Abstract
Rett syndrome (RTT), an X-linked neurological disorder caused by mutations in MECP2, may have a metabolic component. We reported a genetic suppressor screen in a Mecp2-null mouse model to identify pathways for therapeutic improvement of RTT symptoms. Of note, one suppressor mutation implied that cholesterol homeostasis was perturbed in Mecp2 null mice; indeed, cholesterol synthesis was elevated in the brain and body system. Remarkably, the genetic effect of downregulating the cholesterol pathway could be mimicked chemically by statin drugs, improving motor symptoms, and increasing longevity in the mouse. Our work linked cholesterol metabolism to RTT pathology for the first time. Both neurological and systemic effects of perturbed cholesterol homeostasis overlap with many RTT symptoms. Here we show in patients that peripheral cholesterol, triglycerides, and/or LDLs may be elevated early in RTT disease onset, providing a biomarker for patients that could be aided by therapeutic interventions that modulate lipid metabolism.
Rett syndrome (RTT) is an X-linked disorder characterized by progressive development of neurological and motor dysfunction. The prevalence of RTT is 1 in 10 000 female births, classifying it as a rare disease; however, it is one of the most common forms of severe disability in females. RTT was originally thought to be a metabolic disease based on symptom presentation and progression and was first called “cerebral atrophic hyperammonemia.”Citation1 Hyperammonemia was abandoned as a potential cause because elevated blood ammonia was observed in only a minority of patients.Citation2 Decades later, mutations in methyl CpG binding protein 2 (MECP2) were identified as the primary cause of RTT.Citation3 Lifespan and disease severity vary greatly in RTT patients, due in part to random X-chromosome inactivation, mutation type, and other unknown factors. Despite the fact that MECP2 is expressed ubiquitously and that neuronal function is intimately linked with systemic metabolism, there has been little interest in potential metabolic manifestations of RTT, including the possible impact such manifestations may have on neuronal function and disease progression.
Mechanistically, MECP2 binds to methylated DNA to regulate gene transcription, associating with chromatin-remodeling complexes that contain type I histone deacetylases (HDACs).Citation4 Therefore, the elimination of MECP2 results in massive changes in gene expression, making it difficult to pinpoint key pathways in pathology and making treatment strategies difficult. The disease was considered neurodevelopmental with little hope for recovery. In a landmark finding, the re-introduction of Mecp2 into mice that were already neurologically impaired rescued symptoms, suggesting that MECP2 was involved in the maintenance of neuronal function and suggesting that RTT symptoms could be reversed in severely affected patients.Citation5
A Suppressor Screen Points to Cholesterol Metabolism in Mecp2 Mutant Mice
We recently reported a genetic suppressor screen in which we identified a mutation that improves Rett-like motor symptoms and overall health in Mecp2 mutant hemizygous male (Mecp2 null) mice: a premature nonsense mutation in the gene encoding squalene epoxidase (SQLE), a rate limiting cholesterol biosynthesis enzyme.Citation6 This finding suggested that cholesterol metabolism would be perturbed in the Mecp2 mouse model; upon examination, we found that lipid metabolism was widely affected in Mecp2 null mice, both in the brain and systemically. Mecp2 null mice first displayed complex dysregulation of the pathway in the brain: elevated cholesterol synthesis early in disease, followed by a sharp downregulation of cholesterol synthesis as symptoms progressed. Systemically, mice showed progressive worsening of metabolic symptoms over time, including increased serum cholesterol and triglycerides and buildup of triglycerides and other neutral lipids in the liver.Citation6 These data suggest that aspects of symptom progression in RTT may be due to systemic effects of perturbed lipid metabolism.
Commonly, attention is placed on high circulating cholesterol in the blood because it is associated with increased incidence of cardiovascular disease. However, cholesterol has many indispensible functions in neural tissues including membrane trafficking, signal transduction, myelin formation, dendrite remodeling, neuropeptide formation, and synaptogenesis.Citation7 Therefore, perturbations in cholesterol homeostasis may play a role in the neurological symptoms associated with RTT. Cholesterol cannot cross the blood brain barrier, thus all cholesterol needed in the brain must be manufactured there. Cholesterol biosynthesis takes place in the endoplasmic reticulum requiring mitochondria as an energy source. Abnormal mitochondrial function and morphology has been described in RTT patients and Mecp2 mouse models.Citation8 Additionally, mitochondrial dysfunction is increased in metabolic syndrome and nonsyndromic autism spectrum disorders (ASD). Reactive oxygen species (ROS) accumulate through mitochondrial dysfunction, and individuals with autism are subject to higher levels of oxidative stress.Citation9 In the brain, cholesterol must be turned over rapidly and renewed; otherwise, ROS may oxidize cholesterol, leading to inflammation and a shut down in cholesterol synthesis, placing extreme importance on the tight regulation of brain cholesterol homeostasis. Emerging evidence suggests that lipid synthesis, storage, and recycling are involved in many neurological diseases.Citation10 Perturbations in biosynthesis and intracellular trafficking of cholesterol are responsible for the onset of Smith-Lemli Opitz syndrome and Neimann-Pick type C disease, respectively. Further, Alzheimer, Parkinson, and Huntington diseases, Amyotrophic lateral sclerosis, and Fragile X syndrome have all been linked to aberrant cholesterol homeostasis. RTT has now joined this group: the study of this rare disease may inform other common neurological diseases.
RTT patients go through four stages in disease progression.Citation11 Stage I follows a period of apparently normal development from birth when the child begins to display social and communication deficits, similar to those seen in ASD, between six and 18 mo of age. The child shows delays in developmental milestones, particularly for motor ability, such as sitting and crawling. Stage II begins between one and four years of age with a period of regression in which speech and motor abilities are lost, and stereotypical midline hand movements and gait impairments develop. Breathing irregularities, including apnea and hyperventilation, also appear during this stage. Autistic symptoms are still prevalent. Stage III occurs between ages two and ten when the period of regression ends and symptoms plateau. Social and communication skills may show small improvements during this plateau period, which may last for most of the patients’ lives. In the final stage, Stage IV, motor ability and muscle deterioration continues. Many girls eventually develop severe scoliosis and lose the ability to walk.
Sterols are the precursors of steroid hormones, bile acids, and vitamin D, thus progressive symptoms could be associated with systemic effects of perturbed lipid metabolism, including abnormal responses to stress, high anxiety levels, digestive problems, and bone anomalies.Citation12
Because of the role of cholesterol in cardiovascular disease, HMG CoA reductase inhibitors (statins) are medically prescribed to reduce high cholesterol. Statin drugs can ameliorate neurological symptoms in mouse models of Fragile X syndromeCitation13 and are being used in clinical trials to ameliorate cognitive problems in children with neurofibromatosis Type I.Citation14 Indeed, we also found that statin drugs would ameliorate symptoms of Mecp2 mutation in both male and female mice,Citation6 showing relevance to Rett syndrome pathology and suggesting that lipid metabolism, a highly targetable pathway for drug or dietary intervention, could be exploited for symptom improvement in RTT. Mice, although a powerful mammalian model, are not humans. Humans are a genetically heterogeneous population, whereas mouse models are maintained on an inbred genetic background. Therefore, studying a mutation on an inbred strain of mice helps to understand genetic influences but is akin to studying a single individual. Findings in the mouse must always be supported to ensure they have relevance to human patients.
A Subset of Rett Patients Display Dyslipidemia
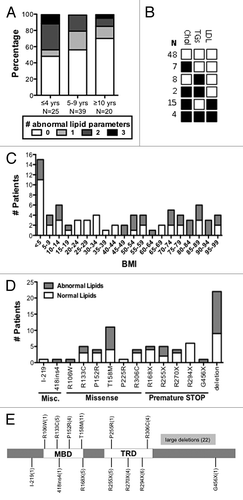
If lipid homeostasis is perturbed in Rett patients as it is in mice lacking functional Mecp2, peripheral dyslipidemia could be a biomarker. The mouse work suggests that peripheral lipid markers will not be elevated in all patients since the different Mecp2-null mouse inbred strains, while consistently displaying perturbations in brain cholesterol metabolism, showed a difference in the presentation of peripheral lipids, as measured in the liver and serum.Citation6 However, only one published study describes a lipid analysis of RTT patients. In this manuscript lipid levels were averaged regardless of age to show that girls with RTT exhibited a serum LDL cholesterol level that was 14.7 mg/dL higher than a healthy control cohort.Citation15 Therefore, we analyzed lipids in consecutive, unselected female patients treated at the Tri-State Rett Syndrome Clinic at Montefiore Medical Center, NY. Serum chemistry panels had been obtained from 84 of these female RTT patients, aged 6 mo to 19 y, as a part of their routine health care monitoring. All patients met the current diagnostic criteria for RTT,Citation11 and all had confirmed mutations in MECP2. Although no other blood parameters were consistently abnormal, pediatric RTT patients commonly showed elevated cholesterol, triglycerides, and LDLs, according to American Academy of Pediatrics guidelinesCitation16 (). Of 25 girls aged 0–4.9 y, 52% had high cholesterol, triglycerides, and/or LDLs. Of 39 girls aged 5–9.9 y, 44% had at least one elevated lipid parameter, and of 20 girls aged 10–19 y, 30% had at least one elevated lipid parameter. High lipids are commonly associated with high body mass index (BMI). Elevated lipids were observed in RTT patients throughout the BMI spectrum, from <5% to 95%, but showed a correlation of only 9% with BMI (R2 = 0.094; ). Patients with large deletions (13 of 22, 59%) and missense mutations in the methyl-binding domain (10 of 20, 50%), especially the T158M mutation (7 of 11, 64%), were more likely to have elevated lipid parameters (). No correlation could be made with diet, although dietary information was available for only 27 patients. Six patients also displayed decreased high-density lipoproteins (HDLs; <35mg/dL); however, for 3 patients low HDL was the only abnormal parameter. Low HDLs were therefore excluded as an abnormal lipid parameter from this analysis. These data were collected retrospectively, without appropriate population and dietary controls, and should be considered preliminary.
Figure 1. RTT patients display dyslipidemia. The American Academy of Pediatrics defines normal lipid parameters for children ages 2–18 as cholesterol < 170 mg/dL, triglycerides < 150 mg/dL, and LDLs < 110 mg/dL based on meta-analysis.Citation16 (A) The lipid panels of 84 patients, ages 0–19, were analyzed for lipid parameters that fall outside normal pediatric ranges. Patients were divided into age groups that represent 0–4.9, 5–9.9, and 10–19. White boxes represent the percentage of patients with no abnormal parameters (or low HDLs, excluded as an abnormal lipid parameter), light gray boxes represent patients with one abnormal parameter, medium gray those with two, and black those with three. Patients at the youngest ages were more likely to exhibit abnormal lipid values. (B) The most common abnormal lipid profile was elevated cholesterol and LDL. Cholesterol and triglycerides were not commonly elevated together. (C) Patient BMI ranged from <5–95th percentile and did not follow a normal curve. BMI was not strongly associated with abnormal lipid parameters (R = 0.307, P = 0.006, Pearson correlation). (D) Mutation status for 78 patients; large MECP2 deletions were most common in this patient population, followed by the T158M missense mutation. (E) Patient mutations mapped to multiple sites on the MECP2 protein.
Implications for Patients and Relevance to Statin Treatment
Excitingly, given the results of this study, a class of FDA-approved cholesterol lowering drugs, the statins, may prove to be useful in preventing symptom progression in RTT patients. Statins have other biological effects that may be independent of their cholesterol-reducing properties, such as enhancing brain-derived neurotrophic factor (BDNF), modulating inflammatory responses, and influencing Ras farnesylation. Therefore, the beneficial effects of statins have been tested in the treatment of multiple sclerosis, lupus, and Parkinson disease, as well as other central nervous system neurodegenerative diseases.Citation17-Citation19 In our Mecp2 mouse study, treatment with statin drugs was more effective than the mutation in Sqle, suggesting that some of these “side effects” of statins were also beneficial in disease amelioration. Lipophilic lovastatin is being used in pediatric trials for neurofibromatosis type ICitation14 and was very effective on the Mecp2 mouse model, suggesting that it is a candidate for clinical trials in RTT patients. Regardless of the possibility of its role in the pathophysiology of RTT, atherosclerosis, and therefore hyperlipidemia, is a pediatric problem. In 2007, the American Heart Association (AHA) recommended drug therapy for high-risk lipid abnormalities in children as young as 8 y, specifically starting therapy with statin medications, a proposal that was subsequently endorsed by the American Academy of Pediatrics.Citation20 Standard treatment for dyslipidemia in childhood consists of lifestyle modifications to diet and exercise regimens and, when these are insufficient, pharmacotherapy—mainly with statins.Citation21 Because of the severity of their motor impairments, as well as almost ubiquitous growth and feeding problems, the role of lifestyle modifications in hyperlipidemic children with RTT is very limited, making pharmacotherapy the most feasible treatment option. Such information suggests that RTT patients with high lipid parameters would be candidates for statin treatment. Clinical trials are under consideration to assess the potential benefits of statins. It is our prediction that statin drugs will prevent lipid accumulation, and therefore, delay the progression or penetrance of co-morbidities.
Considerations for Other Therapies that May Affect Metabolism in RTT Patients
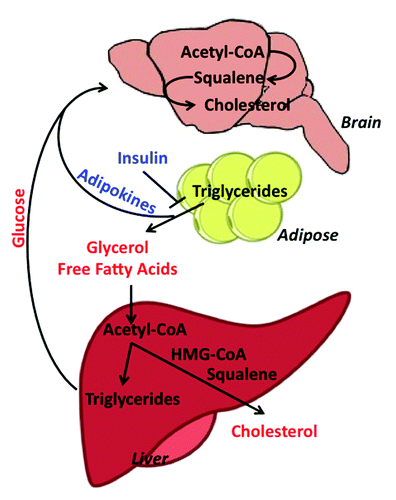
Mecp2-null mice display phenotypes of metabolic syndrome characterized by severe hyperinsulinemia, glucose tolerance, and insulin resistance.Citation22 Metabolism is a highly treatable target that is not limited to statin drugs. Metabolic pathways are complex, interconnected, and regulated at numerous feedback points (), which have been co-opted in pharmaceutical design. While statins treat high cholesterol by slowing the body’s production, cholesterol can also be lowered by inducing the uptake of blood cholesterol by the liver, where it can be converted into bile acids for excretion. Furthermore, glucose metabolism is inextricably linked to diet, cholesterol, and lipid metabolism through the action of adipokines, such as leptin, insulin, and ghrelin, which can be abnormal in RTT patients.Citation23 Neurons rely on energy derived from glucose transported across the blood brain barrier, which is metabolized to lactate by astrocytes. Astrocytes release lactate to neurons for maintenance of glutamatergic synthesis and synaptic activity.Citation24 The high production and utilization of fat may prevent the appropriate utilization of glucose in the Mecp2 mouse model or in RTT patients. In this regard, ketogenic diets—which are designed to force the body to burn fats rather than carbohydrates, allowing the brain to use ketone bodies as energy sources rather than glucose—have been reported to improve RTT symptoms.Citation25 Therefore, pathways involved in brain energy metabolism might be excellent targets in the subsets of RTT patients presenting abnormal lipid parameters. It is possible that combination therapies that reduce lipid accumulation, while improving glucose utilization, will aid RTT patients.
Figure 2. Metabolic crosstalk between the mouse brain, adipose tissue, and liver. Pathway members in red represent biomolecules circulating in the blood and members in blue represent circulating hormones. Loss of insulin sensitivity increases lipolysis in adipose tissue, resulting in the breakdown of stored triglycerides to glycerol and free fatty acids and the release of adipokine signaling molecules, which act on the brain. The increased circulating pool of free fatty acids is taken into the liver, where it is converted to acetyl-CoA and then converted either to triglycerides (stored in lipid droplets) or cholesterol, which is released into the blood. Meanwhile, circulating glucose is taken up by the brain for energy. Glucose absorbed by the brain can also be converted to acetyl-CoA, which enters into the cholesterol pathway and is eventually converted to HMG-CoA, squalene, and finally, cholesterol.
The metabolic symptoms in Mecp2 mutant mice occur concurrently with the development of fatty liver caused by the accumulation of triglycerides and other neutral lipids. MECP2 is required to bridge a corepressor complex containing the class II histone deacetylase HDAC3 to DNA.Citation26 MECP2 missense mutations that prevent the bridge between DNA and the corepressor complex cause Rett syndrome.Citation26 The loss of HDAC3 in mouse liver cells leads to metabolic syndrome and fatty liver disease (FLD), because one of HDAC3’s normal functions is to regulate gluconeogenesis and lipid homeostasis. FLD and the development of metabolic syndrome is accompanied by an increase in expression of enzymes involved in cholesterol and lipid synthesis, including SQLE,Citation27 suggesting that the HDAC3-containing-corepressor complex may directly regulate SQLE, along with other lipid synthesis enzymes. Initially a benign condition, fatty liver results from an imbalance in fatty acid metabolism, which leads to hepatic lipid accumulation. However, FLD can progress to a pathogenic state when there is an increase in oxidative and metabolic stress in the altered tissue.Citation28 Our data showed that statin treatment prevented or delayed the accumulation of lipids in the livers of both male and female Mecp2 mutant mice and would therefore prevent the development of FLD and downstream pathologies. RTT patients often exhibit seizures, which are treated with valproic acid, a weak HDAC3 inhibitor.Citation29 A study of HDAC3 deletion from the mouse liver suggests that treatment with HDAC3 inhibitors may also cause or exacerbate the pathogenesis of FLD.Citation27 In fact, the use of valproic acid is often linked with liver failure,Citation30 therefore its use in RTT patients should be carefully evaluated. Physicians should be aware that any liver involvement in Rett patients could be due to the effect of MECP2 mutation, which could be exacerbated by certain drugs.
Dyslipidemia Should be Considered When Evaluating RTT Patients
Our data suggest that brain cholesterol metabolism is perturbed initially in Mecp2 mutant mice, and then, eventually, lipids accumulate in the peripheral tissues as symptoms progress. The possibility that neuronal cholesterol metabolism is perturbed in RTT patients needs to be examined as well. The degree of peripheral lipid accumulation varies in different mouse inbred strains, even though brain lipids were perturbed regardless of genetic background. Do RTT patients ever exhibit liver disease? No compiled data exist on this matter. However, anecdotally, some RTT patients must have their gall bladder removed; the gall bladder is a sac in the liver that collects and releases bile acids, a product of cholesterol. Parent blogs suggest that some daughters with RTT have been hospitalized for near liver failure and/or liver tumors or lesions that supposedly “have nothing to do with their Rett syndrome” (personal communication). Hyperammonemia can be a consequence of liver failure, and poor liver function has an effect on many body systems, including the brain. In mice, and in humans, lipid metabolism is controlled by many genes. Therefore, whether peripheral lipids accumulate in the liver or elsewhere may depend upon the individual’s genetic makeup—how well they absorb cholesterol in the gut, for example. Interestingly, only a small subset of RTT patients have high BMI, while many are at the extreme low end of the curve, yet we observed evidence for systemic lipid dysregulation across the BMI spectrum. This suggests that abnormal metabolism in RTT will not always involve deposition of fat into adipose tissue but that accumulation of peripheral lipids in organs should also be considered when evaluating patients. Altogether, our studies provide hope for RTT families and patients that the highly treatable metabolic pathway may prove targetable to prevent the ever progressing morbidities associated with Rett syndrome.
| Abbreviations: | ||
| RTT | = | Rett syndrome |
| Mecp2 | = | Methyl CpG binding protein 2 |
| ASD | = | autism spectrum disorder |
| LDL | = | low density lipoprotein |
| HDL | = | high density lipoprotein |
| HDAC | = | histone deacetylase |
| SQLE | = | squalene epoxidase |
| CDKL5 | = | cyclin dependent kinase like 5 |
| FLD | = | fatty liver disease |
| BMI | = | body mass index |
| AST | = | aspartate transasminase |
| ALT | = | alanine transaminase |
| CK | = | creatine kinase |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The Rett Syndrome Research Trust (RSRT) was the primary funder of this work with support provided to both M.J.J. and A.D. M.J.J. also received Angel grant #2608 from the International Rett Syndrome Foundation (IRSF). We thank Sanuja De Costa for helping to compile the patient data. The Rett syndrome community is a unique collaboration, in which families critically read the literature and communicate their personal experiences with their daughters to researchers and physicians to move the field forward. We thank you.
References
- Rett A. [Cerebral atrophic syndrome in hyperammonemia in children]. Landarzt 1968; 44:797 - 801; PMID: 5720544
- Campos-Castelló J, Peral Guerra M, Riviere Gómez A, Oliete García F, Herranz Tanarro J, Toledano Barrero M, Espinar Sierra J, Cristobal Sassot S, Lautre Ecenarro MJ, Franco Carcedo C, et al. [Rett’s syndrome: study of 15 cases]. An Esp Pediatr 1988; 28:286 - 92; PMID: 2456708
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999; 23:185 - 8; http://dx.doi.org/10.1038/13810; PMID: 10508514
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet 2006; 7:415 - 26; http://dx.doi.org/10.1038/nrg1878; PMID: 16708070
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007; 315:1143 - 7; http://dx.doi.org/10.1126/science.1138389; PMID: 17289941
- Buchovecky CM, Turley SD, Brown HM, Kyle SM, McDonald JG, Liu B, Pieper AA, Huang W, Katz DM, Russell DW, et al. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat Genet 2013; 45:1013 - 20; http://dx.doi.org/10.1038/ng.2714; PMID: 23892605
- Pfrieger FW, Ungerer N. Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res 2011; 50:357 - 71; http://dx.doi.org/10.1016/j.plipres.2011.06.002; PMID: 21741992
- Kriaucionis S, Paterson A, Curtis J, Guy J, Macleod N, Bird A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol 2006; 26:5033 - 42; http://dx.doi.org/10.1128/MCB.01665-05; PMID: 16782889
- Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry 2012; 17:389 - 401; http://dx.doi.org/10.1038/mp.2011.165; PMID: 22143005
- Waterham HR. Defects of cholesterol biosynthesis. FEBS Lett 2006; 580:5442 - 9; http://dx.doi.org/10.1016/j.febslet.2006.07.027; PMID: 16876788
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, Leonard H, Bailey ME, Schanen NC, Zappella M, et al, RettSearch Consortium. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 2010; 68:944 - 50; http://dx.doi.org/10.1002/ana.22124; PMID: 21154482
- Percy AK. Rett syndrome: exploring the autism link. Arch Neurol 2011; 68:985 - 9; http://dx.doi.org/10.1001/archneurol.2011.149; PMID: 21825235
- Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, Bear MF. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 2013; 77:243 - 50; http://dx.doi.org/10.1016/j.neuron.2012.01.034; PMID: 23352161
- Ardern-Holmes SL, North KN. Therapeutics for childhood neurofibromatosis type 1 and type 2. Curr Treat Options Neurol 2011; 13:529 - 43; http://dx.doi.org/10.1007/s11940-011-0142-9; PMID: 21850405
- Sticozzi C, Belmonte G, Pecorelli A, Cervellati F, Leoncini S, Signorini C, Ciccoli L, De Felice C, Hayek J, Valacchi G. Scavenger receptor B1 post-translational modifications in Rett syndrome. FEBS Lett 2013; 587:2199 - 204; http://dx.doi.org/10.1016/j.febslet.2013.05.042; PMID: 23711372
- Daniels SR, Greer FR, Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008; 122:198 - 208; http://dx.doi.org/10.1542/peds.2008-1349; PMID: 18596007
- Schreurs BG. The effects of cholesterol on learning and memory. Neurosci Biobehav Rev 2010; 34:1366 - 79; http://dx.doi.org/10.1016/j.neubiorev.2010.04.010; PMID: 20470821
- Roy A, Pahan K. Prospects of statins in Parkinson disease. Neuroscientist 2011; 17:244 - 55; http://dx.doi.org/10.1177/1073858410385006; PMID: 21252380
- Dalla Y, Singh N, Jaggi AS, Singh D. Memory restorative role of statins in experimental dementia: an evidence of their cholesterol dependent and independent actions. Pharmacol Rep 2010; 62:784 - 96; http://dx.doi.org/10.1016/S1734-1140(10)70339-X; PMID: 21098862
- Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents, National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics 2011; 128:Suppl 5 S213 - 56; http://dx.doi.org/10.1542/peds.2009-2107C; PMID: 22084329
- Kwiterovich PO. Clinical and laboratory assessment of cardiovascular risk in children: Guidelines for screening, evaluation, and treatment. J Clin Lipidol 2008; 2:248 - 66; http://dx.doi.org/10.1016/j.jacl.2008.06.003; PMID: 21291741
- Pitcher MR, Ward CS, Arvide EM, Chapleau CA, Pozzo-Miller L, Hoeflich A, Sivaramakrishnan M, Saenger S, Metzger F, Neul JL. Insulinotropic treatments exacerbate metabolic syndrome in mice lacking MeCP2 function. Hum Mol Genet 2013; 22:2626 - 33; http://dx.doi.org/10.1093/hmg/ddt111; PMID: 23462290
- Penning C, van der Wouden JC. Increased plasma leptin levels and valproate use in patients with Rett syndrome. [author reply e.] J Pediatr 2007; 151:e15 - , author reply e15; http://dx.doi.org/10.1016/j.jpeds.2007.06.032; PMID: 17889056
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008; 322:1551 - 5; http://dx.doi.org/10.1126/science.1164022; PMID: 19056987
- Liebhaber GM, Riemann E, Baumeister FA. Ketogenic diet in Rett syndrome. J Child Neurol 2003; 18:74 - 5; http://dx.doi.org/10.1177/08830738030180011801; PMID: 12661945
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci 2013; 16:898 - 902; http://dx.doi.org/10.1038/nn.3434; PMID: 23770565
- Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J 2008; 27:1017 - 28; http://dx.doi.org/10.1038/emboj.2008.51; PMID: 18354499
- Day CP, James OF. Steatohepatitis: a tale of two “hits”?. Gastroenterology 1998; 114:842 - 5; http://dx.doi.org/10.1016/S0016-5085(98)70599-2; PMID: 9547102
- Pardal-Fernandez JM, Jerez-Garcia P, Onsurbe-Ramirez I, Marco-Giner J. Rett syndrome: a case presenting with atypical seizures. Neurophysiological and clinical aspects. Neurophysiologie clinique =. Clin Neurophysiol 2004; 34:49 - 57; http://dx.doi.org/10.1016/j.neucli.2003.08.003
- Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology 1996; 46:465 - 9; http://dx.doi.org/10.1212/WNL.46.2.465; PMID: 8614514