Abstract
Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. CFTR mutations lead to production of non-functional CFTR, reduced amounts of normal functioning CFTR or misfolded CFTR with defects in trafficking or function. For decades, CF treatment has been focused on the symptoms of CF, but pharmacotherapy using small molecules that target the basic defect of CF, the mutant CFTR protein, is now possible for a limited amount of subjects with CF. This raises the exciting possibility that the majority of people with CF may receive effective treatment targeting the different CFTR mutants in the future. We recently described a functional CFTR assay using rectal biopsies from subjects with CF that were cultured in vitro into self-organizing mini-guts or organoids. We here describe how this model may assist in the discovery of new CFTR-targeting drugs, the subjects that may benefit from these drugs, and the mechanisms underlying variability in CFTR genotype-phenotype relations.
Introduction
Cystic fibrosis (CF) is a life-shortening autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that encodes an apically expressed anion channel essential for fluid and electrolyte homeostasis of many mucosal surfaces.Citation1 Subjects with CF display severe pulmonary and gastrointestinal dysfunctions and have a life expectancy of approximately 40 y.
Since the cloning of the CFTR gene 24 y ago, over 1900 mutations have been identifiedCitation2 (http://www.genet.sickkids.on.ca). Based on the mechanism by which mutations affect the CFTR protein, they can be classified into 6 groups: (1) no synthesis, (2) defective protein folding and trafficking, (3) defective channel regulation (gating), (4) reduced Cl- conductance, (5) reduced amounts of normal functioning apical protein (e.g., by altered splicing) or (6) an increased plasma membrane turnover.Citation3 F508del is the most dominant CFTR mutation (~67% of all mutant alleles worldwide) expressed by approximately 90% of subjects with CF (http://www.genet.sickkids.on.ca/). Multiple co- and post-translational folding steps are affected in CFTR-F508del causing retention at the endoplasmic reticulum, rapid degradation and severely reduced expression at the apical membrane.Citation4 At the apical membrane CFTR-F508del further shows defects in gating and plasma membrane retention. Due to the high prevalence of CFTR-F508del in the CF population, it is the prime target for CFTR-directed pharmacotherapy.
Pharmacotherapy of CFTR
Pharmacotherapy using small molecules that target CFTR mutants is an exciting new possibility to treat CF. Effective restoration of mutant CFTR by compounds depends on the defect associated with the CFTR mutation. In addition, it is expected that other subject-specific factors impact the efficacy of the pharmacotherapy as observed for many established drugs.Citation5 Thus far, the CFTR potentiator Kalydeco (VX-770) is the only CFTR-targeting drug commercially available and it is approved for a select subset of CF subjects (< 4%) expressing the gating mutant CFTR-G551D.Citation6,Citation7 This exciting development demonstrates that CFTR-directed pharmacotherapy is feasible, and may possibly be designed for the majority of subjects with CF.
Preclinical and clinical data indicate that robust restoration of CFTR-F508del requires a combination of compounds that both target the folding defect (correctors) and the gating defect (potentiators).Citation8-Citation10 Results from a phase II clinical trial with the corrector Lumacaftor (VX-809) and the potentiator Kalydeco (VX-770) indicated that absolute lung function significantly improved by 6.7% in subjects homozygous for CFTR-F508del (http://investors.vrtx.com/releasedetail.cfm?releaseid=687394). In this trial, treatment effects were observed in over 50% of the subjects, and approximately 25% of the subjects demonstrated 10% lung function increase, and a phase III clinical trial has been initiated for CFTR-F508del homozygous patients. This approach is likely not sufficient to “cure” CF in these subjects, indicating that discovery of novel CFTR-restoring compounds is important.
Lung function improvement in the subjects with a single CFTR-F508del allele was lower and likely relates to the lower expression of Lumacaftor and Kalydeco-responsive mutant CFTR protein. Although the effects for this group as a whole were limited, it could very well be that individual patients may respond to the treatment. Identification of these subjects is important to ensure that subjects who may benefit from potential treatments are not missed, and receive treatment as quickly as possible.
Prediction of Individual CFTR-Restoring Drug Efficacy by Functional Models
The efficacy of CFTR-restoring pharmacotherapy may be predicted ex vivo or in vitro by patient-specific CFTR function measurements. Various methods may have the potential to predict in vivo drug efficacy, and these approaches will likely complement each other. Ex vivo rectal biopsies have been used to study the modulation of CFTR function and the strength of this method is both with its direct relation to the patient and the sensitivity to measure CFTR function.Citation11,Citation12 However, only a limited amount of biopsies (4–8) can be isolated from one patient, so only a small patient-specific data set can be generated. In vitro primary airway cell cultures can be generated in large numbers from lung explants, but cultures set up from patients that can mostly benefit from treatment require invasive procedures and are limited due to loss of differentiation potential upon passaging of basal cells.Citation13,Citation14 In addition, the measurement of CFTR function using this method is time consuming. Recently, progress has been reported in differentiating induced progenitor stem cells (iPSC) into airway cells.Citation15 This approach generates a limitless supply of patient-specific cells, but technical advances are further needed to ensure genomic integrity of iPSC clones and optimal airway differentiation protocols.Citation16
We developed a novel functional CFTR assay using intestinal adult stem cell cultures generated from rectal biopsies after intestinal current measurements.Citation17 Compared with the other available patient-specific models, analysis of CFTR-function in these ‘organoid’ cultures is fast, robust, and accurate and allows for the generation of large data sets from individual subjects within several weeks after isolation. Furthermore, these cultures are (epi)genetically stable and can be stored in liquid nitrogen, features that are essential for biobanking and high-throughput screening.Citation18 The potential weakness of these cultures is due to the intestinal origin of the cells; responses of intestinal cells may not fully represent responses of airway cells.
A Novel Functional CFTR Assay in Intestinal Organoids from CF Subjects
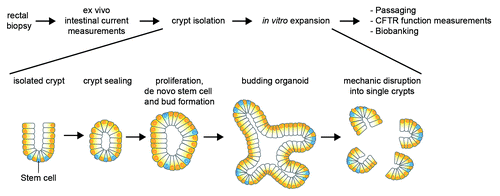
We typically start intestinal organoid cultures from rectal biopsies used for diagnostic purposes of CF.Citation17 Crypts isolated from biopsies are cultured in a three dimensional matrix and self-organize into mini-guts or organoids that recapitulate essential features of the in vivo tissue architecture.Citation19,Citation20 These pure epithelial cultures consist of closed single epithelial layers with the apical membrane facing a central lumen, and multiple crypt domains that harbor the stem cells. A repetitive culture cycle including organoid expansion for 7 d, mechanical disruption into single crypts and passaging to new wells allows for the generation of large amounts of material that can be used for functional CFTR measurements or liquid nitrogen storage ().
Figure 1. Schematic representation of organoid generation from rectal suction biopsies. Crypts are isolated from rectal biopsies of CF subjects that self-organize into closed 3D multicellular structures consisting of a single epithelial layer and an internal lumen termed organoids upon in vitro culture.
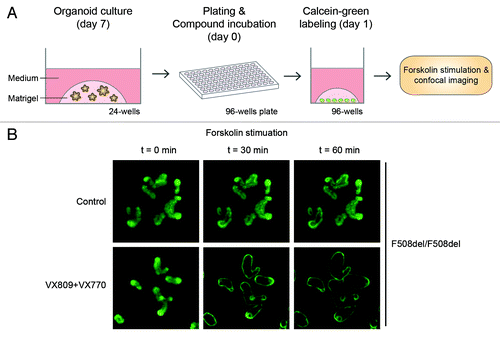
Chemical activation of CFTR by forskolin or cholera toxin rapidly leads to volumetric expansion or swelling of the organoids due to accumulation of fluid in the organoid lumen, a process that is fully CFTR-dependent. To quantify forskolin-induced swelling (FIS), organoids from a 7 d-old culture are passaged into 96-well plates. After 24 h of culturing (with or without CFTR correctors), organoids are labeled with calcein green, stimulated with forskolin (with or without CFTR potentiators) and directly imaged by confocal microscopy (). We observed that FIS was absent in organoids derived from Cftr–/– mice or in human organoids with CFTR null alleles, reduced in organoids expressing mild CFTR mutations and strongly reduced in organoids expressing severe CFTR mutations. Importantly, FIS of CF organoids was restored by treatment of CFTR-restoring compounds ().
Figure 2. CFTR function measurements using intestinal organoids. (A) Schematic representation of the method to measure forskolin-induced organoid swelling. (B) Confocal images of calcein-green labeled F508del/F508del organoids with or without VX-809 + VX-770 treatment at the indicated time points of forskolin stimulation.
We here focus on the implications of CFTR function measurements in these organoids for CFTR-targeting drug development. This model can help to address important questions, including (1) what mechanisms control CFTR genotype-phenotype relations and drug efficacy, (2) can novel CFTR-targeting compounds be developed that restore mutant CFTR function for the majority of subjects with CF?, and (3) can patient-specific in vitro models complement CFTR-genotyping for the stratification of CF patients and CFTR-targeting drugs?
Organoids to Study Genotype-Phenotype Relations in CF
Genetic variation in CFTR and other modifier genes are important for CF disease progression.Citation21-Citation23 FIS of organoids can reveal important relations between genotype and CFTR residual function of individual subjects and how these relate to clinical disease progression, allowing for the identification of mechanisms controlling variability among patients. FIS varied greatly between organoids derived from different CF subjects, and was largest in organoids from patients with milder CF that express CFTR-A455E.Citation17,Citation24 We detected FIS in all organoids expressing CFTR-F508del, also when it was expressed from just a single allele. This indicates that the CFTR-F508del allele clearly confers detectable residual function, albeit very limited compared with wild-type function.
Interestingly, we also detected significant differences in FIS between organoids from CFTR-F508del homozygous subjects, and differences in FIS were stable upon propagation of organoids in culture for at least seven weeks. This indicates that cell-intrinsic mechanisms play a role in controlling individual CFTR-F508del function, and are likely conferred by genetic variability between subjects. These clear phenotypic differences between CFTR-F508del subjects, and the ability to grow large quantities of pure individual epithelium will be important to define cellular mechanisms or molecular and genetic factors associated with residual function.
The variations in the low levels of residual function that we observed in CFTR-F508del homozygous organoids can be clinically relevant as subjects expressing two class I CFTR alleles (no functional CFTR) have reduced lung function compared with subjects with two class II alleles such as CFTR-F508del.Citation25,Citation26 Correlations between clinical disease phenotype and FIS measurements in both cross-sectional and longitudinal studies are required to assess whether subject-specific FIS in organoids is predictive of lung disease progression in CFTR-F508del homozygous subjects. If so, pharmacological targeting of mechanisms that control CFTR-F508del residual function may be important to improve function of mutant CFTR, and possibly efficacy of CFTR-targeting drugs.
CF Organoids for Development of CFTR-Targeting Drugs
The functional CFTR assay in organoids also offers new opportunities for identification of novel CFTR-targeting drugs. The complete dependency of CFTR and robustness of response allow accurate measurements of CFTR function with an excellent signal-to-noise ratio. The simple experimental set up and readout, as well as the capacity to expand cells into large volumes, are all mandatory for high throughput approaches.
Together with Prof GL Lukacs, we recently demonstrated that compounds targeting distinct sites within CFTR-F508del synergize with VX-809 to correct the folding defect.Citation27 This indicates that organoids can be used for identification of novel therapeutic combinations that restore CFTR function. In addition, we successfully used CFTR inhibitors in wild-type CFTR-expressing organoids and showed that other stimuli, such as cholera toxin, could also strongly induce swelling. This points out that potential compounds that inhibit secretory diarrhea may be identified using variations of the swell assay.
In vitro primary airway epithelial cell cultures play an important role for preclinical validation of compounds that modulate mutant CFTR. Thus far, the observations in organoid cultures appear to resemble responses of airway cultures to CFTR-targeting drugs.Citation6,Citation10 We found similar dose-dependencies to VX-809 and VX-770, and a similar hierarchy of CFTR-restoring efficacy of VX-809, VX-770, Corr-4a, and VRT-325. Although the responses to CFTR-restoring drugs are similar between CFTR-F508del airway cultures and organoids, we observed that the relation of responses with wild-type cultures was approximately 2-fold higher in organoids. This likely reflects different relations between CFTR protein function and culture-specific functional readouts.
Together, this indicates that agonist-induced swell assays in organoids are a suited platform for identification of CFTR-targeting drugs, either as a primary screening model or for preclinical validation of previously identified hits.
Organoids for Stratification of CF Subjects
Stratification of subjects for drugs is important to maximize the effective use of therapeutics. As discussed above, clinical trials with CFTR-targeting drugs indicate that these drugs have variable efficacy among individuals, as has been observed for most drugs.Citation5 Especially when treatment efficacy is uncertain for individuals, and when high costs limit empirical approaches for application of drugs, stratification of patients for drugs becomes more important.
Ideal stratification methods should have high sensitivity (fraction of identified true drug-responsive subjects) to identify as much subjects as possible that will benefit from treatment. The specificity of such methods (fraction of identified true non-responsive subjects) is important to limit the risk of potential off-target effects without beneficial therapeutic effects, and to limit the economical burden associated with ineffective treatment. The identification of non-responding subjects becomes more important when multiple treatment options are available and CF disease progresses due to ineffective treatment. As there is only a single CFTR-targeting drug registered at this moment, the main goal for now is to develop methods that have maximal sensitivity.
It is crucial to include subjects that will respond to drugs in clinical trials to demonstrate clinical efficacy. Currently, inclusion of subjects for clinical trials for CFTR-restoring drugs depends mostly on defining CFTR genotypes (or classes) that have been shown to react to drugs in vitro, in combination with specific inclusion and exclusion criteria based on individual clinical disease parameters. In this way, CFTR-targeting drugs can be registered for subgroups of patients expressing particular CFTR genotypes, and this cycle can be repeated to demonstrate therapeutic efficacy for other genotypes. This model was successfully applied to register Kalydeco for CFTR-G551D-expressing subjects, and currently novel genotypes are being selected for Kalydeco treatment.Citation28
Especially for subjects expressing complex or rare CFTR genotypes, in vitro replication of CFTR mutations in ectopic expression models can be difficult (e.g., for splice site mutations). In addition, clinical trials are difficult to perform in subjects with rare genotypes with sufficient power to demonstrate efficacy. This indicates that prediction and in vivo validation of individual responses to CFTR-targeting drugs is important for selection of subjects for drugs that have originally been registered for other genotypes. We found different responses to CFTR-targeting drugs in organoids from subjects with various CFTR genotypes suggesting that this model can be useful to select drug-responsive CFTR genotypes.Citation17
In addition, individual measurements that predict efficacy of drugs can be important to select responding subjects independent of CF-causing mutation. We and others found that CFTR protein expression and function can be different for subjects harboring two CFTR-F508del alleles, as can be their response to CFTR-targeting drugs.Citation6,Citation17,Citation29,Citation30 In organoids, subject-specific drug profiles are stable at multiple culture time points, indicating that cell-intrinsic mechanisms, independent of the CF-causing mutations, further control efficacy of CFTR-restoring drugs. Interestingly, Flume et al. found that sweat chloride levels decreased ≥ 10 mmol/L in approximately 15% of CFTR-F508del homozygous patients upon Kalydeco treatment, suggesting that some homozygous CFTR-F508del individuals may already benefit from potentiator therapy.Citation9 This supports that polymorphisms within CFTR itself, efficacy of transcription, translation, and/or post-translational processing of CFTR-F508del are different between individuals, which can affect in vivo efficacy of CFTR-targeting drugs.
A clear advantage of the organoid-based FIS measurements is the relatively rapid and detailed analysis of subject-specific efficacy of CFTR-modulating agents. Organoid cultures can be biobanked to ensure that material is stored after a single isolation procedure for current and future drug testing. Experiences from our own clinical center and others have indicated that procedures to isolate rectal biopsies are well tolerated by subjects, although repeated isolations of biopsies are less well tolerated.Citation12,Citation31,Citation32
In conclusion, organoids can be cultured from individual subjects without highly invasive procedures, and can play an important role in stratifying subjects for CFTR-restoring drugs. The main advantage of organoid-based stratification over current genetic approaches is that organoid-based measurements also integrate individual factors that interact with the CFTR gene and the CFTR protein and play a role in the efficacy of CFTR-restoring drugs at the level of the individual. A personalized medicine approach for CF may be developed by identifying the most optimal combinations of CFTR-restoring drugs for an individual using the FIS assay when more drugs are registered in the future. For now, FIS in organoids can be important to select individuals (and CFTR-genotypes) for Kalydeco treatment. In addition, individual drug measurements in organoids may play a role in clinical trials with CFTR-restoring drugs to identify responsive individuals. Future direct studies should point out whether responses in organoids are predictive of in vivo drug efficacy.
Final Remarks
In the past decades, tremendous progress has been achieved in the treatment of CF. Targeting the mutant CFTR protein has been proven effective for a limited number of patients, and has ignited an enormous effort by academia and industry to develop CFTR-targeting pharmacotherapy for the majority of CF subjects. Still, robust functional restoration of CFTR-F508del by pharmacotherapy has been proven difficult, and it is likely that a variety of compounds are needed to treat CF effectively in the majority of patients.
We developed a novel functional CFTR assay in organoid cultures grown from rectal biopsies from CF patients. These stem cell-cultures self-organize into mini-guts in vitro and can be greatly expanded and biobanked. CFTR function measurements in this model are highly accurate and sensitive, and offer new opportunities for CFTR-targeting drug discovery and patient stratification for CFTR-targeting drugs. These cultures are also especially suited to study functional interactions between genotype, phenotype, and CFTR-restoring drugs, and the mechanisms that define these interactions at the level of individual subjects.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352:1992 - 2001; http://dx.doi.org/10.1056/NEJMra043184; PMID: 15888700
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245:1066 - 73; http://dx.doi.org/10.1126/science.2475911; PMID: 2475911
- Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration 2000; 67:117 - 33; http://dx.doi.org/10.1159/000029497; PMID: 10773783
- Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med 2012; 18:81 - 91; http://dx.doi.org/10.1016/j.molmed.2011.10.003; PMID: 22138491
- Spear BB, Heath-Chiozzi M, Huff J. Clinical application of pharmacogenetics. Trends Mol Med 2001; 7:201 - 4; http://dx.doi.org/10.1016/S1471-4914(01)01986-4; PMID: 11325631
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009; 106:18825 - 30; http://dx.doi.org/10.1073/pnas.0904709106; PMID: 19846789
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, et al, VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365:1663 - 72; http://dx.doi.org/10.1056/NEJMoa1105185; PMID: 22047557
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012; 67:12 - 8; http://dx.doi.org/10.1136/thoraxjnl-2011-200393; PMID: 21825083
- Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordoñez CL, Geller DE, VX 08-770-104 Study Group. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012; 142:718 - 24; http://dx.doi.org/10.1378/chest.11-2672; PMID: 22383668
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011; 108:18843 - 8; http://dx.doi.org/10.1073/pnas.1105787108; PMID: 21976485
- Roth EK, Hirtz S, Duerr J, Wenning D, Eichler I, Seydewitz HH, Amaral MD, Mall MA. The K+ channel opener 1-EBIO potentiates residual function of mutant CFTR in rectal biopsies from cystic fibrosis patients. PLoS One 2011; 6:e24445; http://dx.doi.org/10.1371/journal.pone.0024445; PMID: 21909392
- Derichs N, Sanz J, Von Kanel T, Stolpe C, Zapf A, Tümmler B, Gallati S, Ballmann M. Intestinal current measurement for diagnostic classification of patients with questionable cystic fibrosis: validation and reference data. Thorax 2010; 65:594 - 9; http://dx.doi.org/10.1136/thx.2009.125088; PMID: 20627915
- Karp PH, Moninger TO, Weber SP, Nesselhauf TS, Launspach JL, Zabner J, Welsh MJ. An in vitro model of differentiated human airway epithelia. Methods for establishing primary cultures. Methods Mol Biol 2002; 188:115 - 37; PMID: 11987537
- Neuberger T, Burton B, Clark H, Van Goor F. Use of primary cultures of human bronchial epithelial cells isolated from cystic fibrosis patients for the pre-clinical testing of CFTR modulators. Methods Mol Biol 2011; 741:39 - 54; http://dx.doi.org/10.1007/978-1-61779-117-8_4; PMID: 21594777
- Wong AP, Bear CE, Chin S, Pasceri P, Thompson TO, Huan LJ, Ratjen F, Ellis J, Rossant J. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat Biotechnol 2012; 30:876 - 82; http://dx.doi.org/10.1038/nbt.2328; PMID: 22922672
- Liang G, Zhang Y. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell 2013; 13:149 - 59; http://dx.doi.org/10.1016/j.stem.2013.07.001; PMID: 23910082
- Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, Brandsma AM, de Jong NW, Bijvelds MJ, Scholte BJ, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med 2013; 19:939 - 45; http://dx.doi.org/10.1038/nm.3201; PMID: 23727931
- Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 2013; 340:1190 - 4; http://dx.doi.org/10.1126/science.1234852; PMID: 23744940
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009; 459:262 - 5; http://dx.doi.org/10.1038/nature07935; PMID: 19329995
- Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011; 141:1762 - 72; http://dx.doi.org/10.1053/j.gastro.2011.07.050; PMID: 21889923
- Tsui L-C. The spectrum of cystic fibrosis mutations. Trends Genet 1992; 8:392 - 8; PMID: 1279852
- Vanscoy LL, Blackman SM, Collaco JM, Bowers A, Lai T, Naughton K, Algire M, McWilliams R, Beck S, Hoover-Fong J, et al. Heritability of lung disease severity in cystic fibrosis. Am J Respir Crit Care Med 2007; 175:1036 - 43; http://dx.doi.org/10.1164/rccm.200608-1164OC; PMID: 17332481
- Weiller CA, Drumm ML. Genetic influences on cystic fibrosis lung disease severity. Front Pharmacol 2013.
- Gan KH, Veeze HJ, van den Ouweland AM, Halley DJ, Scheffer H, van der Hout A, Overbeek SE, de Jongste JC, Bakker W, Heijerman HG. A cystic fibrosis mutation associated with mild lung disease. N Engl J Med 1995; 333:95 - 9; http://dx.doi.org/10.1056/NEJM199507133330204; PMID: 7539891
- Cleveland RH, Zurakowski D, Slattery D, Colin AA. Cystic fibrosis genotype and assessing rates of decline in pulmonary status. Radiology 2009; 253:813 - 21; http://dx.doi.org/10.1148/radiol.2533090418; PMID: 19952026
- Geborek A, Hjelte L. Association between genotype and pulmonary phenotype in cystic fibrosis patients with severe mutations. J Cyst Fibros 2011; 10:187 - 92; http://dx.doi.org/10.1016/j.jcf.2011.01.005; PMID: 21354377
- Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, Roldan A, Verkman AS, Kurth M, Simon A, et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat Chem Biol 2013; 9:444 - 54; http://dx.doi.org/10.1038/nchembio.1253; PMID: 23666117
- Yu H, Burton B, Huang CJ, Worley J, Cao D, Johnson JP Jr., Urrutia A, Joubran J, Seepersaud S, Sussky K, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros 2012; 11:237 - 45; http://dx.doi.org/10.1016/j.jcf.2011.12.005; PMID: 22293084
- van Meegen MA, Terheggen-Lagro SWJ, Koymans KJ, van der Ent CK, Beekman JM. Apical CFTR expression in human nasal epithelium correlates with lung disease in cystic fibrosis. PLoS One 2013; 8:e57617; http://dx.doi.org/10.1371/journal.pone.0057617; PMID: 23483918
- Penque D, Mendes F, Beck S, Farinha C, Pacheco P, Nogueira P, Lavinha J, Malhó R, Amaral MD. Cystic fibrosis F508del patients have apically localized CFTR in a reduced number of airway cells. Lab Invest 2000; 80:857 - 68; http://dx.doi.org/10.1038/labinvest.3780090; PMID: 10879737
- De Boeck K, Derichs N, Fajac I, de Jonge HR, Bronsveld I, Sermet I, Vermeulen F, Sheppard DN, Cuppens H, Hug M, et al, ECFS Diagnostic Network Working Group, EuroCareCF WP3 Group on CF diagnosis. New clinical diagnostic procedures for cystic fibrosis in Europe. J Cyst Fibros 2011; 10:Suppl 2 S53 - 66; http://dx.doi.org/10.1016/S1569-1993(11)60009-X; PMID: 21658643
- Servidoni MF, Sousa M, Vinagre AM, Cardoso SR, Ribeiro MA, Meirelles LR, de Carvalho RB, Kunzelmann K, Ribeiro AF, Ribeiro JD, et al. Rectal forceps biopsy procedure in cystic fibrosis: technical aspects and patients perspective for clinical trials feasibility. BMC Gastroenterol 2013; 13:91; http://dx.doi.org/10.1186/1471-230X-13-91; PMID: 23688510