Abstract
Severe aplastic anemia is a rare bone marrow failure disease with the majority of cases caused by aberrant immune destruction of blood progenitors. Although the Th1-mediated pathology of aplastic anemia is well-described, the molecular mechanisms that drive disease progression remain ill-defined. The NOTCH signaling pathway mediates Th1 differentiation in the presence of polarizing cytokines, an action requiring enzymatic processing of NOTCH receptors by γ- secretase. We used a mouse model of aplastic anemia to demonstrate that expression both of intracellular NOTCH1 (NOTCH1IC) and T-BET, a key transcription factor regulating Th1 differentiation, were increased in T cells in the spleen and bone marrow during active disease. Conditionally deleting NOTCH1 or administering γ-secretase inhibitors (GSI) in vivo, attenuated disease and rescued mice from lethal bone marrow failure. In peripheral T cells from patients with untreated aplastic anemia, NOTCH1IC was significantly elevated and was detected at the TBX21 promoter, showing NOTCH1 directly regulates the gene encoding T-BET. Treating patients’ cells ex vivo with GSI lowered NOTCH1IC levels, decreased the level of NOTCH1 detectable at the TBX21 promoter, and also decreased T-BET expression, indicating NOTCH1 signaling is responsive to GSI during active disease. Collectively, these results identify NOTCH1 signaling as a primary driver of Th1-mediated pathogenesis in aplastic anemia and may represent a novel target for therapeutic intervention.
Severe aplastic anemia is a rare acquired bone marrow failure syndrome.Citation1 Evidence in the majority of cases suggests a breakdown in self-tolerance leads to infiltration of destructive T helper type-1 (Th1) cells into the bone marrow where they target hematopoietic stem cells and compromise stromal cells through bystander effects.Citation1,Citation2 As a result, the population of self-renewing progenitors in the bone marrow is destroyed. Without the ability to replenish platelets, red and white blood cells, patients with aplastic anemia are at increased risk of bleeding episodes, hypoxia and infection. If left untreated, severe aplastic anemia is uniformly fatal.Citation3
The NOTCH family is an evolutionarily-conserved group of transmembrane receptors and ligands. In mammals, it is comprised of four receptors, NOTCH1–4, and five ligands, designated Jagged1, 2, and Delta-like-1, -3, and -4. Signaling is initiated when NOTCH receptors engage cognate ligands, leading to sequential cleavage events that culminate in release from the cell membrane of the intracellular, signaling-competent form of NOTCH (NOTCH1IC).Citation4 Once liberated from the inner-membrane, NOTCH1IC translocates to the nucleus and regulates the transcription of numerous genes either through its canonical nuclear binding partner, CBF1/Supressor of Hairless/Lag1 (CSL), or through interaction with non-canonical partners, such as members of the nuclear factor-kappaB (NFκB) family of transcriptional regulators.Citation5 The final process that untethers NOTCH receptors from the cell membrane is mediated by the enzymatic action of γ-secretase and can be blocked with pharmacological inhibitors.Citation6,Citation7 γ-Secretase inhibitors (GSI) successfully prevent this final enzymatic step that is required to cleave and activate all four NOTCH receptors, and can block NOTCH signaling in vitro and in vivo.Citation8,Citation9 As a result, the use of GSI as a therapeutic modality is the focus of substantial and growing interest. Numerous clinical trials are underway with several chemically distinct GSI, primarily in oncology. Despite initial concerns that dose-limiting toxicity may reduce the clinical usefulness of these drugs, intermittent administration of GSI is safe and well-tolerated. Two Phase 1 clinical trials in early-stage and metastatic ER+ breast cancer with endocrine therapy/GSI combinations have shown safety, tolerability and preliminary suggestions of efficacy.Citation31
NOTCH1IC has been shown to directly regulate genes involved in T cell activation, cell cycle progression, differentiation, cytokine production and effector cell function.Citation4,Citation10-Citation12 We and others have demonstrated that NOTCH signaling can facilitate differentiation of naïve T cells to a Th1 phenotype in mice.Citation8,Citation13-Citation15 One way NOTCH1 drives Th1 polarization is by influencing expression of the T-box transcription factor, Tbx21, the gene encoding T-BET, which is a master transcriptional regulator of Th1 differentiation.Citation16 T-BET is often upregulated in patients with aplastic anemia and mouse models utilizing cells deficient in Tbx21 show less severe disease induction.Citation17 Furthermore, T-BET expression may serve as a biomarker for response to immunosuppressive therapy (IST), since high levels of T-BET have been observed in patients who are refractory to IST, while, for those who respond to IST and remain in remission, T-BET expression in circulating peripheral blood mononuclear cells (PBMCs) is below detectable limits.Citation18
In our recently published study, we used a mouse model to show that the cleaved, active form of NOTCH1 (NOTCH1IC), but not NOTCH2 or NOTCH3, is increased in the T cells of mice with aplastic anemia.Citation19 This high level of NOTCH1 expression was accompanied by characteristic symptoms of aplastic anemia: hypocellular bone marrow, peripheral pancytopenia and elevated levels of circulating proinflammatory cytokines, IFNγ and TNF. When we abrogated NOTCH1 signaling using genetic or pharmacological approaches, bone marrow cellularity in treated animals was higher, as were the numbers of red and white blood cells in the circulation. Additionally, the levels of pro-inflammatory cytokines were diminished and the percentages of CD4 and CD8 T cells infiltrating the bone marrow were greatly decreased. As a result of ameliorating the symptoms of disease, the lifespan of these diseased mice was significantly lengthened.
For patients with aplastic anemia, a bone marrow transplant from an HLA-matched donor is curative. However, only about 25% of patients have a matched sibling donor. For the majority of patients, an immunosuppressive regimen of horse anti-thymocyte globulin and cyclosporin A is necessary. Should a patient require bone marrow transplantation after IST, it is critically important to know that the IST will not adversely affect the engraftment and long-term hematopoiesis of the transplanted bone marrow. Using serial bone marrow transplantations, we determined that, at a dose that was efficacious in attenuating the symptoms of BM failure, extended GSI treatment showed no adverse effects on engraftment or long-term hematopoiesis.
We further demonstrated that in PBMCs from patients with untreated aplastic anemia, NOTCH1IC is increased, can be detected bound to the TBX21 promoter, and is lost from the promoter following GSI treatment. Collectively, our findings demonstrate that NOTCH1 is a critical mediator of Th1 pathology in aplastic anemia through its direct regulation of TBX21 and is responsive to the inhibitory actions of GSI, both in vitro and in vivo.
Detailing a role for NOTCH1 as a driver of pathogenesis in aplastic anemia brings us one step closer to understanding the molecular mechanisms that facilitate disease progression, but much of what causes the pathogenesis of this autoimmune condition remains unclear. One aspect to consider centers on the role additional T helper subsets may play in aplastic anemia and how they are influenced by NOTCH signaling. Studies from the Young lab suggest Th17 cells may also contribute to disease, albeit to a lesser extent than Th1 cells.Citation20 Although we did not observe a direct contribution of Th17 cells or significant upregulation of IL-17 in our mouse model, there may well be a population of effector Th17 cells present in some human patients. Where does NOTCH signaling come into play then? Recent reports have defined a role for NOTCH3 in regulating Th17 differentiation and IL-17 production (Osborne BA, personal communication).Citation21,Citation22 As such, it may be important to examine NOTCH3 levels in aplastic anemia patients, particularly in those patients who are refractory to standard IST. There exists considerable plasticity between Th1 and Th17 cells that is only now being recognized.Citation23,Citation24 Furthermore, since Th1 and Th17 responses act to cross-regulate each other, could dampening the Th1 cytokine millieu with the current IST serve to “de-repress” Th17 responses in some patients, making them refractory to treatment? This phenomenon is not without precedence. Inhibiting the induction of Th1 responses in a mouse model of experimental autoimmune neuritis, unexpectedly, exacerbated disease mediated by the unchecked Th17 response that ensued.Citation25 If this were the case in that subset of aplastic anemia patients who do not respond to standard immunosuppressive protocols, therapeutic use of GSI may be all the more relevant. GSI targeting both of NOTCH1 and NOTCH3 would prevent an amplified Th17 response in the absence of a modulating Th1 environment. Comparing and contrasting NOTCH expression levels in patients who respond to IST and those who do not may lay the ground work for a clearer understanding of how these cohorts are fundamentally different in their disease pathogenesis and perhaps even allow for stratification into different treatment categories.
Another unexplained aspect of aplastic anemia is why the bone marrow is the organ targeted for destruction. Unlike other autoimmune conditions such as type I diabetes or multiple sclerosis, the inciting self-antigen(s) that precipitate aplastic anemia have not been identified. Prevailing thought suggests that self-antigen(s) or neoantigen(s) expressed on bone marrow stem cells are revealed during the course of an infectious event,Citation26 but the identity of this bone marrow-resident antigen(s) awaits discovery.
In the absence of knowing the precipitating antigenic stimulus, what do we know about how effector T cells traffic to the bone marrow? We found that using GSI greatly reduced NOTCH1 expression in the spleen of diseased mice and reduced the bone marrow-infiltrating T cell population, as well. Since NOTCH1 can be upregulated in T cells following an infectious event that elicits a Th1 response, does failure to downregulate NOTCH1 contribute to disease initiation? Does NOTCH receptor-ligand interaction influence trafficking of CD4 and CD8 effector cells to the bone marrow? If so, what are the NOTCH ligands that are expressed on bone marrow stem or stromal cells that may mediate homing to the bone marrow? Do expression patterns of NOTCH ligands differ between aplastic and normal bone marrow and does GSI treatment act to restore a more “normal” ligand expression pattern? Ligands of the Delta-like family, especially Dll-1 and Dll-4, have been shown to be strong drivers of Th1 differentiationCitation27,Citation28. Furthermore, both Delta-like and Jagged family ligands are processed in a gamma-secretase-dependent fashion to signal in the ligand-expressing cell population.Citation29,Citation30 A greater understanding of changes in the temporal expression of NOTCH ligands in the bone marrow, both during disease progression and in response to GSI treatment, may provide answers to these questions, as well as identify new targets for therapeutic intervention.
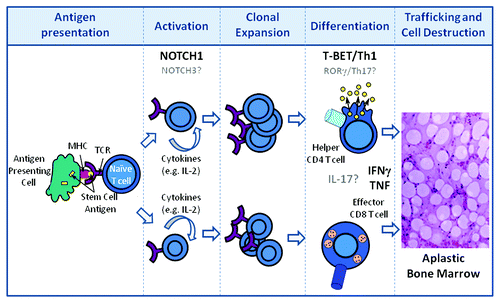
Alternatively, are there other surface receptors found in abundance on effector T cells that mediate trafficking to the bone marrow? Are these NOTCH-regulated? Might these be differentially expressed on T cells during the initiation or expansion phases of aplastic anemia, thus explaining why effector cells traffic to the bone marrow and not to other organs? If so, what are these receptors and what might be their bone marrow-resident cognate ligands? Much more work is required to define in greater detail the molecular mechanisms responsible for the initiation and activation, the expansion and differentiation, and ultimately the homing and cellular destruction that manifest in immune-mediated bone marrow failure ().
Figure 1. Disease progression in aplastic anemia. Following antigen presentation of HSC self- or neoantigen(s) by antigen presenting cells during the initiation phase of the immune response, T cells become activated and release growth factors (IL-2) which result in the clonal expansion of CD4 and CD8 T cells. Expanded T cells differentiate into Th1 helper (CD4) and cytolytic (CD8) T cells. CD4 and CD8 T cells traffic to the bone marrow and produce pro-inflammatory cytokines which are directly toxic to HSCs. NOTCH1 contributes to Th1 differentiation through its direct regulation of T-BET. HSC destruction is also mediated directly by effector CD8 T cells through mechanisms involving FAS-FASL interactions as well as cytolytic granule release and, indirectly, through loss of supportive stromal cells through “by-stander” effects of the pro-inflammatory microenvironment. Putative contributions of NOTCH3, rorγ, and IL-17 are indicated in gray.
Aplastic anemia is a rare and somewhat mystifying autoimmune bone marrow failure disease. The paucity of cells found in the bone marrow at time of diagnosis presents a significant barrier to more fully characterizing the molecular mechanisms that drive disease. Representative animal models of disease, as well as strong collaborative efforts between clinicians and basic scientists to extend research findings and establish their clinical relevance, will be essential to furthering our understanding of aplastic anemia pathogenesis and devising novel and effective therapeutic strategies for patients in the future.
| Abbreviations: | ||
| T-BET | = | T-box protein expressed in T cells |
| Th1 | = | T helper type-1 |
| Th17 | = | T helper type 17 |
| IFNγ | = | interferon gamma |
| TNF | = | tumor necrosis factor |
| ER+ | = | estrogen receptor positive |
| CD4 | = | Cluster designation 4 |
| CD8 | = | cluster designation 8 |
| IL-17 | = | interleukin 17 |
Citation: Minter LM. NOTCH signaling in immune-mediated bone marrow failure of aplastic anemia. Rare Diseases 2013; 1:e26764;
Addendum to:
Disclosure of Potential Conflicts of Interest
The author declares no conflicts of interest.
References
- Young NS, Scheinberg P, Calado RT. Aplastic anemia. Curr Opin Hematol 2008; 15:162 - 8; http://dx.doi.org/10.1097/MOH.0b013e3282fa7470; PMID: 18391779
- Chen J, Brandt JS, Ellison FM, Calado RT, Young NS. Defective stromal cell function in a mouse model of infusion-induced bone marrow failure. Exp Hematol 2005; 33:901 - 8; http://dx.doi.org/10.1016/j.exphem.2005.04.008; PMID: 16038782
- Dezern AE, Brodsky RA. Clinical management of aplastic anemia. Expert Rev Hematol 2011; 4:221 - 30; http://dx.doi.org/10.1586/ehm.11.11; PMID: 21495931
- Osborne BA, Minter LM. Notch signalling during peripheral T-cell activation and differentiation. Nat Rev Immunol 2007; 7:64 - 75; http://dx.doi.org/10.1038/nri1998; PMID: 17170755
- Minter LM, Osborne BA. Canonical and non-canonical Notch signaling in CD4⁺ T cells. Curr Top Microbiol Immunol 2012; 360:99 - 114; http://dx.doi.org/10.1007/82_2012_233; PMID: 22695917
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999; 398:518 - 22; http://dx.doi.org/10.1038/19083; PMID: 10206645
- Shih IeM, Wang TL. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res 2007; 67:1879 - 82; http://dx.doi.org/10.1158/0008-5472.CAN-06-3958; PMID: 17332312
- Minter LM, Turley DM, Das P, Shin HM, Joshi I, Lawlor RG, Cho OH, Palaga TP, Gottipati S, Telfer JC, et al. Inhibitors of gamma-secretase block invivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21.. Nat Immunol 2005; 7:680 - 8; http://dx.doi.org/10.1038/ni1209
- Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, Tsaparikos K, Jani JP, Hosea N, Sands M, et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther 2010; 9:1618 - 28; http://dx.doi.org/10.1158/1535-7163.MCT-10-0034; PMID: 20530712
- Adler SH, Chiffoleau E, Xu L, Dalton NM, Burg JM, Wells AD, Wolfe MS, Turka LA, Pear WS. Notch signaling augments T cell responsiveness by enhancing CD25 expression. J Immunol 2003; 171:2896 - 903; PMID: 12960312
- Palaga T, Miele L, Golde TE, Osborne BA. TCR-mediated Notch signaling regulates proliferation and IFN-gamma production in peripheral T cells. J Immunol 2003; 171:3019 - 24; PMID: 12960327
- Joshi I, Minter LM, Telfer J, Demarest RM, Capobianco AJ, Aster JC, Sicinski P, Fauq A, Golde TE, Osborne BA. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood 2009; 113:1689 - 98; http://dx.doi.org/10.1182/blood-2008-03-147967; PMID: 19001083
- Maekawa Y, Tsukumo S-I, Chiba S, Hirai H, Hayashi Y, Okada H, Kishihara K, Yasutomo K. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity 2003; 19:549 - 59; http://dx.doi.org/10.1016/S1074-7613(03)00270-X; PMID: 14563319
- Skokos D, Nussenzweig MC. CD8- DCs induce IL-12-independent Th1 differentiation through Delta 4 Notch-like ligand in response to bacterial LPS. J Exp Med 2007; 204:1525 - 31; PMID: 17576775
- Zhang Y, Sandy AR, Wang J, Radojcic V, Shan GT, Tran IT, Friedman A, Kato K, He S, Cui S, et al. Notch signaling is a critical regulator of allogeneic CD4+ T-cell responses mediating graft-versus-host disease. Blood 2011; 117:299 - 308; http://dx.doi.org/10.1182/blood-2010-03-271940; PMID: 20870902
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000; 100:655 - 69; http://dx.doi.org/10.1016/S0092-8674(00)80702-3; PMID: 10761931
- Tang Y, Desierto MJ, Chen J, Young NS. The role of the Th1 transcription factor T-bet in a mouse model of immune-mediated bone-marrow failure. Blood 2010; 115:541 - 8; http://dx.doi.org/10.1182/blood-2009-03-211383; PMID: 19903901
- Solomou EE, Keyvanfar K, Young NS. T-bet, a Th1 transcription factor, is up-regulated in T cells from patients with aplastic anemia. Blood 2006; 107:3983 - 91; http://dx.doi.org/10.1182/blood-2005-10-4201; PMID: 16434488
- Roderick JE, Gonzalez-Perez G, Kuksin CA, Dongre A, Roberts ER, Srinivasan J, Andrzejewski CA Jr., Fauq AH, Golde TE, Miele L, et al. Therapeutic targeting of NOTCH signaling ameliorates immune-mediated bone marrow failure of aplastic anemia. J Exp Med 2013; 210:1311 - 29; http://dx.doi.org/10.1084/jem.20112615; PMID: 23733784
- de Latour RP, Visconte V, Takaku T, Wu C, Erie AJ, Sarcon AK, Desierto MJ, Scheinberg P, Keyvanfar K, Nunez O, et al. Th17 immune responses contribute to the pathophysiology of aplastic anemia. Blood 2010; 116:4175 - 84; http://dx.doi.org/10.1182/blood-2010-01-266098; PMID: 20733158
- Jiao Z, Wang W, Xu H, Wang S, Guo M, Chen Y, Gao J. Engagement of activated Notch signalling in collagen II-specific T helper type 1 (Th1)- and Th17-type expansion involving Notch3 and Delta-like1. Clin Exp Immunol 2011; 164:66 - 71; http://dx.doi.org/10.1111/j.1365-2249.2010.04310.x; PMID: 21235539
- Jurynczyk M, Jurewicz A, Raine CS, Selmaj K. Notch3 inhibition in myelin-reactive T cells down-regulates protein kinase C theta and attenuates experimental autoimmune encephalomyelitis. J Immunol 2008; 180:2634 - 40; PMID: 18250475
- Basu R, Hatton RD, Weaver CT. The Th17 family: flexibility follows function. Immunol Rev 2013; 252:89 - 103; http://dx.doi.org/10.1111/imr.12035; PMID: 23405897
- Henriques A, Gomes V, Duarte C, Pedreiro S, Carvalheiro T, Areias M, Caseiro A, Gabriel AJ, Laranjeira P, Pais ML, et al. Distribution and functional plasticity of peripheral blood Th(c)17 and Th(c)1 in rheumatoid arthritis. Rheumatol Int 2013; 33:2093 - 9; http://dx.doi.org/10.1007/s00296-013-2703-6; PMID: 23412693
- Zhang HL, Azimullah S, Zheng XY, Wang XK, Amir N, Mensah-Brown EP, Al Shamsi M, Shahin A, Press R, Zhu J, et al. IFN-γ deficiency exacerbates experimental autoimmune neuritis in mice despite a mitigated systemic Th1 immune response. J Neuroimmunol 2012; 246:18 - 26; http://dx.doi.org/10.1016/j.jneuroim.2012.02.011; PMID: 22445739
- Young NS. Acquired aplastic anemia. JAMA 1999; 282:271 - 8; http://dx.doi.org/10.1001/jama.282.3.271; PMID: 10422997
- Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell 2004; 117:515 - 26; http://dx.doi.org/10.1016/S0092-8674(04)00451-9; PMID: 15137944
- Sun J, Krawczyk CJ, Pearce EJ. Suppression of Th2 cell development by Notch ligands Delta1 and Delta4. J Immunol 2008; 180:1655 - 61; PMID: 18209061
- Ikeuchi T, Sisodia SS. The Notch ligands, Delta1 and Jagged2, are substrates for presenilin-dependent “gamma-secretase” cleavage. J Biol Chem 2003; 278:7751 - 4; http://dx.doi.org/10.1074/jbc.C200711200; PMID: 12551931
- LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem 2003; 278:34427 - 37; http://dx.doi.org/10.1074/jbc.M302659200; PMID: 12826675
- Albain et al. Thirty-Fourth Annual CTRC-AACR San Antonio Breast Cancer Symposium. Abstr. S1–5; Means-Powell et al. 2012. Thirty-Fifth Annual CTRC-AACR San Antonio Breast Cancer Symposium. Abstr. P2–14–04.