Abstract
Recently, loss-of-function mutations in PTPN11 were linked to the cartilage tumor syndrome metachondromatosis (MC), a rare inherited disorder featuring osteochondromas, endochondromas and skeletal deformation. However, the underlying molecular and cellular mechanism for MC remained incompletely understood. By studying the role of the Src homology-2 domain-containing protein tyrosine phosphatase Shp2 (encoded by mouse Ptpn11) in cathepsin K-expressing cells, we identified a novel cell population in the perichondrial groove of Ranvier. In the absence of Shp2, these cells exhibit elevated Indian hedgehog (Ihh) signaling, proliferate excessively and cause ectopic cartilage formation and tumors. Our findings establish a critical role for a protein-tyrosine phosphatase (PTP) family member, in addition to the well-known roles of receptor tyrosine kinases (RTKs), in cartilage development and homeostasis. However, whether Shp2 deficiency in other epiphyseal chondroid cells and whether signaling pathways in addition to the IHH/Parathyroid Hormone–related Peptide (PTHrP) axis attribute to the formation of enchondromas and osteochondromas remains elusive. Understanding how chondrogenic events are regulated by SHP2 could aid in the development of novel therapeutic approaches to prevent and treat cartilage diseases, such as MC and osteoarthritis (OA).
Introduction
Metachondromatosis (MC) is a rare skeletal disorderCitation1-Citation3 characterized by cartilaginous cysts (enchondromas) and osteocartilaginous exostosis-like lesions (osteochondromas).Citation4,Citation5 The enchondromas in MC mainly affect the iliac crests and metaphyses of tubular bones of the lower extremities, while osteochondromas are typically seen in the hands and feet (primarily in the digits and toes). Occasionally, there is the vertebral and flat bone involvement,Citation6 and avascular necrosis of the femoral epiphysis is another common feature.Citation7 These lesions can regress spontaneouslyCitation8 or transform to chondrosarcoma.Citation9 The actual prevalence of MC is unknown, although more than 30 cases have been reported. Diagnosis is made on the basis of clinical signs, radiographic findings and familial history,Citation8 and treatment relies primarily on surgical intervention to ameliorate symptoms and/or skeletal deformity.Citation4,Citation5
MC was known to show autosomal dominant inheritance, but the molecular basis for this skeletal disorder had been obscure. Recently, mutations in the PTPN11 gene (located on 12q24) were linked to MC.Citation1,Citation2 PTPN11 encodes SHP2, one of two vertebrate Src homology-2 domain containing PTPs. SHP2 is expressed ubiquitously and is required for RAS/ERK pathway activation in most RTK, cytokine receptor and integrin signaling pathways.Citation10,Citation11 The RAS/ERK pathway regulates a broad spectrum of biological responses including, but not limited to, cell viability, proliferation, differentiation and migration. Not surprisingly, alterations in RAS/ERK pathway function contribute to various diseases, affecting multiple organs and tissues.Citation12,Citation13
The PTPN11 mutations reported in MC families include deletions, nonsense mutations and splice site mutations.Citation1,Citation2 In contrast to the gain-of-function PTPN11 mutations associated with other human neoplasms (e.g., juvenile myelomonocytic leukemia (JMML), acute myelogenous (AML) and lymphoblastic leukemia (ALL), neuroblastoma and other solid tumors),Citation14 MC mutations result in functionally null alleles and represent a new category of human disease-associated PTPN11 mutations. Intriguingly, we found that transgenic mice lacking Shp2 in a specific subset of chondroid progenitor cells recapitulate most of the features of MC.Citation15 Our mouse MC model, along with published data in human MC, establishes an important role for SHP2 in cartilage development and homeostasis. These studies also provide the first insight into PTP function in this cell type, in contrast to the well-studied roles for specific RTKs in this system.
PTPN11 Mutations in Metachondromatosis
PTPN11 is essential for the development and/or homeostasis of multiple organs/tissues. Global Ptpn11 deletion results in early embryonic lethality.Citation16,Citation17 Germ line and somatic mutations in PTPN11 have been identified and linked with human diseases.Citation10,Citation11PTPN11 mutations that encode catalytically defective (but not inactive) forms of SHP2 and impair the activation of the RAS/ERK signaling pathway cause LEOPARD syndrome (LS),Citation18 a rare genetic disorder characterized by lentigines, ECG abnormalities; ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retarded growth and deafness. In contrast to LS mutations, PTPN11 mutations that lower the threshold for activation of SHP2 cause Noonan Syndrome (NS),Citation19 which typically presents with some combination of facial abnormalities, short stature, heart defects, cognitive deficits, bleeding problems and/or skeletal malformations. NS-associated SHP2 mutants increase RAS/ERK signaling in several different cell types/tissues, which result in the observed phenotypic abnormalities.Citation20-Citation22 Somatic PTPN11 mutations that cause constitutive activation of SHP2 contribute to the pathogenesis of certain types of myeloproliferative disorders and leukemias, most prominently JMML, but also AML and ALL, as well as some solid tumors.Citation11,Citation14,Citation23
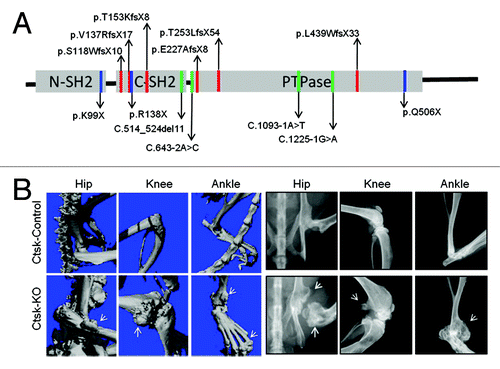
The PTPN11 mutations in MC are distinct from those seen in NS and LS. Using whole-genome sequencing and partial linkage analysis, Sobreira et al.Citation2 first reported that nonsense and deletion mutations that cause premature translation termination co-segregate with the MC phenotype. Shortly thereafter, additional heterozygous loss-of-function mutations in PTPN11 were identified in MC patients by linkage analysis with high-density SNP arrays.Citation1 These included frameshift, nonsense and splice-site mutations, as well as deletions.Citation1 A diagram showing all mutations identified in human MC can be found in . Clearly, their location and predicted effects on SHP2 suggested that MC arises from PTPN11 loss-of-function/null alleles, which were not found in Ollier disease and Maffucci syndrome,Citation1 the other two cartilage tumor syndromes in human. However, whether PTPN11 loss-of-function mutations exist in other cartilage neoplasms (e.g., chondrosarcomas) remains unknown.
Figure 1. (A) PTPN11 mutations identified in MC patients. The locations of MC mutations in the corresponding SHP2 structure are indicated by red, blue and green colored lines, representing frameshift, nonsense and splice-site mutations, respectively. Predicted protein changes are indicated with arrows. Please see references Citation1 and Citation2 for original references. (B) Micro-CT and Faxitron images demonstrate the existence of osteochondromas and enchondromas (arrows) at hip, knee and ankles of 12-week-old mice lacking SHP2 in cathepsin K-expressing cells (CtskCre;Ptpn11fl/fl, Ctsk-KO) but not its littermate controls (CtskCre;Ptpn11fl/+, Ctsk-Control).
Cell-of-Origin of Metachondromatosis
The cell-of-origin for the osteochondroma(s) and enchondroma(s) in MC also had been unclear/controversial.Citation24 Anatomically, MC lesions appear adjacent to the growth plate, suggesting cells from the growth plate, periosteum, perichondrium, or groove of Ranvier as candidates. Osteochondromas morphologically resemble growth plate cartilage and preferentially occur at the metaphyses of tubular bones; hence, epiphyseal chondrocytes were assumed to be their cell-of-origin.Citation25 Indeed, Jones et al.Citation26 appeared to have proved this hypothesis by means of a mouse model in which Ext1, which encodes a glycosyltransferase involved in heparan sulfate (HS) biosynthesis, was deleted in Col2α1-expressing cells, but not periosteum. However, because that Col2α1 promoter also is active in cells residing in the groove, the involvement of groove cells in the initiation of osteochondromas was not excluded by these studies.Citation27
In an attempt to study Shp2 function in osteoclast development in vivo using cathepsin K (Ctsk)-Cre-directed Ptpn11 deletion, we unexpected observed large numbers of enchondromas and osteochondromas (), a phenotype strongly resembling the clinical features of human MC. Further analysis demonstrated that the skeletal disease in these mutant mice was not transferrable by bone marrow transplantation, and thus could not arise from osteoclasts. Subsequent lineage tracing studies using Rosa26LSLlacZ and Rosa26LSLYFP alleles revealed the presence of a previously undefined subset of Ctsk-expressing cells residing in the groove of Ranvier. Moreover, the absence of Shp2 in these cells caused their excessive proliferation, chondrogenic differentiation and eventually, cartilage tumor formation. Phenotypically these cells shared markers for mesenchymal stem cells (MSC), such as CD44, CD90, Stro-1 and CD166 and, under appropriate conditions, could undergo differentiation into bone, fate and cartilage ex vivo.Citation15 Based on these properties, we proposed these cells be renamed “Cathepsin K-expressing Chondroid Progenitors” (CCPs). Much remains to be learned about the property and physiology of CCPs; in particular, it will be informative to define the relationship between CCPs and bona fide MSCs.
To ask whether growth plate chondrocytes participate in osteochondroma development, as reported by Jones et al.Citation26 for Ext1 deletion, or enchondromas, we crossed our Ptpn11 floxed allele to Col2CreERT2 and Col10a1-Cre mice to delete Ptpn11 specifically in proliferating (after tamoxifen induction) or hypertrophic chondrocytes, respectively. In marked contrast to the effects of Ctsk-Cre-evoked deletion, no osteochondromas were observed in mice lacking Shp2 in Col2α1-expressing cells over 6 mo of observation. Occasionally, solitary enchondromas developed in a few mice that lacked Shp2 in Col2α1- or Col10α1-expressing cells (Yu Y and Yang W, manuscript in preparation). Given that Ctsk+ groove of Ranvier cells could migrate into articular and growth plate cartilage, it is unclear whether those enchondromas arose from “native” physeal chondrocytes or from progeny of the Ctsk+ groove of Ranvier cells; further studies are needed to address these issues. Taken together, though, our data clearly show that the cell-of-origin for MC is CCPs, and the effects of Ext1 and Ptpn11 deletion suggest a different cell-of-origin for hereditary osteochromas and MC, respectively. It remains unclear (and an important topic for future research) whether Col2α1- and/or Col10α1Cre-expressing cells derive from Ctsk-Cre-expressing cells or represent a distinct lineage.
Molecular Pathogenesis of MC
Perichondrial cells originate from the outer edges of the mesenchymal condensation, and these cells have distinct properties.Citation28,Citation29 During skeletal development and homeostasis, perichondrial cells can send and receive signals from the adjacent growth plate. In turn, these signals can influence the perichondrium and surrounding chondroid cells.Citation28 The growth plate comprises cartilaginous cells in sequential resting, proliferating, pre-hyperthrophic and hyperthrophic stages. At the distal end of the plate, hypertrophic chondrocytes undergo apoptosis and serve as scaffolds to facilitate new bone formation by invading osteoblasts.Citation30 Skeletal growth is achieved by the progressive replenishment and replacement of the growth plate cartilage by chondrocytes and bone cells acting in concert.Citation31,Citation32
Enchondromas and osteochondromas arise at the nexus of the growth plate and epiphyseal cartilage of tubular bones, suggesting that deregulation of signaling pathways that modulate growth plate chondrocytes and perichondrial cells could contribute to the pathogenesis of these lesions.Citation25 Multiple signaling pathways, including the TGFβ, BMP, FGF, IGF1, IHH, PTHrP, and Wnt/β-catenin pathways, regulate chondrogenesis and endochondral bone formation.Citation31,Citation33-Citation37 Among these, the FGFR3, PTHrP and IHH signaling pathways are critical. FGF18 is expressed predominantly in the perichondrium and signals through FGFR3 and the ERK MAPK pathway to negatively regulate IHH expression by pre-hypertrophic and hypertrophic chondrocytes.Citation38,Citation39 IHH, in turn, can induce PTHrP expression in the periarticular perichondrium. IHH signaling normally stimulates chondrocyte proliferation, whereas IHH/PTHrP, acting together, intitiate a negative feedback loop that inhibits the differentiation of proliferating chondrocytes and sustains their proliferation.Citation40 Enchondromas are assumed to result from a failure in terminal differentiation of growth plate chondrocytes,Citation25 a notion supported by the observation that constitutively active Hedgehog or PTHrP signaling causes enchondromasCitation41,Citation42 and osteochondromas.Citation15 Moreover, altered HH signaling is observed in osteochondromas arising from mutations in EXT1 or EXT2.Citation5,Citation43 In the growth plate, FGF and IHH signaling are modulated by HS, and thus by EXT-1/2, which limit the range of the signal by restricting FGF and IHH diffusion, and thereby facilitate normal endochondrial bone formation. Any change in the strength or territory of FGF and IHH signaling can influence the polarization of physeal chondroid cells, allowing them to grow in the wrong direction or location. IHH signaling also is critical for ossification of the perichondrium, which couples chondrogenesis to osteogenesisCitation44 at the bony collar adjacent to the growth plate; hence, deregulated IHH signaling might also cause defective ossification of the perichondrium and facilitate osteochondroma outgrowth.
In the light of the requirement for SHP2 in RTK-evoked RAS/ERK pathway activation and the role of ERK in repressing IHH/PTHrP production, we suspected that defective Erk activation in CCPs lacking Shp2 might regulate Ihh production. Indeed, Erk activation was compromised in Shp2-deficient CCPs and in a chondroprogenitor cell line after Ptpn11 knockdown; consequently, these cells showed increased Ihh and PTHrP expression and enhanced chondrogenic differentiation.Citation15 Collectively, our data demonstrate that deregulation of the IHH/PTHrP pathway due to SHP2 deficiency in CCPs contributes to MC development. However, we do not exclude the potential involvement of other signaling pathway(s) in MC pathogenesis; indeed, whether additional pathway(s) (e.g., Wnt/β-catenin signaling) contribute to the pathogenesis of MC remains an important topic for future research.
Hedgehog Pathway Antagonists for the Treatment of MC and Other Cartilage Tumors
The HH proteins comprise a group of secreted proteins that regulate cell growth, differentiation and survival.Citation45 HH signaling is modulated by the Patched and Smoothened molecules, which together form a signaling complex that controls downstream targets such Gli family transcription factors. Abnormal HH signaling is implicated in the development of a variety of cancers, including cartilage tumors.Citation25,Citation46-Citation48 Elevated IHH production and IHH/PTHrP signaling are reported in enchondromas and osteochondromas,Citation49 and blockade of HH signaling in our mouse model of MC ameliorates the severity of the disease.Citation15 These findings strongly suggest that HH pathway antagonists could potentially be used to prevent and/or treat enchondromas and osteochondromas.
Several HH pathway inhibitors are in development as therapeutic agents.Citation50 These act on the proteins involved in HH pathway regulation (PTCH1, SMO and GLI). Some of these agents are undergoing pre-clinical or clinical studies as anti-cancer agents, particularly for Gorlin syndrome, an inherited tumor syndrome characterized by widespread basal cell carcinomas.Citation51 In our mouse model of MC, animals were treated with PF-04449913, a small molecule Smoothened inhibitor from Pfizer, to inhibit SMO activity and test whether HH pathway blockade could ameliorate symptom severity. Remarkably, this compound significantly slowed the progression of MC, reduced cartilaginous tumor burden and dramatically improved the mobility of affected mice.Citation15 While these findings are quite exciting, obviously, human clinical trials will be needed to test whether this compound (and/or others of its class) is effective in MC patients. Other candidates for future clinical trials include Novartis LDE-225, Millennium Pharmaceuticals TAK-441, Genentech IPI-926 and Bristol-Myers BMS-833923.Citation50 Given the importance of the HH pathway in normal embryonic and skeletal development, caution must be taken in administering these agents to children or pregnant MC patients. It could be difficult to obtain a therapeutic index, although notably, at the doses of PF-04449913 employed in our mouse MC studiesCitation15 and those of IPI-926 for human basal cell carcinoma treatment,Citation52 linear growth was not affected, although there were some common adverse effects from the latter, including abdominal pain, diarrhea, hyponatremia and arthralgia.Citation52
CCPs in Cartilage Development and Homeostasis
Articular cartilage, when damaged by trauma or diseases such as OA, is difficult to repair due to its relative lack of blood vessels and low number of stem/progenitor cells. The ability of CCPs to migrate toward articular cartilage suggests a potential role in articular cartilage development, homeostasis and injury repair (). Morko et al.Citation53 showed earlier that a low number of Ctsk+ cells exist in healthy articular cartilage of adult mice, and their number increased during OA progression, mainly in clusters of proliferating chondrocytes. Conceivably, Ctsk production by cells within these clusters could degrade extracellular matrix and contribute to OA pathogenesis. Alternatively, cluster formation could arise from a unique Ctsk+ subpopulation of stem cells/progenitors. This hypothesis are actually supported by the notion that stem cell-like progenitors present in adult articular cartilage and also in the OA cell clusters,Citation54,Citation55 they coincidently express high level of stem cell markers, such as Stro1, VCAM-1 and Notch.Citation56 Intriguingly, migratory cartilage stem cells were recently reported to exist in OA cartilage; they resemble cells that are seen in the OA clusters.Citation57 The origin of these cells and whether they express Ctsk are unclear. Further characterization of CCP function during articular cartilage development, homeostasis and disease is clearly indicated.
Figure 2. Location and potential functions of CCPs in epiphyseal cartilage Diagrams showing the location of perichondrial groove of Ranvier (G.R) (shaded in red) in the knee joint. CCPs and their differentiated progeny are denoted by dots of black and gray color, respectively. Note that CCPs reside primarily in the groove of Ranvier (i, black), but they can migrate toward, and live in, epiphyseal (ii) and articular cartilage (iii). In these alternative niches, they could either live quiescently (black) or undergo chondrocytic differentiation (gray) and replenish cartilage. We hypothesize that under physiological conditions, CCPs might be required for epiphyseal cartilage development and homeostasis. During cartilage injury (such as osteoarthritis and trauma) or disease conditions (mutations, etc.), these cells respond to pathogenic insults and start to expand to repair cartilage damage or cause tumorigenesis.
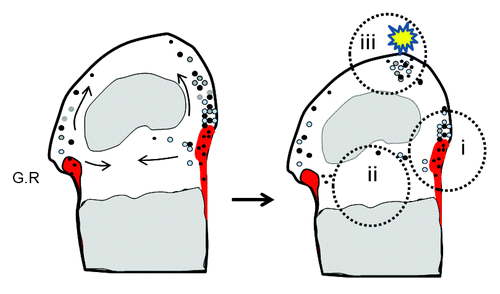
The groove of Ranvier is a wedge-shaped tissue surrounding the periphery of the epiphyseal cartilage. Previous studies suggested that it serves as a reservoir of cartilaginous stem cells/progenitors for the germinal layer of the growth plateCitation58 and possibly for the superficial layer of articular cartilage.Citation59 However, the number of these progenitors is low and no specific markers were available, making it hard to discern their function in chondrogenesis. Our identification of CCPs and the finding that these cells can expand and form cartilage in the absence of SHP2 raises the exciting possibility that manipulation of signaling pathway(s) in these cells could potentially be used to prevent and/or treat degenerative cartilage diseases and cartilage injury.
Historically we always gain more than we expect by studying rare diseases, particularly medications based on the discoveries from rare disease research.Citation29,Citation60 Understanding the molecular and cellular mechanisms underlying MC development has led to the discovery of an important anti-oncogenic function for PTPN11, heretofore viewed as a proto-oncogene, in cartilage. The double-edged sword effects of PTPN11 are determined by both the specific cell type and the particular “wiring” of the signaling pathways therein. In certain types of cartilaginous cells, PTPN11 controls the threshold for IHH and PTHrP production to sustain normal chondrogenesis; in such cells, excessive IHH and PTHrP due to PTPN11 inactivation can cause cartilage tumor formation. This finding could also be relevant to oncogenic mechanisms in other tissues and/or explain the effects of PTPN11 deletion on tissues where HH signaling is particularly important (e.g., in nervous system development). The discovery of CCPs residing within the groove of Ranvier, and possibly in articular cartilage, could also shed new light on cartilage development/homeostasis and lead to novel therapeutic approaches to cartilage diseases.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
We thank Dr Richard M Terek for review of the manuscript. This publication was made possible by the National Institutes of Health (NIH) and the National Institute for General Medicine Sciences (NIGMS) grant no. 8P20GM103468. This work was also funded by NIH R21AR57156 (to Yang W) and R37 CA49132 (to Neel BG), the Rhode Island Hospital Orthopedic Foundation and a grant from the Pediatric Orthopedic Society of North America and the Orthopedic Research and Education Foundation (to Yang W). Neel BG is a Canada Research Chair, Tier 1, and is also supported in part by the Ontario Ministry of Health and Long-term Care and the Princess Margaret Cancer Foundation.
http:dx.doi.org/10.4161/rdis.26657
References
- Bowen ME, Boyden ED, Holm IA, Campos-Xavier B, Bonafé L, Superti-Furga A, Ikegawa S, Cormier-Daire V, Bovée JV, Pansuriya TC, et al. Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet 2011; 7:e1002050; http://dx.doi.org/10.1371/journal.pgen.1002050; PMID: 21533187
- Sobreira NL, Cirulli ET, Avramopoulos D, Wohler E, Oswald GL, Stevens EL, Ge D, Shianna KV, Smith JP, Maia JM, et al. Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet 2010; 6:e1000991; http://dx.doi.org/10.1371/journal.pgen.1000991; PMID: 20577567
- Kennedy LA. Metachondromatosis. Radiology 1983; 148:117 - 8; PMID: 6602353
- Pansuriya TC, Kroon HM, Bovée JV. Enchondromatosis: insights on the different subtypes. Int J Clin Exp Pathol 2010; 3:557 - 69; PMID: 20661403
- Bovée JV. Multiple osteochondromas. Orphanet J Rare Dis 2008; 3:3; http://dx.doi.org/10.1186/1750-1172-3-3; PMID: 18271966
- Hunter AG, Kozlowski K, Hochberger O. Metachondromatosis. Can Assoc Radiol J 1995; 46:202 - 8; PMID: 7538882
- Keret D, Bassett GS. Avascular necrosis of the capital femoral epiphysis in metachondromatosis. J Pediatr Orthop 1990; 10:658 - 61; http://dx.doi.org/10.1097/01241398-199009000-00017; PMID: 2394820
- Bassett GS, Cowell HR. Metachondromatosis. Report of four cases. J Bone Joint Surg Am 1985; 67:811 - 4; PMID: 3873457
- Mavrogenis AF, Skarpidi E, Papakonstantinou O, Papagelopoulos PJ. Chondrosarcoma in metachondromatosis: a case report. J Bone Joint Surg Am 2010; 92:1507 - 13; http://dx.doi.org/10.2106/JBJS.I.00693; PMID: 20516327
- Neel BG, Chan G, Dhanji S. SH2 Domain-Containing Protein-Tyrosine Phosphatases. Handbook of Cell Signaling, 771-809, (2009).
- Grossmann KS, Rosário M, Birchmeier C, Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res 2010; 106:53 - 89; http://dx.doi.org/10.1016/S0065-230X(10)06002-1; PMID: 20399956
- Blenis PPRJ. MAPK Signaling in Human Diseases. Vol. 2 (Humana Press, 2007).
- Matsushita T, Murakami S.The ERK MAPK Pathway in Bone and Cartilage Formation. In: Da Silva Xavier G, ed., Protein Kinases: 2012.
- Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev 2008; 27:179 - 92; http://dx.doi.org/10.1007/s10555-008-9126-y; PMID: 18286234
- Yang W, Wang J, Moore DC, Liang H, Dooner M, Wu Q, Terek R, Chen Q, Ehrlich MG, Quesenberry PJ, et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 2013; 499:491 - 5; http://dx.doi.org/10.1038/nature12396; PMID: 23863940
- Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, Feng GS, Pawson T. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J 1997; 16:2352 - 64; http://dx.doi.org/10.1093/emboj/16.9.2352; PMID: 9171349
- Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, George EL, Neel BG. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell 2006; 10:317 - 27; http://dx.doi.org/10.1016/j.devcel.2006.01.002; PMID: 16516835
- Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, Fryns JP. PTPN11 mutations in LEOPARD syndrome. J Med Genet 2002; 39:571 - 4; http://dx.doi.org/10.1136/jmg.39.8.571; PMID: 12161596
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 2001; 29:465 - 8; http://dx.doi.org/10.1038/ng772; PMID: 11704759
- Nakamura T, Gulick J, Pratt R, Robbins J. Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. Proc Natl Acad Sci U S A 2009; 106:15436 - 41; http://dx.doi.org/10.1073/pnas.0903302106; PMID: 19706403
- Fragale A, Tartaglia M, Wu J, Gelb BD. Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum Mutat 2004; 23:267 - 77; http://dx.doi.org/10.1002/humu.20005; PMID: 14974085
- Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, Brunner HG, Bertola DR, Crosby A, Ion A, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 2002; 70:1555 - 63; http://dx.doi.org/10.1086/340847; PMID: 11992261
- Li S, Hsu DD, Wang H, Feng GS. Dual faces of SH2-containing protein-tyrosine phosphatase Shp2/PTPN11 in tumorigenesis. Front Med 2012; 6:275 - 9; http://dx.doi.org/10.1007/s11684-012-0216-4; PMID: 22869052
- Shapiro F, Holtrop ME, Glimcher MJ. Organization and cellular biology of the perichondrial ossification groove of ranvier: a morphological study in rabbits. J Bone Joint Surg Am 1977; 59:703 - 23; PMID: 71299
- Bovée JV, Hogendoorn PC, Wunder JS, Alman BA. Cartilage tumours and bone development: molecular pathology and possible therapeutic targets. Nat Rev Cancer 2010; 10:481 - 8; http://dx.doi.org/10.1038/nrc2869; PMID: 20535132
- Jones KB, Piombo V, Searby C, Kurriger G, Yang B, Grabellus F, Roughley PJ, Morcuende JA, Buckwalter JA, Capecchi MR, et al. A mouse model of osteochondromagenesis from clonal inactivation of Ext1 in chondrocytes. Proc Natl Acad Sci U S A 2010; 107:2054 - 9; http://dx.doi.org/10.1073/pnas.0910875107; PMID: 20080592
- Bovée JV. EXTra hit for mouse osteochondroma. Proc Natl Acad Sci U S A 2010; 107:1813 - 4; http://dx.doi.org/10.1073/pnas.0914431107; PMID: 20133829
- Kronenberg HM. The role of the perichondrium in fetal bone development. Ann N Y Acad Sci 2007; 1116:59 - 64; http://dx.doi.org/10.1196/annals.1402.059; PMID: 18083921
- Brunham LR, Hayden MR. Hunting human disease genes: lessons from the past, challenges for the future. Hum Genet 2013; 132:603 - 17; http://dx.doi.org/10.1007/s00439-013-1286-3; PMID: 23504071
- de Crombrugghe B, Lefebvre V, Nakashima K. Regulatory mechanisms in the pathways of cartilage and bone formation. Curr Opin Cell Biol 2001; 13:721 - 7; http://dx.doi.org/10.1016/S0955-0674(00)00276-3; PMID: 11698188
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 2002; 16:1446 - 65; http://dx.doi.org/10.1101/gad.990702; PMID: 12080084
- Nakashima K, de Crombrugghe B. Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet 2003; 19:458 - 66; http://dx.doi.org/10.1016/S0168-9525(03)00176-8; PMID: 12902164
- Goldring MB, Tsuchimochi K, Ijiri K. The control of chondrogenesis. J Cell Biochem 2006; 97:33 - 44; http://dx.doi.org/10.1002/jcb.20652; PMID: 16215986
- Kronenberg HM. Developmental regulation of the growth plate. Nature 2003; 423:332 - 6; http://dx.doi.org/10.1038/nature01657; PMID: 12748651
- Pogue R, Lyons K. BMP signaling in the cartilage growth plate. Curr Top Dev Biol 2006; 76:1 - 48; http://dx.doi.org/10.1016/S0070-2153(06)76001-X; PMID: 17118262
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993; 75:59 - 72; PMID: 8402901
- Day TF, Yang Y. Wnt and hedgehog signaling pathways in bone development. J Bone Joint Surg Am 2008; 90:Suppl 1 19 - 24; http://dx.doi.org/10.2106/JBJS.G.01174; PMID: 18292352
- Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev 2002; 16:859 - 69; http://dx.doi.org/10.1101/gad.965602; PMID: 11937493
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet 1996; 12:390 - 7; http://dx.doi.org/10.1038/ng0496-390; PMID: 8630492
- St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 1999; 13:2072 - 86; http://dx.doi.org/10.1101/gad.13.16.2072; PMID: 10465785
- Hopyan S, Gokgoz N, Poon R, Gensure RC, Yu C, Cole WG, Bell RS, Jüppner H, Andrulis IL, Wunder JS, et al. A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat Genet 2002; 30:306 - 10; http://dx.doi.org/10.1038/ng844; PMID: 11850620
- Bovée JV, van den Broek LJ, Cleton-Jansen AM, Hogendoorn PC. Up-regulation of PTHrP and Bcl-2 expression characterizes the progression of osteochondroma towards peripheral chondrosarcoma and is a late event in central chondrosarcoma. Lab Invest 2000; 80:1925 - 34; http://dx.doi.org/10.1038/labinvest.3780202; PMID: 11140704
- Benoist-Lasselin C, de Margerie E, Gibbs L, Cormier S, Silve C, Nicolas G, LeMerrer M, Mallet JF, Munnich A, Bonaventure J, et al. Defective chondrocyte proliferation and differentiation in osteochondromas of MHE patients. Bone 2006; 39:17 - 26; http://dx.doi.org/10.1016/j.bone.2005.12.003; PMID: 16476576
- Chung UI, Schipani E, McMahon AP, Kronenberg HM. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J Clin Invest 2001; 107:295 - 304; http://dx.doi.org/10.1172/JCI11706; PMID: 11160153
- Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal 2012; 5:re6; http://dx.doi.org/10.1126/scisignal.2002906; PMID: 23074268
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841 - 51; http://dx.doi.org/10.1016/S0092-8674(00)81268-4; PMID: 8681379
- Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol 2012; 44:2144 - 51; http://dx.doi.org/10.1016/j.biocel.2012.08.022; PMID: 22981632
- Shahi MH, Rey JA, Castresana JS. The sonic hedgehog-GLI1 signaling pathway in brain tumor development. Expert Opin Ther Targets 2012; 16:1227 - 38; http://dx.doi.org/10.1517/14728222.2012.720975; PMID: 22992192
- Tiet TD, Hopyan S, Nadesan P, Gokgoz N, Poon R, Lin AC, Yan T, Andrulis IL, Alman BA, Wunder JS. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am J Pathol 2006; 168:321 - 30; http://dx.doi.org/10.2353/ajpath.2006.050001; PMID: 16400033
- Hadden MK. Hedgehog pathway inhibitors: a patent review (2009--present). Expert Opin Ther Pat 2013; 23:345 - 61; http://dx.doi.org/10.1517/13543776.2013.757304; PMID: 23294277
- Sekulic A, Mangold AR, Northfelt DW, LoRusso PM. Advanced basal cell carcinoma of the skin: targeting the hedgehog pathway. Curr Opin Oncol 2013; 25:218 - 23; PMID: 23493193
- Dubey AK, Dubey S, Handu SS, Qazi MA. Vismodegib: the first drug approved for advanced and metastatic basal cell carcinoma. J Postgrad Med 2013; 59:48 - 50; http://dx.doi.org/10.4103/0022-3859.109494; PMID: 23525058
- Morko J, Kiviranta R, Mulari MT, Ivaska KK, Väänänen HK, Vuorio E, Laitala-Leinonen T. Overexpression of cathepsin K accelerates the resorption cycle and osteoblast differentiation in vitro. Bone 2009; 44:717 - 28; http://dx.doi.org/10.1016/j.bone.2008.11.019; PMID: 19118660
- Alsalameh S, Amin R, Gemba T, Lotz M. Identification of mesenchymal progenitor cells in normal and osteoarthritic human articular cartilage. Arthritis Rheum 2004; 50:1522 - 32; http://dx.doi.org/10.1002/art.20269; PMID: 15146422
- Lotz MK, Otsuki S, Grogan SP, Sah R, Terkeltaub R, D’Lima D. Cartilage cell clusters. Arthritis Rheum 2010; 62:2206 - 18; http://dx.doi.org/10.1002/art.27528; PMID: 20506158
- Grogan SP, Miyaki S, Asahara H, D’Lima DD, Lotz MK. Mesenchymal progenitor cell markers in human articular cartilage: normal distribution and changes in osteoarthritis. Arthritis Res Ther 2009; 11:R85; http://dx.doi.org/10.1186/ar2719; PMID: 19500336
- Koelling S, Kruegel J, Irmer M, Path JR, Sadowski B, Miro X, Miosge N. Migratory chondrogenic progenitor cells from repair tissue during the later stages of human osteoarthritis. Cell Stem Cell 2009; 4:324 - 35; http://dx.doi.org/10.1016/j.stem.2009.01.015; PMID: 19341622
- Fenichel I, Evron Z, Nevo Z. The perichondrial ring as a reservoir for precartilaginous cells. In vivo model in young chicks’ epiphysis. Int Orthop 2006; 30:353 - 6; http://dx.doi.org/10.1007/s00264-006-0082-2; PMID: 16652202
- Karlsson C, Thornemo M, Henriksson HB, Lindahl A. Identification of a stem cell niche in the zone of Ranvier within the knee joint. J Anat 2009; 215:355 - 63; http://dx.doi.org/10.1111/j.1469-7580.2009.01115.x; PMID: 19563472
- Heemstra HE, van Weely S, Büller HA, Leufkens HG, de Vrueh RL. Translation of rare disease research into orphan drug development: disease matters. Drug Discov Today 2009; 14:1166 - 73; http://dx.doi.org/10.1016/j.drudis.2009.09.008; PMID: 19818412