsuka M, Oguni H, Liang JS, Ikeda H, Imai K, Hirasawa K, Imai K, Tachikawa E, Shimojima K, Osawa M, et al. STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome--result of Japanese cohort study. Epilepsia 2010; 51:2449 - 52; http://dx.doi.org/10.1111/j.1528-1167.2010.02767.x; PMID: 21204804
- Paciorkowski AR, Thio LL, Rosenfeld JA, Gajecka M, Gurnett CA, Kulkarni S, Chung WK, Marsh ED, Gentile M, Reggin JD, et al. Copy number variants and infantile spasms: evidence for abnormalities in ventral forebrain development and pathways of synaptic function. Eur J Hum Genet 2011; 19:1238 - 45; http://dx.doi.org/10.1038/ejhg.2011.121; PMID: 21694734
- http://decipher.sanger.ac.uk