Abstract
Congenital skeletal anomalies are rare disorders, with a subset affecting both the cranial and appendicular skeleton. Two categories, craniosynostosis syndromes and chondrodysplasias, frequently result from aberrant regulation of the fibroblast growth factor (FGF) signaling pathway. Our recent work has implicated FGF signaling in a third category: ciliopathic skeletal dysplasias. In this work, we have used mouse mutants in two ciliopathy genes, Fuzzy (Fuz) and orofacial digital syndrome-1 (Ofd-1), to demonstrate increase in Fgf8 gene expression during critical stages of embryogenesis. While the mechanisms underlying FGF dysregulation differ in the different syndromes, our data raise the possibility that convergence on FGF signal transduction may underlie a wide range of skeletal anomalies. Here, we provide additional evidence of the skeletal phenotypes from the Fuz mouse model and highlight similarities between human ciliopathies and FGF-related syndromes.
Keywords: :
Fibroblast growth factors are well-studied signaling molecules that are critical for embryonic development.Citation1,Citation2 In humans, 22 structurally related FGF ligands have been identified; most of these are secreted proteins that bind with varying affinities to tyrosine kinase receptors (FGFR1–4). The majority of FGF ligands can bind promiscuously to multiple different FGFRs; further complexity is generated by alternative splicing of FGFR1, 2 and 3. Ligand binding induces receptor dimerization and cross-phosphorylation, which then initiates a broad range of intracellular signaling cascades such as PI3 kinase (PI3K), MAPK, phospholipase C (PLCγ) or JAK/STAT. Activation of the pathway elicits diverse cellular responses, including proliferation and differentiation of multiple cell types. In recent years, developmental studies, as well as identification of human alleles, has made it clear that precise temporal and spatial activation of FGF signaling is necessary for normal development.
The FGF signaling pathway is a key regulator of skeletal development. In humans, mutations in the FGF receptors (FGFRs) are a hallmark of two classes of skeletal anomalies: craniosynostoses and chondrodysplasias (reviewed by Ornitz et al.).Citation1 Craniosynostosis syndromes are characterized by premature fusion of the cranial sutures and are frequently accompanied by malformations of the axial skeleton. In contrast, chondrodysplasias feature truncation of the appendicular skeleton, due to an increase in FGF signaling during endochondral ossification. Both craniosynostosis and chondrodysplasia syndromes are associated with autosomal dominant mutations of the FGF receptors, which result in increased activation of the signaling pathway.Citation1 The array of phenotypes correlates with specific mutations in different receptors, but the net effect appears to be that sustained or increased FGF signaling tips the balance between critical steps in osteoblast differentiation.Citation1,Citation2
Ciliopathies are a heterogeneous group of disorders that arise from abnormal formation or function of the cilium.Citation3 Cilia are finger-like organelles at the cell surface comprised of a microtubule axoneme attached to a basal body. Depending on the microtubule arrangement, a cilium may be motile or immotile. Disorders of the motile cilia frequently involve fluid flow; for example, patients with primary ciliary dyskinesia have difficulty clearing mucus from their lungs due to defects in the multi-ciliated epithelium.Citation4 Immotile or primary cilia stand alone and are required for function of many signaling pathways.Citation5 To date, ciliopathic skeletal phenotypes have mainly been attributed to changes in primary cilia-dependent transduction of Hedgehog signals.Citation6 This has been best studied in the long bones; however, the phenotypic range of ciliopathies is extremely broad. As a consequence, it can be difficult to diagnose or treat ciliopathies, and the underlying etiology is often unclear.
Ciliopathies affecting the skeleton are rare syndromic anomalies. Some patients exhibit limb phenotypes such as syndactyly or polydactyly [in the case of Bardet-Biedl syndrome (MIM #209900)].Citation7,Citation8 Dysplasia of the ribs, and occasional shortened limbs, are also seen, as in Jeune asphyxiating thoracic dystrophy (MIM #208500).Citation9 Frequently, ciliary defects also lead to changes in the craniofacial skeleton in humans and mice,Citation10-Citation12 with craniosynostosis observed in Sensenbrenner syndrome or cranioectodermal dysplasia (MIM #614378).Citation13 The broad range of systems affected suggests varied molecular causes. Thus, grouping similar skeletal phenotypes across multiple disorders, and comparison with animal models, may provide useful insight into the underlying molecular events.
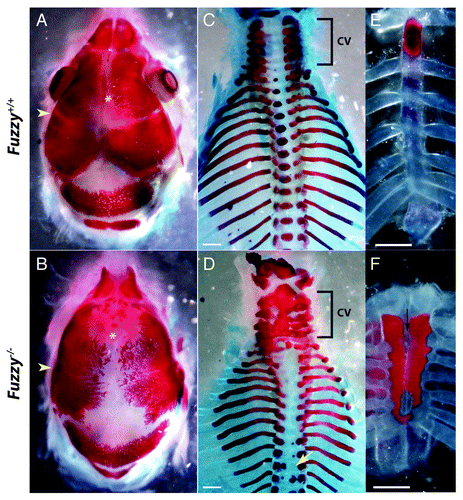
The Fuzzy gene is associated with neural tube defects and has previously been shown to be a ciliopathy gene.Citation14-Citation17 In our current work, we examine the requirements for Fuz in development of the craniofacial structures.Citation18 Craniofacial defects include craniosynostosis and facial anomalies ().Citation18 As documented in Tabler et al., Fuz mutant mice have a complete synostosis of the coronal suture, as well as an open anterior fontanelle, reminiscent of Apert syndrome synostoses (MIM #101200) ().Citation19 Our analysis of the Fuzzy mutant also showed broader defects of the skeleton. Consistent with ciliopathic Hedgehog phenotypes, Fuz mutants have polydactyly and shortened long bones.Citation17 Most interesting, we also found anomalous elements in the axial skeleton. The cervical vertebra (cv) were frequently fused (, bracket), while ossification of the centrum in the thoracic vertebra is generally absent. Occasionally, small islands of ectopic ossification are seen (, yellow arrow). As in ciliopathies such as Jeune Syndrome (MIM #208500), the ribs are shorter. Surprisingly, the sternum is hyperossified, shorter, and bifid (, black arrow marks cleft). In earlier stages, cartilaginous joints are formed (data not shown); by embryonic day (E)18.5, the sternal joints have been obliterated ().
Figure 1. Skeletal preparations of wild-type and Fuz−/− embryos at E18.5. Alizarin red staining marks the bone. Alcian blue staining marks the cartilage. (A and B) Dorsal views of the skull. (A) Control. (B) Mutant mice display synostosis of the coronal suture (yellow arrowhead) as well as an open anterior fontanelle (yellow asterix). (C and D) Dorsal view of the axial skeleton. (C) Control. (D) In mutant animals, the cervical vertebra (cv, bracket) are fused. Ossification of the centrum in thoracic vertebra is lost or aberrant (yellow arrow). (E and F) Frontal views of the sternum. (E) Control. (F) In mutants, the sternum is shorter, hyperossified and cleft/bifid (black arrow).
This array of phenotypes suggested a similarity to several classes of FGFR-dependent skeletal anomalies, including craniosynostosis syndromes and chondrodysplasias.Citation20 All of these syndromes arise from dysregulation of FGF receptors;Citation1 however, the status of FGF signaling in ciliopathy mutants has not been well explored. catalogs the skeletal malformations ciliopathies and FGF related syndromes. We found significant overlap across the range of disorders.
Table 1. Skeletal phenotypes observed in ciliopathies and FGF syndromes. Included are selected human disorders and animal models. Unfortunately, due to space constraints, we regret that we are unable to cite all relevant papers.
Many craniosynostosis patients have a progressive fusion of the cervical spine, with two thirds of Apert patients exhibiting complex fusions in the C5-C6 segment.Citation21 C2-C3 fusions are also quite common in these syndromes, with additional reports from Saethre-Chotzen (MIM #101400) and Pfeiffer syndromes (MIM#101600).Citation22-Citation24 To our knowledge, FGF-induced chondrodysplasias are not associated with spinal stenosis; however, cases have been reported in rhizomelic chondrodysplasia punctata patients.Citation25 Interestingly, congenital scoliosis due to vertebral defects has also been linked to aberrant FGF signaling during development.Citation26 In addition, in a mouse model of Pfeiffer syndrome, vertebral homeotic transformations have been noted.Citation27 Thus, phenotypes seen in the different mouse models, combined with some reported anomalies of the cervical vertebra in Joubert Syndrome (MIM #213300), suggest that ciliopathy patients could be assessed for spinal aberrations.Citation28
Sternal abnormalities, a common feature of ciliopathies and FGF syndromes, are another striking phenotype seen in Fuz mutants. Premature or ectopic fusion of the sternum is seen in a number of mouse models, including Apert/Pfeiffer Syndrome and achondroplasia mice.Citation27,Citation29-Citation31 The current data suggest that hyperactivation of FGF receptors leads to an impairment in sternal joint formation and subsequent hyperossification. In humans, premature ossification of the sternum is a hallmark of Noonan syndrome.Citation32 The causative mutation in Noonan syndrome is PTPN11, which encodes SHP-2, a key regulator of the FGF-Ras-MAPK pathway.Citation33 Finally, sternal anomalies are also observed in ciliopathic animal models.Citation34,Citation35
Taken together, our data suggest that the skeletal anomalies described may all converge on deregulation of the FGF signaling pathway. Indeed, we found that a subset of phenotypes in our ciliopathic mouse mutants, Fuz and OFD-1, are attributable to increased Fgf8 gene expression and genetic reduction of FGF8 rescued these phenotypes.Citation18
Our approach of cataloguing human phenotypes, and comparison to animal models, led us to a surprising role for FGFs in ciliopathies. However, clearly, FGFs alone, or in combination with Hedgehog signaling, cannot be the sole molecular players in skeletal dysplasias. FGF signaling plays roles at multiple steps in both endochondral and intramembranous ossification.Citation1 For example, during long bone formation, signaling via FGFR2 and FGFR3 promotes chondrocyte condensation and differentiation respectively. Later in this process, FGFR3 is needed to limit the amounts of proliferative pre-hypertrophic chondrocytes. In intramembranous ossification, as seen in the calvaria, FGFs are involved in every step of osteoblast differentiation and subsequent ossification. FGFR1 and 2 are expressed at the osteogenic front, and are necessary for osteoblast differentiation. FGF2 is expressed in the sutures, and signals to the adjacent osteogenic cells. Thus, specific phenotypes observed in the human syndromes described can be attributed to inappropriate temporal or spatial activation of the pathway. For example, hyperactivation of FGFR3 severely reduces regions of pre-hypertrophic chondrocyte proliferation resulting in short long bones. Conversely, FGFR1 and 2 dysregulation leads to premature osteoblast differentiation and craniosynostosis.
Dysregulation of Gli processing is also known to cause a variety of skeletal defects, notably in the long bones, vertebra and sternum. Gli2 mouse mutants have shortened long bones and absence or malformation of vertebral bodies, while Gli3 mutants have slightly shortened long bones accompanied by polydactyly, as well as fusions of the cervical vertebra and bifid, hyperossified sterna.Citation36 Thus, there is substantial phenotypic overlap between Gli mutants and other animal models of skeletal syndromes. As described above, it is likely that correct timing and location of a suite of signals is critical for shaping the skeleton. Because pathological mutations can lead to changes at multiple levels during development, we propose that further comparison of human phenotypes and animal models can provide important insights into the genetic networks governing overlapping disease phenotypes.
| Abbreviations: | ||
| C | = | cleft |
| CF | = | cervical fusion |
| CS | = | coronal synostosis |
| HA | = | high arch |
| PD | = | polydactyly |
| SD | = | syndactyly |
| SL | = | short limbs |
| SS | = | sagittal synostosis |
Acknowledgments
We thank the Liu lab for discussions and Marc Dionne for critical reading of the manuscript. This work was funded by grants from the Wellcome Trust, BBSRC, and an MRC studentship.
Citation: Yannakoudakis BZ, Liu KJ. Common skeletal features in rare diseases: New links between ciliopathies and FGF-related syndromes. Rare Diseases 2013; 1:e27109
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 2002; 16:1446 - 65; http://dx.doi.org/10.1101/gad.990702; PMID: 12080084
- Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 2005; 16:107 - 37; http://dx.doi.org/10.1016/j.cytogfr.2005.01.008; PMID: 15863029
- Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol 2009; 111:39 - 53; http://dx.doi.org/10.1159/000208212; PMID: 19276629
- Rossman CM, Forrest JB, Lee RM, Newhouse MT. The dyskinetic cilia syndrome. Ciliary motility in immotile cilia syndrome. Chest 1980; 78:580 - 2; http://dx.doi.org/10.1378/chest.78.4.580; PMID: 6448132
- Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol 2007; 23:345 - 73; http://dx.doi.org/10.1146/annurev.cellbio.23.090506.123249; PMID: 17506691
- Haycraft CJ, Serra R. Cilia involvement in patterning and maintenance of the skeleton. Curr Top Dev Biol 2008; 85:303 - 32; http://dx.doi.org/10.1016/S0070-2153(08)00811-9; PMID: 19147010
- David A, Bitoun P, Lacombe D, Lambert JC, Nivelon A, Vigneron J, Verloes A. Hydrometrocolpos and polydactyly: a common neonatal presentation of Bardet-Biedl and McKusick-Kaufman syndromes. J Med Genet 1999; 36:599 - 603; PMID: 10465109
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet 1999; 36:437 - 46; PMID: 10874630
- Baujat G, Huber C, El Hokayem J, Caumes R, Do Ngoc Thanh C, David A, Delezoide AL, Dieux-Coeslier A, Estournet B, Francannet C, et al. Asphyxiating thoracic dysplasia: clinical and molecular review of 39 families. J Med Genet 2013; 50:91 - 8; http://dx.doi.org/10.1136/jmedgenet-2012-101282; PMID: 23339108
- Brugmann SA, Cordero DR, Helms JA. Craniofacial ciliopathies: A new classification for craniofacial disorders. Am J Med Genet A 2010; 152A:2995 - 3006; http://dx.doi.org/10.1002/ajmg.a.33727; PMID: 21108387
- Macca M, Franco B. The molecular basis of oral-facial-digital syndrome, type 1. Am J Med Genet C Semin Med Genet 2009; 151C:318 - 25; http://dx.doi.org/10.1002/ajmg.c.30224; PMID: 19876934
- Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A, et al. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet 2010; 86:949 - 56; http://dx.doi.org/10.1016/j.ajhg.2010.04.012; PMID: 20493458
- Levin LS, Perrin JC, Ose L, Dorst JP, Miller JD, McKusick VA. A heritable syndrome of craniosynostosis, short thin hair, dental abnormalities, and short limbs: cranioectodermal dysplasia. J Pediatr 1977; 90:55 - 61; http://dx.doi.org/10.1016/S0022-3476(77)80764-6; PMID: 830894
- Seo JH, Zilber Y, Babayeva S, Liu J, Kyriakopoulos P, De Marco P, Merello E, Capra V, Gros P, Torban E. Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans. Hum Mol Genet 2011; 20:4324 - 33; http://dx.doi.org/10.1093/hmg/ddr359; PMID: 21840926
- Park TJ, Haigo SL, Wallingford JB. Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nat Genet 2006; 38:303 - 11; http://dx.doi.org/10.1038/ng1753; PMID: 16493421
- Heydeck W, Zeng H, Liu A. Planar cell polarity effector gene Fuzzy regulates cilia formation and Hedgehog signal transduction in mouse. Dev Dyn 2009; 238:3035 - 42; http://dx.doi.org/10.1002/dvdy.22130; PMID: 19877275
- Gray RS, Abitua PB, Wlodarczyk BJ, Szabo-Rogers HL, Blanchard O, Lee I, Weiss GS, Liu KJ, Marcotte EM, Wallingford JB, et al. The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis and mouse embryonic development. Nat Cell Biol 2009; 11:1225 - 32; http://dx.doi.org/10.1038/ncb1966; PMID: 19767740
- Tabler JM, Barrell WB, Szabo-Rogers HL, Healy C, Yeung Y, Perdiguero EG, Schulz C, Yannakoudakis BZ, Mesbahi A, Wlodarczyk B, et al. Fuz mutant mice reveal shared mechanisms between ciliopathies and FGF-related syndromes. Dev Cell 2013; 25:623 - 35; http://dx.doi.org/10.1016/j.devcel.2013.05.021; PMID: 23806618
- Cohen MM Jr., Kreiborg S. Skeletal abnormalities in the Apert syndrome. Am J Med Genet 1993; 47:624 - 32; http://dx.doi.org/10.1002/ajmg.1320470509; PMID: 8266987
- Hennekam R, et al. Gorlin's Syndromes of the Head and Neck Oxford University Press, USA, 2010.
- Hemmer KM, McAlister WH, Marsh JL. Cervical spine anomalies in the craniosynostosis syndromes. Cleft Palate J 1987; 24:328 - 33; PMID: 3479279
- Moore MH, Cantrell SB, Trott JA, David DJ. Pfeiffer syndrome: a clinical review. Cleft Palate Craniofac J 1995; 32:62 - 70; http://dx.doi.org/10.1597/1545-1569(1995)032<0062:PSACR>2.3.CO;2; PMID: 7727489
- Moore MH, Lodge ML, Clark BE. Spinal anomalies in Pfeiffer syndrome. Cleft Palate Craniofac J 1995; 32:251 - 4; http://dx.doi.org/10.1597/1545-1569(1995)032<0251:SAIPS>2.3.CO;2; PMID: 7605793
- Anderson PJ, Hall CM, Evans RD, Hayward RD, Harkness WJ, Jones BM. The cervical spine in Saethre-Chotzen syndrome. Cleft Palate Craniofac J 1997; 34:79 - 82; http://dx.doi.org/10.1597/1545-1569(1997)034<0079:TCSISC>2.3.CO;2; PMID: 9003917
- Khanna AJ, Braverman NE, Valle D, Sponseller PD. Cervical stenosis secondary to rhizomelic chondrodysplasia punctata. Am J Med Genet 2001; 99:63 - 6; http://dx.doi.org/10.1002/1096-8628(20010215)99:1<63::AID-AJMG1117>3.0.CO;2-9; PMID: 11170096
- Sparrow DB, Chapman G, Smith AJ, Mattar MZ, Major JA, O’Reilly VC, Saga Y, Zackai EH, Dormans JP, Alman BA, et al. A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell 2012; 149:295 - 306; http://dx.doi.org/10.1016/j.cell.2012.02.054; PMID: 22484060
- Hajihosseini MK, Lalioti MD, Arthaud S, Burgar HR, Brown JM, Twigg SR, Wilkie AO, Heath JK. Skeletal development is regulated by fibroblast growth factor receptor 1 signalling dynamics. Development 2004; 131:325 - 35; http://dx.doi.org/10.1242/dev.00940; PMID: 14668415
- Juric-Sekhar G, Adkins J, Doherty D, Hevner RF. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol 2012; 123:695 - 709; http://dx.doi.org/10.1007/s00401-012-0951-2; PMID: 22331178
- Hajihosseini MK, Wilson S, De Moerlooze L, Dickson C. A splicing switch and gain-of-function mutation in FgfR2-IIIc hemizygotes causes Apert/Pfeiffer-syndrome-like phenotypes. Proc Natl Acad Sci U S A 2001; 98:3855 - 60; http://dx.doi.org/10.1073/pnas.071586898; PMID: 11274405
- Wang Y, Xiao R, Yang F, Karim BO, Iacovelli AJ, Cai J, Lerner CP, Richtsmeier JT, Leszl JM, Hill CA, et al. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development 2005; 132:3537 - 48; http://dx.doi.org/10.1242/dev.01914; PMID: 15975938
- Wang Q, Green RP, Zhao G, Ornitz DM. Differential regulation of endochondral bone growth and joint development by FGFR1 and FGFR3 tyrosine kinase domains. Development 2001; 128:3867 - 76; PMID: 11585811
- Hoeffel JC, Pernot C, Juncker P. Radiologic patterns of the sternum in Noonan’s syndrome with congenital heart defect. Am J Dis Child 1981; 135:1044 - 6; PMID: 6794358
- Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, Brunner HG, Bertola DR, Crosby A, Ion A, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 2002; 70:1555 - 63; http://dx.doi.org/10.1086/340847; PMID: 11992261
- Zhang Q, Murcia NS, Chittenden LR, Richards WG, Michaud EJ, Woychik RP, Yoder BK. Loss of the Tg737 protein results in skeletal patterning defects. Dev Dyn 2003; 227:78 - 90; http://dx.doi.org/10.1002/dvdy.10289; PMID: 12701101
- Cui C, Chatterjee B, Francis D, Yu Q, SanAgustin JT, Francis R, Tansey T, Henry C, Wang B, Lemley B, et al. Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel-Gruber syndrome. Dis Model Mech 2011; 4:43 - 56; http://dx.doi.org/10.1242/dmm.006262; PMID: 21045211
- Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 1997; 124:113 - 23; PMID: 9006072
- Waters A, et al. Bardet-Biedl Syndrome. In: RA P, et al., eds. GeneReviews. Seattle (WA): University of Washington, Seattle, 2011.
- Ho NC, Francomano CA, van Allen M. Jeune asphyxiating thoracic dystrophy and short-rib polydactyly type III (Verma-Naumoff) are variants of the same disorder. Am J Med Genet 2000; 90:310 - 4; http://dx.doi.org/10.1002/(SICI)1096-8628(20000214)90:4<310::AID-AJMG9>3.0.CO;2-N; PMID: 10710229
- Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, Leh SM, Midtbø M, Filhol E, Bole-Feysot C, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet 2011; 89:634 - 43; http://dx.doi.org/10.1016/j.ajhg.2011.10.001; PMID: 22019273
- Sensenbrenner JA, Dorst JP, Owens RP. New syndrome of skeletal, dental and hair anomalies. Birth Defects Orig Artic Ser 1975; 11:372 - 9; PMID: 1227553
- Young ID. Cranioectodermal dysplasia (Sensenbrenner’s syndrome). J Med Genet 1989; 26:393 - 6; http://dx.doi.org/10.1136/jmg.26.6.393; PMID: 2661822
- Smith HL, Hand AM. Chondroectodermal dysplasia (Ellis-van Creveld syndrome); report of two cases. Pediatrics 1958; 21:298 - 307; PMID: 13505024
- Weatherbee SD, Niswander LA, Anderson KV. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet 2009; 18:4565 - 75; http://dx.doi.org/10.1093/hmg/ddp422; PMID: 19776033
- Pacheco M, Valencia M, Caparrós-Martín JA, Mulero F, Goodship JA, Ruiz-Perez VL. Evc works in chondrocytes and osteoblasts to regulate multiple aspects of growth plate development in the appendicular skeleton and cranial base. Bone 2012; 50:28 - 41; http://dx.doi.org/10.1016/j.bone.2011.08.025; PMID: 21911092
- Ruiz-Perez VL, Blair HJ, Rodriguez-Andres ME, Blanco MJ, Wilson A, Liu YN, Miles C, Peters H, Goodship JA. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development 2007; 134:2903 - 12; http://dx.doi.org/10.1242/dev.007542; PMID: 17660199
- Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dollé P, Franco B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet 2006; 38:112 - 7; http://dx.doi.org/10.1038/ng1684; PMID: 16311594
- Bimonte S, De Angelis A, Quagliata L, Giusti F, Tammaro R, Dallai R, Ascenzi MG, Diez-Roux G, Franco B. Ofd1 is required in limb bud patterning and endochondral bone development. Dev Biol 2011; 349:179 - 91; http://dx.doi.org/10.1016/j.ydbio.2010.09.020; PMID: 20920500
- Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, Reijns MA, Keighren M, Bahlo M, Bromhead CJ, Budd P, et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet 2011; 88:508 - 15; http://dx.doi.org/10.1016/j.ajhg.2011.03.015; PMID: 21473986
- Rynearson RD. Case report: orthodontic and dentofacial orthopedic considerations in Apert’s syndrome. Angle Orthod 2000; 70:247 - 52; PMID: 10926435
- Anderson PJ, Hall C, Evans RD, Harkness WJ, Hayward RD, Jones BM. The cervical spine in Crouzon syndrome. Spine (Phila Pa 1976) 1997; 22:402 - 5; http://dx.doi.org/10.1097/00007632-199702150-00009; PMID: 9055367
- Eswarakumar VP, Ozcan F, Lew ED, Bae JH, Tomé F, Booth CJ, Adams DJ, Lax I, Schlessinger J. Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc Natl Acad Sci U S A 2006; 103:18603 - 8; http://dx.doi.org/10.1073/pnas.0609157103; PMID: 17132737
- Perlyn CA, DeLeon VB, Babbs C, Govier D, Burell L, Darvann T, Kreiborg S, Morriss-Kay G. The craniofacial phenotype of the Crouzon mouse: analysis of a model for syndromic craniosynostosis using three-dimensional MicroCT. Cleft Palate Craniofac J 2006; 43:740 - 8; http://dx.doi.org/10.1597/05-212; PMID: 17105336
- Bellus GA, McIntosh I, Smith EA, Aylsworth AS, Kaitila I, Horton WA, Greenhaw GA, Hecht JT, Francomano CA. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet 1995; 10:357 - 9; http://dx.doi.org/10.1038/ng0795-357; PMID: 7670477
- Elejalde BR, de Elejalde MM. Thanatophoric dysplasia: fetal manifestations and prenatal diagnosis. Am J Med Genet 1985; 22:669 - 83; http://dx.doi.org/10.1002/ajmg.1320220404; PMID: 4073120
- Georgoulis G, Alexiou G, Prodromou N. Achondroplasia with synostosis of multiple sutures. Am J Med Genet A 2011; 155A:1969 - 71; http://dx.doi.org/10.1002/ajmg.a.33744; PMID: 21739570
- Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev 2004; 18:290 - 305; http://dx.doi.org/10.1101/gad.1179104; PMID: 14871928
- Chen L, Adar R, Yang X, Monsonego EO, Li C, Hauschka PV, Yayon A, Deng CX. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest 1999; 104:1517 - 25; http://dx.doi.org/10.1172/JCI6690; PMID: 10587515