Abstract
Myotonic dystrophy type 1 (DM1), an incurable, neuromuscular disease, is caused by the expansion of CTG repeats within the 3′ UTR of DMPK on chromosome 19q. In DM1 patients, mutant DMPK transcripts deregulate RNA metabolism by altering CUG RNA-binding proteins. Several approaches have been proposed for DM1 therapy focused on specific degradation of the mutant CUG repeats or on correction of RNA-binding proteins, affected by CUG repeats. One such protein is CUG RNA-binding protein (CUGBP1). The ability of CUGBP1 to increase or inhibit translation depends on phosphorylation at Ser302, which is mediated by cyclin D3-CDK4. The mutant CUG repeats increase the levels of CUGBP1 protein and inhibit Ser302 phosphorylation, leading to the accumulation of CUGBP1 isoforms that repress translation (i.e., CUGBP1REP). Elevation of CUGBP1REP in DM1 is caused by increased GSK3β kinase, which reduces the cyclin D3-CDK4 pathway and subsequent phosphorylation of CUGBP1 at Ser302. In this review, we discuss our recent discovery showing that correction of GSK3β activity in the DM1 mouse model (i.e., HSALR mice) reduces DM1 muscle pathology. These findings demonstrate that GSK3β is a novel therapeutic target for treating DM1.
Introduction
Myotonic dystrophy type 1 (DM1) is an autosomal dominant inherited disease that affects many tissues. In skeletal muscle, this condition leads to weakness, atrophy, and myotonia.Citation1 In addition to skeletal muscle symptoms, patients with DM1 develop arrythmias and cardiomyopathy in the heart, neurological abnormalities, and insulin resistance. DM1 is caused by a mutation in the dystrophia myotonica protein kinase (DMPK) gene, which results in an expansion of CTG trinucleotide repeats within its 3′ UTR.Citation2,Citation3 CTG expansions are highly unstable and vary in length in normal individuals from 5 to 35 repeats. In patients with DM1, the length of CTG expansions, which correlates with the severity of clinical symptoms, varies from 50 to several thousand repeats.
Development of DM1 therapy requires elucidation of the molecular mechanisms by which CTG repeats lead to DM1 pathology. Studies have established that CTG repeats lead to DM1 pathology by causing the accumulation of mutant DMPK mRNA transcripts containing an expanded array of CUG repeats that target RNA-binding proteins, thereby changing their activities and altering RNA homeostasis in DM1 cells.Citation4-Citation8 The most studied RNA-binding proteins affected by CUG repeats are CELF1 (also known as CUGBP1) and MBNL1. CUGBP1 is a member of the family of CUGBP and ELAV-like factors, whereas MBNL1 belongs to the muscleblind-like (MBNL) family of proteins.Citation9-Citation11
The mutant CUG repeats primarily cause two toxic events in DM1 cells. First, the aggregation of mutant DMPK transcripts, possibly due to disruption of their subsequent processing,Citation11,Citation12 can lead to sequestration of MBNL1 proteins, thereby reducing their splicing activityCitation11,Citation13 (). Second, the mutant CUG repeats increase CUGBP1 stability, leading to elevated levels of this protein in DM1 cellsCitation14,Citation15 (). This stabilization is mediated by an increase in PKC-mediated phosphorylation of CUGBP1 and its subsequent binding to soluble CUG repeats.Citation14,Citation16
Figure 1. Toxic events caused by mutant CUG repeats in DM1 cells and possible therapeutic approaches for their correction. Mutant CUG repeats cause three toxic molecular events: (1) sequestration of MBNL1, (2) higher levels of CUGBP1, leading to elevation of the active form of CUGBP1 (CUGBP1ACT), and (3) elevation of active GSK3β, which reduces cyclin D3 and converts a portion of CUGBP1ACT to CUGBP1REP. Thus, both CUGBP1 forms are elevated in DM1. Reduced MBNL1 and increased CUGBP1ACT lead to deregulation of mRNA splicing, translation, and stability. Futhermore, increased CUGBP1REP may reduce mRNA translation in stress granules. Taken together, these molecular changes lead to myotonia, weakness, and muscle atrophy. Administration of GSK3β inhibitors reduces DM1 muscle histopathology, weakness, and myotonia similar to degradation of mutant CUG repeats by AONs.
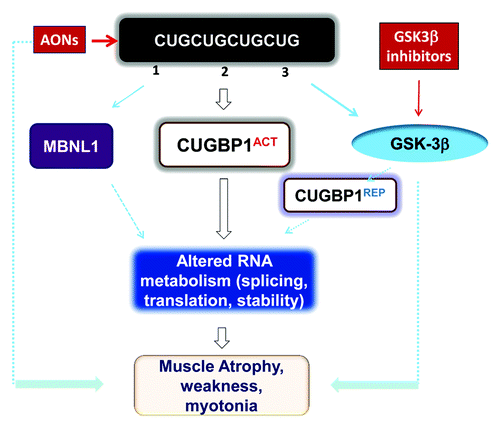
However, these mutant CUG repeats also disrupt CUGBP1 phosphorylation by other kinases. CUGBP1 contains a number of serine and threonine residues that regulate its activity and intracellular localization. Ser28 is normally phosphorylated by Akt to control the nuclear-cytoplasmic distribution of CUGBP1.Citation17 Another important phosphorylation site is Ser302, which is located within the spacer between RNA-binding domains 2 and 3. Ser302 phosphorylation, which is mediated by cyclin D3-dependent kinase 4 (CDK4),Citation17 promotes CUGBP1 binding to eukaryotic translation initiation factor 2α (eIF2α) and the subsequent association of CUGBP1 with polysomes.Citation17-Citation19 Because of its positive role in activating the translation of several mRNAs, encoding p21, HDAC1, MEF2A, and cyclin D1, phospho-Ser302 CUGBP1 was designated as CUGBP1ACT. Although CUGBP1ACT binds to eIF2α, it does not affect global translation.
We found that a significant proportion of CUGBP1 in DM1 cells is un-phosphorylated at Ser302.Citation17 In contrast to the phosphorylated form, un-phosphorylated Ser302 CUGBP1 binds to inactive (phospho-Ser51) eIF2α, thereby forming the inactive CUGBP1REP-eIF2α complex. This repressor complex is localized within stress granules (SGs) to trap mRNAs and delay their translation.Citation17,Citation20 We found that one CUGBP1 target, MRG15, which encodes a protein, involved in DNA repair and chromatin remodeling, is bound by CUGBP1REP in DM1 cells, leading to reduced MRG15 protein levels.Citation17 These data showed that CUGBP1REP may repress the translation of many additional mRNAs in cytoplasmic aggregates or SGs, and that Ser302 phosphorylation of CUGBP1 by cyclin D3-CDK4 may act to switch the function of CUGBP1 from repressor to activator of translation. Interestingly, both forms of CUGBP1 are elevated in DM1. In addition to regulating translation, CUGBP1 controls mRNA splicing and stability.Citation17-Citation19,Citation21-Citation24 Therefore, elevation of CUGBP1ACT in DM1 may increase the translation of some mRNAs and change the splicing and stability of CUGBP1 targets.
Our recently published work showed that the increase in CUGBP1REP in DM1 cells is due to elevated levels of activated GSK3β25. Consistent with this, GSK3β levels are also increased in the skeletal muscle of HSALR mice, which express tracks of 250 CUG repeats within the 3′ UTR of human skeletal actin and is commonly used model of CUG repeat RNA mediated pathology in DM1.Citation5,Citation25 GSK3β has many substrates, including cyclin D3.Citation26 Phosphorylation of cyclin D3 by GSK3β in DM1 patients and HSALR mice results in reduced cyclin D3 protein levels because phosphorylated cyclin D3 is targeted for degradation. In agreement with this reduction in cyclin D3 in DM1 cells, CUG repeats convert CUGBP1ACT to CUGBP1REP.
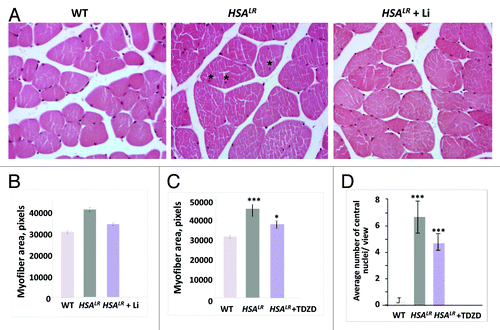
Once alterations to the GSK3β pathway were discovered, we investigated whether GSK3 inhibitors can correct the pathology of DM1. Analysis of HSALR mice treated with inhibitors of GSK3, namely lithium and TDZD-8, showed that elevated GSK3β plays a crucial role in DM1 pathology. We found that both inhibitors significantly increased grip strength in HSALR mice and improved DM1 muscle histology.Citation25 Moreover, treatment of HSALR mice with lithium or TDZD-8 significantly reduced variability in myofiber size (), as well as the number of central nuclei in HSALR muscleCitation25 (). Based on these data, we concluded that the elevated GSK3β is the third major toxic event in DM1 pathogenesis ().
Figure 2. Correction of muscle histopathology in HSALR mice treated with GSK3 inhibitors, namely lithium and TDZD-8. (A) Hematoxylin and eosin staining of muscle sections (gastrocnemius) from age-matched 6-mo-old WT, untreated HSALR mice, and lithium-treated HSALR mice. Internal nuclei in the muscle of untreated HSALR mice are indicated by asterisks. (B) Lithium reduces myofiber size variability in HSALR mice. Myofiber area was compared in gastrocnemius from age-matched 6-mo-old WT mice and HSALR mice untreated and treated for 2 weeks with lithium. The y-axis shows the average myofiber area in pixels. p < 3.36 × 10−16 (untreated HSALR mice vs. WT mice); p < 1.10 × 10−7 (treated vs. untreated HSALR mice). (C) TDZD-8 treatment reduces myofiber size variability in HSALR muscle. Myofiber area was increased in 4-mo-old HSALR mice relative to WT mice (***p < 5.88 × 10−6). The myofiber area was reduced in HSALR mice after TDZD-8 treatment. *p < 0.02677 (treated HSALR mice vs. untreated). (D) Treatment of HSALR mice with TDZD-8 reduces the number of central nuclei in the skeletal muscle (gastrocnemius) of HSALR mice. The number of central nuclei was counted in six randomly selected 20 × views and the average values are shown. The average number of central nuclei per view was increased in HSALR mice (***p < 4.616 × 10−8) relative to WT mice. However, treatment with TDZD-8 reduced the number of central nuclei. ***p < 0.000387 (treated HSALR mice vs. untreated).
Initially, we expected that deregulation of the GSK3β-cyclin D3-CDK4-CUGBP1 pathway may be responsible for muscle atrophy in DM1. However, treatment with lithium and TDZD-8 also reduced myotonia in HSALR mice.Citation25 Thus, it is possible that the correction of CUGBP1 activity may not only “normalize” the translational activity of CUGBP1, but also its splicing activity. This correction may lead to the correction of the splicing of one myotonia-associated target of CUGBP1, the chloride channel.Citation23 Although we found that lithium converts CUGBP1REP into CUGBP1ACT in HSALR muscle, further investigation is necessary to determine whether the GSK3β-cyclin D3-CDK4 pathway plays a major role in reducing myotonia in DM1 mice or whether GSK3β inhibition affects other pathways in these mice. Taken together, we found that GSK3 inhibitors reduce DM1 pathology in HSALR mice similar to the degradation of mutant CUG repeats by antisense oligonucleotides (AONs)Citation27-Citation29 ().
Our initial experiments in which 6-mo-old HSALR mice were treated with GSK3 inhibitors were based on protocols using short treatment durations, specifically 2–7 d for TDZD-8 or 2 weeks for lithium.Citation25 We noticed that grip strength increased and that myotonia was reduced in the treated mice immediately after treatment. However, once treatment was completed, HSALR mice developed muscle weakness again. These results suggest that, to maintain “normalized” grip strength in adult HSALR mice, GSK3 inhibitors must be administered continuously if the treatment protocols tested in our recent study are used.Citation25 In contrast, myotonia in these mice remained lower for several weeks after cessation of treatment.Citation25 The mechanisms responsible for the long beneficial effect of the GSK3 inhibitors on myotonia in HSALR mice remain to be studied. Interestingly, the effects of these inhibitors on grip weakness were more pronounced in 6-week-old HSALR mice. Treatment of 6-week-old HSALR mice with TDZD-8 for 1 week significantly increased the grip strength in these mice when evaluated at 3 mo of age.Citation25 These data suggest that inhibition of GSK3β in HSALR mice at a pre-symptomatic age may be beneficial to reducing the incidence of DM1 pathology in adult HSALR mice.
Conclusions
In summary, our study found increased levels of GSK3β in the skeletal muscle of DM1 patients and HSALR mice.Citation25 This alteration seems to be a key event in DM1 pathology because GSK3 inhibitors can reduce dysfunctional muscle pathology in the DM1 mouse model. It remains to be determined whether GSK3β plays a role in the development of other DM1 symptoms, such as insulin resistance and defects in cardiac and neurological functions. Studies have demonstrated that GSK3β is increased in other conditions, including type 2 diabetes mellitus.Citation30 Therefore, elevated GSK3β may contribute to the development of insulin resistance in DM1. Since GSK3 inhibitors are used to treat neurological disorders such as bipolar disease,Citation30 these compounds may also improve neurological dysfunction in patients with DM1 in addition to correcting skeletal muscle defects.
| Abbreviations: | ||
| DMI | = | myotonic dystrophy type 1 |
| GSK3β | = | glycogen-synthase kinase-3β |
| DMPK | = | dystrophia myotonica protein kinase |
| TDZD-8 | = | 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione |
Disclosure of Potential Conflict of Interest
No potential conflict of interest was disclosed.
Acknowledgments
The authors are thankful to Dr Charles Thornton (University of Rochester) for providing HSALR mice. LT is supported by grants from the National Institute of Health (2R01-AR044387–13, 2R01-AR052791–07, and R21-NS078659). NAT is supported by NIH grants GM551888, CA100070, AG039885, AG028865, and CA159942.
References
- Harper PS. Myotonic Dystrophy. London: WB Saunders, 2001.
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992; 68:799 - 808; http://dx.doi.org/10.1016/0092-8674(92)90154-5; PMID: 1310900
- Fu YH, Pizzuti A, Fenwick RG Jr., King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992; 255:1256 - 8; http://dx.doi.org/10.1126/science.1546326; PMID: 1546326
- Timchenko LT. Myotonic dystrophy: the role of RNA CUG triplet repeats. Am J Hum Genet 1999; 64:360 - 4; http://dx.doi.org/10.1086/302268; PMID: 9973273
- Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000; 289:1769 - 73; http://dx.doi.org/10.1126/science.289.5485.1769; PMID: 10976074
- Seznec H, Agbulut O, Sergeant N, Savouret C, Ghestem A, Tabti N, Willer JC, Ourth L, Duros C, Brisson E, et al. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum Mol Genet 2001; 10:2717 - 26; http://dx.doi.org/10.1093/hmg/10.23.2717; PMID: 11726559
- Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci 2006; 29:259 - 77; http://dx.doi.org/10.1146/annurev.neuro.29.051605.113014; PMID: 16776586
- Schoser B, Timchenko L. Myotonic dystrophies 1 and 2: complex diseases with complex mechanisms. Curr Genomics 2010; 11:77 - 90; http://dx.doi.org/10.2174/138920210790886844; PMID: 20885816
- Timchenko LT, Timchenko NA, Caskey CT, Roberts R. Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: implications for myotonic dystrophy. Hum Mol Genet 1996; 5:115 - 21; http://dx.doi.org/10.1093/hmg/5.1.115; PMID: 8789448
- Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res 1996; 24:4407 - 14; http://dx.doi.org/10.1093/nar/24.22.4407; PMID: 8948631
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 2000; 19:4439 - 48; http://dx.doi.org/10.1093/emboj/19.17.4439; PMID: 10970838
- Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol 1995; 128:995 - 1002; http://dx.doi.org/10.1083/jcb.128.6.995; PMID: 7896884
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science 2003; 302:1978 - 80; http://dx.doi.org/10.1126/science.1088583; PMID: 14671308
- Timchenko NA, Cai Z-J, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem 2001; 276:7820 - 6; http://dx.doi.org/10.1074/jbc.M005960200; PMID: 11124939
- Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 2001; 29:40 - 7; http://dx.doi.org/10.1038/ng704; PMID: 11528389
- Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell 2007; 28:68 - 78; http://dx.doi.org/10.1016/j.molcel.2007.07.027; PMID: 17936705
- Huichalaf C, Sakai K, Jin B, Jones K, Wang G-L, Schoser B, Schneider-Gold C, Sarkar P, Pereira-Smith OM, Timchenko N, et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J 2010; 24:3706 - 19; http://dx.doi.org/10.1096/fj.09-151159; PMID: 20479119
- Timchenko NA, Wang G-L, Timchenko LT. RNA CUG-binding protein 1 increases translation of 20-kDa isoform of CCAAT/enhancer-binding protein beta by interacting with the alpha and beta subunits of eukaryotic initiation translation factor 2. J Biol Chem 2005; 280:20549 - 57; http://dx.doi.org/10.1074/jbc.M409563200; PMID: 15788409
- Timchenko LT, Salisbury E, Wang G-L, Nguyen H, Albrecht JH, Hershey JWB, Timchenko NA. Age-specific CUGBP1-eIF2 complex increases translation of CCAAT/enhancer-binding protein beta in old liver. J Biol Chem 2006; 281:32806 - 19; http://dx.doi.org/10.1074/jbc.M605701200; PMID: 16931514
- Anderson P, Kedersha N. RNA granules. J Cell Biol 2006; 172:803 - 8; http://dx.doi.org/10.1083/jcb.200512082; PMID: 16520386
- Paillard L, Omilli F, Legagneux V, Bassez T, Maniey D, Osborne HB. EDEN and EDEN-BP, a cis element and an associated factor that mediate sequence-specific mRNA deadenylation in Xenopus embryos. EMBO J 1998; 17:278 - 87; http://dx.doi.org/10.1093/emboj/17.1.278; PMID: 9427761
- Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998; 280:737 - 41; http://dx.doi.org/10.1126/science.280.5364.737; PMID: 9563950
- Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA, Charlet-BN. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell 2002; 10:45 - 53; http://dx.doi.org/10.1016/S1097-2765(02)00572-5; PMID: 12150906
- Rattenbacher B, Beisang D, Wiesner DL, Jeschke JC, von Hohenberg M, St Louis-Vlasova IA, Bohjanen PR. Analysis of CUGBP1 targets identifies GU-repeat sequences that mediate rapid mRNA decay. Mol Cell Biol 2010; 30:3970 - 80; http://dx.doi.org/10.1128/MCB.00624-10; PMID: 20547756
- Jones K, Wei C, Iakova P, Bugiardini E, Schneider-Gold C, Meola G, Woodgett J, Killian J, Timchenko NA, Timchenko LT. GSK3β mediates muscle pathology in myotonic dystrophy. J Clin Invest 2012; 122:4461 - 72; http://dx.doi.org/10.1172/JCI64081; PMID: 23160194
- Naderi S, Gutzkow KB, Låhne HU, Lefdal S, Ryves WJ, Harwood AJ, Blomhoff HK. cAMP-induced degradation of cyclin D3 through association with GSK-3beta. J Cell Sci 2004; 117:3769 - 83; http://dx.doi.org/10.1242/jcs.01210; PMID: 15252116
- Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 2009; 325:336 - 9; http://dx.doi.org/10.1126/science.1173110; PMID: 19608921
- Mulders SA, van den Broek WJ, Wheeler TM, Croes HJ, van Kuik-Romeijn P, de Kimpe SJ, Furling D, Platenburg GJ, Gourdon G, Thornton CA, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci U S A 2009; 106:13915 - 20; http://dx.doi.org/10.1073/pnas.0905780106; PMID: 19667189
- Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, Thornton CA. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012; 488:111 - 5; http://dx.doi.org/10.1038/nature11362; PMID: 22859208
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 2003; 116:1175 - 86; http://dx.doi.org/10.1242/jcs.00384; PMID: 12615961