Abstract
Alkaptonuria (AKU) is a rare disorder of autosomal recessive inheritance. It is caused by a mutation in a gene that results in the accumulation of homogentisic acid (HGA). Characteristically, the excess HGA means sufferers pass dark urine, which upon standing turns black. This is a feature present from birth. Over time patients develop other manifestations of AKU, due to deposition of HGA in collagenous tissues, namely ochronosis and ochronotic osteoarthropathy.
Although this condition does not reduce life expectancy, it significantly affects quality of life. The natural history of this condition is becoming better understood, despite gaps in knowledge. Clinical assessment of the condition has also improved along with the development of a potentially disease-modifying therapy. Furthermore, recent developments in AKU research have led to new understanding of the disease, and further study of the AKU arthropathy has the potential to influence therapy in the management of osteoarthritis.
Introduction
AKU is an iconic disease in medicine, historically used by Archibald Garrod in his Croonian lectures of 1908, to demonstrate the theory behind “inborn errors of metabolism”. It was one of the first disorders in humans found to conform with the principles of Mendelian recessive inheritance.Citation1 It is a hereditary disorder and results from absence of homogentisate 1,2 dioxygenase (HGD), the enzyme, predominantly produced by hepatocytes in the liver and within the kidney, is responsible for the breakdown of HGA; an intermediate in the tyrosine degradation pathway ().Citation2
Figure 1. Adapted from reference Citation3. The diagram illustrates the normal phenylalanine and tyrosine degradation pathway, enzymes involved and defects that can occur (highlighted blue). The metabolism of HGA occurs in the liver, and HGD is an enzyme expressed in this organ.
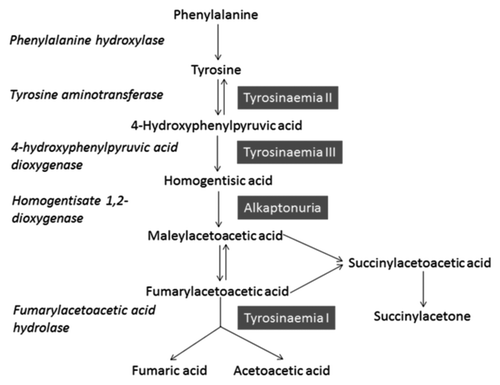
Deficient HGD activity within the liver causes HGA levels to rise systemically. Large (gram) quantities of HGA are removed by urinary excretion on a daily basis.Citation3 Other incidental observations of HGA in bodily fluids and tissues has been documented, based on the darkening of fluids or pathological deposition in tissues.Citation4,Citation5 However, the removal of HGA by urinary excretion is not sufficient to completely remove it from bodily tissues and fluids.
Characteristic early clinical presentation is the observation that urine darkens on standing. This is because the HGA polymerizes but can also be observed upon the addition of alkali substances.Citation2 This is the only symptom seen in the pediatric age group.Citation6 Over time the HGA polymer is deposited within connective tissues, causing ochronosis (a darkening of collagenous tissues).Citation7 Long-term ochronosis results in the development of ochronotic osteoarthropathy, often misdiagnosed as early onset osteoarthritis (OA),Citation2 unless observation of darkened urine has been seen before joint manifestations. Previous work has summarized the human disease, the genetics, and its manifestations.Citation8
There is currently no approved cure for AKU. However, research surrounding the mechanism of disease progression has highlighted that HGA pigment deposition occurs within structurally intact cartilage. The research identified that ochronotic osteoarthropathy and subsequent joint failure appear to arise following initial changes in the calcified cartilage and subchondral bone becoming susceptible to damage following focal change. This has led to suggestions that there could be a large overlap between the pathogenesis of OA and ochronotic osteoarthropathy, increasing the potential avenues of research.Citation9
Epidemiology
The worldwide prevalence of AKU is 1 case in 250 000–1 000 000 births.Citation2 So far, 950 AKU sufferers have been identified in 40 countries ().Citation10,Citation11 It is a condition that is reported to be more prevalent in Slovakia, the Dominican Republic, India, and Jordan. The highest prevalence is in Slovakia where up to 1 in 19 000 are affected.Citation10 Analysis of the affected families revealed they typically live within isolated hamlets, leading to conclusions that the usually high incidence was predominantly due to the founder effect (loss of genetic variation) as a result of genetic isolation.Citation12 Although difficult to perform with such a rare condition, there is also no genotype-phenotype correlation; all mutations lead to the development of ochronosis.Citation13
Figure 2. Taken from reference Citation11. The map illustrates the current number of AKU patients identified worldwide.
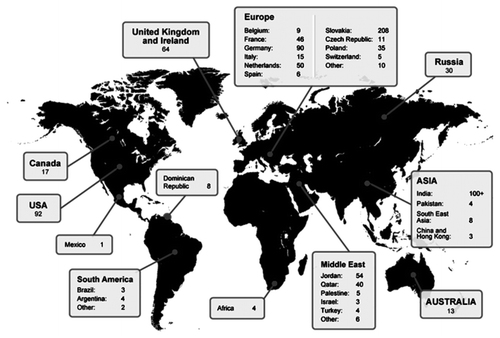
Research is still ongoing to fully understand the mechanism of disease progression; literature suggests the reporting of new cases has increased due to a raised profile of features associated with the disease.Citation10 However, even with the increase in reporting of new cases, the number of individuals documented as suffering from the disease is well below what would be expected based on the incidence.
Clinical Features
AKU has three distinct clinical features; homogentisic aciduria, ochronosis, and ochronotic osteoarthropathy.Citation14-Citation17 Each feature presents at various stages in life, the earliest being detection of HGA in urine.Citation18 The passing of black urine is the only manifestation of the condition known in pediatrics, leading to 21% of patients being diagnosed with AKU before 1 year of age.Citation2,Citation6 Although newborn screening is not undertaken, incidental discoveries have been observed during other screening programs.Citation19 HGA plasma levels in AKU sufferers range between 0.018–0.165 mM in comparison to non-AKU sufferers plasma levels of 0.014–0.071 µM, a thousand fold difference.Citation2,Citation20 The darkening of urine occurs because the HGA pigment oxidizes to Benzoquinoneacetate (BQA), which forms a melanin-like polymer that slowly turns urine black.Citation2
Ochronosis develops as the BQA accumulates both intra- and extra-cellularly in connective tissue. This feature is commonly observed in the third through to fifth decades of life. Typically the pigment is seen clearly in the eyes and ears of patients but is also present in bodily fluids, including perspiration, which often results in skin discoloration.Citation21 Organs affected are: large joints, cardiovascular system, kidney, skin, and glands.Citation4,Citation7,Citation22-Citation24 Other manifestations include: renal, prostate, gall bladder, and salivary gland stones, ruptures of tendons and ligaments, osteopenia, and fractures.Citation4,Citation25-Citation27
Cardiac manifestations of the disease are not uncommon. Aortic valve stenosis is a frequent finding, often requiring surgical replacement.Citation23 It may be that mechanical forces play a key role in mediating ochronosis given that there appears to be little pigmentation in the venous component of the circulation compared with the arterial side.Citation17 Interestingly, while HGD expression and HGA metabolism occur in the liver, there are no reports to demonstrate that these tissues—along with those of the pancreas, gastrointestinal, lymphoreticular, or endocrine organs—develop ochronosis.Citation17
The development of ochronotic arthropathy is the result of deposition of the HGA polymer within hyaline articular cartilage. Pigmentation is widespread, with all tissues of the joint organ being affected (). The affected tissues often become weak, brittle, and prone to chipping, fracturing, and cracking, causing rapid joint degeneration, which means that patients can be left profoundly disabled at a young age.Citation7 Although AKU does not affect longevity, it significantly affects quality of life due to these secondary pathologies.
Table 1. Summarizes the location of HGA pigment deposition through all layers of an articulating joint.
Patients with ochronotic arthropathy usually present with lumbar pain as the initial joint manifestation. Larger weight bearing joints tend to be affected later in the progression of the condition.Citation28 Quite often this complaint is misdiagnosed as an early form of osteoarthritis (OA) or ankylosing spondylitis. The differences between the two conditions are summarized in and . It is interesting to note that the ossification of tissues in ochronosis is different from that observed in other forms of pathological ossification. This is because the ossification of tissues in ochronosis is seen with a variety of calcium crystals.Citation29,Citation30 Furthermore, biomechanical studies have shown that ochronotic cartilage calcification is harder than normal cartilage calcification.Citation17,Citation31,,Citation32
Table 2. Adapted from reference 21 The table shows the differences between peripheral ochronotic arthropathy and osteoarthritis.Citation31
Table 3. Adapted from reference Citation21. The table shows the radiographic differences between ankylosing spondylitis and ochronotic spondylarthropathy.
History of AKU
Garrod’s use of AKU in the Croonian lectures brought the condition into the spotlight in 1908. Yet many descriptions of the triad of features associated with AKU date back long before this.Citation2 Documentation of the condition began in the 16th and 17th centuries.Citation24 The earliest clinical case of AKU was found in the Egyptian mummy Harwa, which is believed to date back as far as 1500 BC.Citation33 The name Alkaptonuria is derived from the Arabic word “alkali” (meaning alkali) and the Greek word meaning “to suck up oxygen greedily in alkali”.Citation24 The name was created by Boedeker in 1859 after he discovered unusual reducing properties in the urine of a patient.Citation14,Citation15
Ochronosis was first described and named by Virchow in 1866, because under microscopy the HGA pigment appeared to be ochre (yellow/brown) in color.Citation34 In 1891 HGA was identified as the causative component and named so due to its close structural relationship with gentisic acid, a derivative of benzoic acid.Citation1,Citation21,Citation35,Citation36 By 1995 the genetic defect was discovered, cloned, and mapped to chromosome 3q21-q23.Citation22,Citation37,Citation38
Therapies
Many therapies have been tried. However, currently as there is no effective therapy, the management of AKU remains palliative and involves physiotherapy, joint replacement surgery, and pain control. Ascorbic acid (ASC), more commonly known as vitamin C, is an antioxidant believed to reduce the conversion of HGA to BQA via oxidation. However, investigation revealed that although ASC reduced the HGA to BQA conversion, it did not affect HGA urinary excretion.Citation39 Furthermore, it was found to increase HGA production, contributing to the formation of renal oxalate stones. This is concerning, as AKU patients are already at high risk for developing renal calculi.Citation40,Citation41 An additional study highlighted that vitamin C is a co-factor for 4-hydroxyphenylpyruvate dioxygenase, which causes increased HGA production. In the cases of young infants there were profound increases in urinary levels of HGA, leading to conclusions that this is a highly unsuitable treatment.Citation42
A low protein diet, although logical, is not sustainable in the long-term for many patients. Approximately 6% of dietary protein is degraded via the HGA pathway, and intensive supervision is required with younger patients, especially during growth periods.Citation43,Citation44 Also, regardless of restrictions on dietary intake of tyrosine, tissue catabolism is likely to contribute to raised HGA plasma levels within individuals with AKU. There is also evidence to suggest that liver transplantation is a successful way to eradicate HGA from the body.Citation45 Many therapies have been trialed and are summarized in .
Table 4. Adapted from reference Citation44. Information on current and future therapies available from references Citation5 and Citation46.
Other, more promising therapy includes a triketone herbicide, Nitisinone (NTBC), that inhibits 4-hydroxyphenylpyruvate, an enzyme involved in the conversion of hydroxyphenylpyruvate to HGA (). It significantly reduces urinary excretion of HGA in both murine models and humans.Citation47-Citation49 Initially approved by the Food and Drug Administration (FDA) for the treatment of hereditary tyrosinaemia type I (HT1), there are, however, known side effects, including elevated plasma tyrosine levels causing corneal irritation. This can be ameliorated by reducing dietary intake of tyrosine. Other serious adverse events associated with elevated plasma tyrosine levels include thrombocytopenia, leukopenia, and porphyria.Citation49 However, complications of using this therapy are the development of hereditary tyrosinaemia type III (HT3) and rarely a deficiency of 4-hydroxyphenylpyruvate. These conditions cause neurological complications; tremor, ataxia, delayed development, and intellectual impairment.Citation2,Citation50
Enzyme replacement would be an ideal therapy for AKU, consisting of immediate replacement of HGD in the tyrosine degradation pathway. However, there are potentially fatal complications associated with this therapy. It is imperative that the HGD enzyme is delivered to the exact location of tyrosine metabolism within the hepatocytes of the liver. If not, spontaneous formation of succinylacetone (formed from the production of MAA and FAA in tyrosine metabolism) would occur, which is toxic and highly mutagenic. Build-up of this substance in the body and bloodstream would create complications more serious than the ochronosis of AKU.Citation51
Models of AKU
Many spontaneous reports of AKU in animals have been documented: crab-eating macaque, chimpanzee, orangutan, Dalmatians, cattle, horses, dogs, and rabbits. All the reports identify the appearance of dark urine but rarely identify ochronosis or joint involvement.Citation52-Citation56 Early attempts to study AKU in animal models were undertaken by intraperitoneal or intravenous and intra-articular injection of HGA into rabbits.Citation57 The study demonstrated the damaging effects of HGA. Animals that received multiple injections exhibited darkening of urine along with ochronosis of joint tissues, and those injected with HGA via intraperitoneal or intravenous routes did not exhibit ochronosis.Citation56 The first model study was made using rats fed on a diet of 8% l-tyrosine for at least a 9 month period. This appeared to induce ochronosis and osteoarthropathy.Citation58
The first murine model of AKU was generated in 1994 and proved to be key in aiding to map the location of the HGD mutation to chromosome 16. The report stated that although the mice excreted high levels of urinary HGA, there was no evidence that they developed ochronosis or arthropathy. This was hypothesized to be the result of the endogenous production of ascorbic acid in the digestive tract of the mouse, thus inhibiting the polymerization of HGA.Citation59
Other studies have developed murine models of AKU, also concluding that despite elevated levels of HGA, the mice did not exhibit the typical phenotypic ochronosis observed in the human presentation. Hypotheses as to why this occurred, aside from the production of ascorbic acid in mice, have stated that the mice do not live long enough for ochronosis to occur and that urinary excretion is efficient so that tissues are not exposed to the high concentration of HGA as seen in humans.Citation60,Citation61 Other animal models have produced similar reports, although arguably not as reliable.Citation54
Contrary to the above reports, Taylor et al. produced the first data of tissue ochronosis in a murine model. The data from this study demonstrated ochronosis in tissues and joints of mice with the AKU genotype, similar to the presentation seen in humans.Citation62 This was a novel finding and has subsequently enabled better understanding of the molecular pathology of the condition.
In more recent murine models, initiation and progression of pigmentation has been documented. At 15 weeks, pigmentation is visible, suggesting that ochronosis begins at an early age. Furthermore, ochronosis was seen in all mice, and pigmentation of chondrocytes also increased with mouse age.Citation48 These models, along with others, have been used to investigate the effects of NTBC. The trials have been successful and demonstrate the delayed progression of ochronosis after drug administration. The conclusions from these studies suggest beginning therapy at an early stage in the development of ochronosis results in a more beneficial outcome.Citation48,Citation61
Initial trials of low dose NTBC in two patients with AKU showed a 69% reduction in urinary HGA excretion. However, there was a significant elevation in plasma tyrosine levels, increasing the risk of corneal crystal formation, corneal epithelial damage, and photophobia.Citation2 A larger scale trial of nine patients was performed and also discovered a 95% reduction in urinary HGA excretion but an 11-fold increase in plasma tyrosine over a four month period. Although the tyrosinaemia did not cause eye complications in these patients, this is a concern when considering the efficacy of NTBC as a therapy for AKU.Citation3 A three year trial of NTBC performed on a cohort of 40 patients also demonstrated similar results.Citation47
AKU as a Model of Osteoarthritis
The development of murine and in vitro models of AKU have enabled a better understanding of the pathophysiology involved in the progression of ochronosis and the related osteoarthropathy. So far, research has identified that HGA is present in healthy cartilage but only becomes susceptible to degeneration following focal change.Citation9 The potential overlap between the pathogenesis of OA and ochronotic arthropathy has stemmed from a better understanding of the importance of subchondral bone in the pathogenesis of OACitation63,Citation64 and determining factors that influence the integrity of articular cartilage: subchondral bone turnover, chondrocyte function, and biomechanical stresses, all of which are affected in the arthropathy of AKU.Citation63,Citation65 Furthermore, AKU has already been documented as causing premature OA and mimicking the typical disease process; this extreme phenotype of OA could provide insights into OA.Citation66,Citation67
Summary
It has been over 100 years since Garrod first described AKU as an “inborn error of metabolism”. Within this time, the causative molecule has been isolated and identified, the enzymatic defect located, and the genetic mutation mapped. Yet there is still no effective treatment for this iconic condition. Interest in AKU research has increased recently, generating hope that potential disease modifying therapy is within reach.
Furthermore, it appears that the arthropathy of AKU may have similarities to OA—not only in its presentation but also its pathogenesis—leading to speculation that it could prove a useful extreme phenotype in highlighting missing knowledge in understanding OA. This would not be the first time that a rare disease has brought more knowledge to a common condition. For example in musculoskeletal conditions, the discovery of sclerostin and its importance in the Wnt signaling pathway, highlighted by the high bone mass phenotypes of those with van Buchem Disease (VBD), has led to therapies for osteoporosis.Citation68,Citation69
It is encouraging to consider that further research into this condition may contribute to the understanding and treatment of joint degeneration not only in AKU but also in OA.
| Abbreviations: | ||
| AKU | = | alkaptonuria, BQA, benzoquinoneacetate |
| HGA | = | homogentisic acid |
| HGD | = | homogentisate 1,2-dioxygenase |
| OA | = | osteoarthritis |
| ASC | = | ascorbic acid |
| NTBC | = | 2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione |
| FDA | = | Food and Drug Administration |
| HT1 | = | hereditary tyrosinaemia type 1 |
| HT3 | = | hereditary tyrosinaemia type 3 |
| MAA | = | maleylacetoacetic acid |
| FAA | = | fumarylacetoacetic acid |
| VBD | = | van Buchem disease |
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
The authors would like to thank the AKU society, Rosetrees Trust, and University Hospitals of Morecambe Bay NHS Foundation trust for funding.
References
- Garrod AE, Oxon MD. The incidence of alkaptonuria: a study in chemical individuality. 1902. Mol Med 1996; 2:274 - 82; PMID: 8784780
- Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, Fitzpatrick DL, Anderson PD, Huizing M, Anikster Y, Gerber LH, et al. Natural history of alkaptonuria. N Engl J Med 2002; 347:2111 - 21; http://dx.doi.org/10.1056/NEJMoa021736; PMID: 12501223
- Suwannarat P, O’Brien K, Perry MB, Sebring N, Bernardini I, Kaiser-Kupfer MI, Rubin BI, Tsilou E, Gerber LH, Gahl WA. Use of nitisinone in patients with alkaptonuria. Metabolism 2005; 54:719 - 28; http://dx.doi.org/10.1016/j.metabol.2004.12.017; PMID: 15931605
- Taylor AM, Wilson PJ, Ingrams DR, Helliwell TR, Gallagher JA, Ranganath LR. Calculi and intracellular ochronosis in the submandibular tissues from a patient with alkaptonuria. J Clin Pathol 2010; 63:186 - 8; http://dx.doi.org/10.1136/jcp.2009.071365; PMID: 20154043
- Garcia SF, Egbert B, Swetter SM. Hereditary ochronosis: hyperpigmented skin overlying cartilaginous structures. Cutis 1999; 63:337 - 8; PMID: 10388955
- Peker E, Yonden Z, Sogut S. From darkening urine to early diagnosis of alkaptonuria. Indian J Dermatol Venereol Leprol 2008; 74:700; http://dx.doi.org/10.4103/0378-6323.45142; PMID: 19180686
- Keller JM, Macaulay W, Nercessian OA, Jaffe IA. New developments in ochronosis: review of the literature. Rheumatol Int 2005; 25:81 - 5; http://dx.doi.org/10.1007/s00296-004-0498-1; PMID: 15322814
- Kraus VB. Rare osteoarthritis: Ochronosis, Kashin-Beck disease and Mseleni joint disease. In: Hochberg MC, Smolen SJ, Weinblatt ME, Weisman MH, ed. Rheumatology. 5th ed. Philadelphia (PA): Mosby; 2011 p1825-1837.
- Taylor AM, Boyde A, Wilson PJ, Jarvis JC, Davidson JS, Hunt JA, Ranganath LR, Gallagher JA. The role of calcified cartilage and subchondral bone in the initiation and progression of ochronotic arthropathy in alkaptonuria. Arthritis Rheum 2011; 63:3887 - 96; http://dx.doi.org/10.1002/art.30606; PMID: 22127706
- Ranganath L, Taylor AM, Shenkin A, Fraser WD, Jarvis J, Gallagher JA, Sireau N. Identification of alkaptonuria in the general population: a United Kingdom experience describing the challenges, possible solutions and persistent barriers. J Inherit Metab Dis 2011; 34:723 - 30; http://dx.doi.org/10.1007/s10545-011-9282-z; PMID: 21311977
- Develop AKUre. 950 AKU patients [Internet]. Cambridge (UK): The AKU Society and the DevelopAKUre Consortium: c2014 [cited 15 May 2013]. Available from: http://www.developakure.eu/950-aku-patients/
- Srsen S, Müller CR, Fregin A, Srsnova K. Alkaptonuria in Slovakia: thirty-two years of research on phenotype and genotype. Mol Genet Metab 2002; 75:353 - 9; http://dx.doi.org/10.1016/S1096-7192(02)00002-1; PMID: 12051967
- Vilboux T, Kayser M, Introne W, Suwannarat P, Bernardini I, Fischer R, O’Brien K, Kleta R, Huizing M, Gahl WA. Mutation spectrum of homogentisic acid oxidase (HGD) in alkaptonuria. Hum Mutat 2009; 30:1611 - 9; http://dx.doi.org/10.1002/humu.21120; PMID: 19862842
- Boedeker C.. Ueber das Alcapton; ein neuer Beitrag zur Frage: Welche Stoffe des Hams kannen Kupferreduction bewirken?. Ztschr f rat Med 1859; 7:130
- Boedeker C. Das Alkapton; ein Beitrag zur Frage:Welche Stoffe des Harns Konnen aus einer alkalischen Kupferoxydlosung Kupferoxydul reduciren?. Ann Chem Pharmacol 1861; 117:98; http://dx.doi.org/10.1002/jlac.18611170106
- Gaines JJ Jr.. The pathology of alkaptonuric ochronosis. Hum Pathol 1989; 20:40 - 6; http://dx.doi.org/10.1016/0046-8177(89)90200-1; PMID: 2643557
- Helliwell TR, Gallagher JA, Ranganath L. Alkaptonuria--a review of surgical and autopsy pathology. Histopathology 2008; 53:503 - 12; PMID: 18336562
- Peker E, Yonden Z, Sogut S, Peker E, Yonden Z, Sogut S. From darkening of urine is the early sign of alkaptonuria. Indian J Dermatol Ve 2008; 74:705
- Bradley DM. Screening for inherited metabolic disease in Wales using urine-impregnated filter paper. Arch Dis Child 1975; 50:264 - 8; http://dx.doi.org/10.1136/adc.50.4.264; PMID: 1147666
- Deutsch JC, Santhosh-Kumar CR, Deutsch J, Santhoshkumar C. Quantitation of homogentisic acid in normal human plasma. J Chromatogr B Biomed Appl 1996; 677:147 - 51; http://dx.doi.org/10.1016/0378-4347(95)00442-4; PMID: 8925087
- Klippel JH, Dieppe PA. Rheumatology - Volume Two. London, UK: Mosby, 1998.
- Fernández-Cañón JM, Granadino B, Beltrán-Valero de Bernabé D, Renedo M, Fernández-Ruiz E, Peñalva MA, Rodríguez de Córdoba S. The molecular basis of alkaptonuria. Nat Genet 1996; 14:19 - 24; http://dx.doi.org/10.1038/ng0996-19; PMID: 8782815
- Fisher AA, Davis MW. Alkaptonuric ochronosis with aortic valve and joint replacements and femoral fracture: a case report and literature review. Clin Med Res 2004; 2:209 - 15; http://dx.doi.org/10.3121/cmr.2.4.209; PMID: 15931360
- O’Brien WM, La Du BN, Bunim JJ. Biochemical, pathologic and clinical aspects of alkaptonuria, ochronosis and ochronotic arthropathy. Am J Med 1963; 34:813 - 38; http://dx.doi.org/10.1016/0002-9343(63)90089-5
- Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br 2003; 85:883 - 6; PMID: 12931812
- Introne WJ, Phornphutkul C, Bernardini I, McLaughlin K, Fitzpatrick D, Gahl WA. Exacerbation of the ochronosis of alkaptonuria due to renal insufficiency and improvement after renal transplantation. Mol Genet Metab 2002; 77:136 - 42; http://dx.doi.org/10.1016/S1096-7192(02)00121-X; PMID: 12359141
- Krízek V. Urolithiasis and prostatolithiasis in alcaptonuria with ochronosis. Int Urol Nephrol 1971; 3:245 - 50; http://dx.doi.org/10.1007/BF02081762; PMID: 5154571
- Gaines JJ Jr., Tom GD, Khankhanian N. An ultrastructural and light microscopic study of the synovium in ochronotic arthropathy. Hum Pathol 1987; 18:1160 - 4; http://dx.doi.org/10.1016/S0046-8177(87)80385-4; PMID: 3679190
- Reginato AJ, Schumacher HR, Martinez VA. Ochronotic arthropathy with calcium pyrophosphate crystal deposition. A light and electron microscopic study. Arthritis Rheum 1973; 16:705 - 14; http://dx.doi.org/10.1002/art.1780160602; PMID: 4757869
- McClure J, Smith PS, Gramp AA. Calcium pyrophosphate dihydrate (CPPD) deposition in ochronotic arthropathy. J Clin Pathol 1983; 36:894 - 902; http://dx.doi.org/10.1136/jcp.36.8.894; PMID: 6308064
- Milch RA. Biochemical studies on the pathogenesis of collagen tissue changes in alcaptonuria. Clin Orthop 1962; 24:213 - 29; PMID: 14473824
- Perry MB, Suwannarat P, Furst GP, Gahl WA, Gerber LH. Musculoskeletal findings and disability in alkaptonuria. J Rheumatol 2006; 33:2280 - 5; PMID: 16981292
- Stenn FF, Milgram JW, Lee SL, Weigand RJ, Veis A. Biochemical identification of homogentisic acid pigment in an ochronotic egyptian mummy. Science 1977; 197:566 - 8; http://dx.doi.org/10.1126/science.327549; PMID: 327549
- Virchow R. Ein Fall von allgemeiner Ochronose der Knorpel und knorpelahnlichen Theile. Arch Path Anat 1866; 37:212 - 9
- Wolkow M, Baumann E. Ueber das Wesen der Alkaptonurie. Z Phys Chem 1891; 15:228 - 85
- Scriver CR. Garrod’s Croonian Lectures (1908) and the charter ‘Inborn Errors of Metabolism’: albinism, alkaptonuria, cystinuria, and pentosuria at age 100 in 2008. J Inherit Metab Dis 2008; 31:580 - 98; http://dx.doi.org/10.1007/s10545-008-0984-9; PMID: 18850300
- La Du BN, Zannoni VG, Laster L, Seegmiller JE. The nature of the defect in tyrosine metabolism in alcaptonuria. J Biol Chem 1958; 230:251 - 60; PMID: 13502394
- Pollak MR, Chou YH, Cerda JJ, Steinmann B, La Du BN, Seidman JG, Seidman CE. Homozygosity mapping of the gene for alkaptonuria to chromosome 3q2. Nat Genet 1993; 5:201 - 4; http://dx.doi.org/10.1038/ng1093-201; PMID: 8252048
- Sealock RR, Gladstone M, Steele JM. Administration of ascorbic acid to an alkaptonuric patient. Proc Soc Exp Biol Med 1940; 44:580 - 3; http://dx.doi.org/10.3181/00379727-44-11534
- Lorenzini S, Mannoni A, Selvi E. Alkaptonuria. N Engl J Med 2003; 348:1408 - , author reply 1408; http://dx.doi.org/10.1056/NEJM200304033481421; PMID: 12672874
- Forslind K, Wollheim FA, Akesson B, Rydholm U. Alkaptonuria and ochronosis in three siblings. Ascorbic acid treatment monitored by urinary HGA excretion. Clin Exp Rheumatol 1988; 6:289 - 92; PMID: 3180550
- Wolff JA, Barshop B, Nyhan WL, Leslie J, Seegmiller JE, Gruber H, Garst M, Winter S, Michals K, Matalon R. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res 1989; 26:140 - 4; http://dx.doi.org/10.1203/00006450-198908000-00015; PMID: 2771520
- de Haas V, Carbasius Weber EC, de Klerk JB, Bakker HD, Smit GP, Huijbers WA, Duran M, Poll-The BT. The success of dietary protein restriction in alkaptonuria patients is age-dependent. J Inherit Metab Dis 1998; 21:791 - 8; http://dx.doi.org/10.1023/A:1005410416482; PMID: 9870204
- Ranganath LR, Jarvis JC, Gallagher JA. Recent advances in management of alkaptonuria (invited review; best practice article). J Clin Pathol 2013; 66:367 - 73; http://dx.doi.org/10.1136/jclinpath-2012-200877; PMID: 23486607
- Kobak AC, Oder G, Kobak S, Argin M, Inal V. Ochronotic arthropathy: disappearance of alkaptonuria after liver transplantation for hepatitis B-related cirrhosis. J Clin Rheumatol 2005; 11:323 - 5; http://dx.doi.org/10.1097/01.rhu.0000191157.25894.55; PMID: 16371803
- Alkaptonuria Society [Internet]. Cambridge (UK): Alkaptonuria Society Ltd; c2012 [cited 15 May 2013]. Available from: http://www.akusociety.org/index.html
- Introne WJPM, Perry MB, Troendle J, Tsilou E, Kayser MA, Suwannarat P, O’Brien KE, Bryant J, Sachdev V, Reynolds JC, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab 2011; 103:307 - 14; http://dx.doi.org/10.1016/j.ymgme.2011.04.016; PMID: 21620748
- Preston AJ, Keenan CM, Sutherland H, Wilson PJ, Wlodarski B, Taylor AM, Williams DP, Ranganath LR, Gallagher JA, Jarvis JC. Ochronotic osteoarthropathy in a mouse model of alkaptonuria, and its inhibition by nitisinone. Ann Rheum Dis 2014; 73:284 - 9; http://dx.doi.org/10.1136/annrheumdis-2012-202878; PMID: 23511227
- Medscape Reference Nitisinone (Rx) - Orfadin; adverse effects [Internet]. New York (NY): Medscape, LLC; c2014 [cited 15 May 2013]. Available from: http://reference.medscape.com/drug/orfadin-nitisinone-342859#4
- Ellaway CJ, Holme E, Standing S, Preece MA, Green A, Ploechl E, Ugarte M, Trefz FK, Leonard JV. Outcome of tyrosinaemia type III. J Inherit Metab Dis 2001; 24:824 - 32; http://dx.doi.org/10.1023/A:1013936107064; PMID: 11916315
- Taylor AM. An investigation of the pathogenesis of ochronosis in Alkaptonuria (AKU) [dissertation]. Liverpool: University of Liverpool, 2011.
- Johnson EH, Miller RL. Alkaptonuria in a cynomolgus monkey (Macaca fascicularis). J Med Primatol 1993; 22:428 - 30; PMID: 8169945
- Watkins SP, Benley H, Shulman NR. 2nd conference on experimental medicine and surgery in primates, New York NY. September 1969. Medical primatology 1970.: Basel: Karger.
- Keeling ME, McClure HM, Kibler RF. Alkaptonuria in an orangutan (Pongo pygmaeus). Am J Phys Anthropol 1973; 38:435 - 8; http://dx.doi.org/10.1002/ajpa.1330380245; PMID: 4689767
- Lonsdale D. Why I left orthodox medicine: Healing for the 21st century. Charlottesville, VA: Hampton Roads Publishing Company, 1994.
- Lewis JH. Alcaptonuria in a rabbit. J Biol Chem 1926; 70:659
- Moran TJ, Yunis EJ. Studies on ochronosis. 2. Effects of injection of homogentisic acid and ochronotic pigment in experimental animals. Am J Pathol 1962; 40:359 - 69; PMID: 14475795
- Blivaiss BB, Rosenberg EF, Kutuzov H, Stoner R. Experimental ochronosis. Induction in rats by long-term feeding with L-tyrosine. Arch Pathol 1966; 82:45 - 53; PMID: 5938449
- Montagutelli X, Lalouette A, Coudé M, Kamoun P, Forest M, Guénet JL. aku, a mutation of the mouse homologous to human alkaptonuria, maps to chromosome 16. Genomics 1994; 19:9 - 11; http://dx.doi.org/10.1006/geno.1994.1004; PMID: 8188247
- Manning K, Fernández-Cañón JM, Montagutelli X, Grompe M. Identification of the mutation in the alkaptonuria mouse model. Mutations in brief no. 216. Online. Hum Mutat 1999; 13:171; http://dx.doi.org/10.1002/(SICI)1098-1004(1999)13:2<171::AID-HUMU15>3.0.CO;2-W; PMID: 10094559
- Suzuki Y, Oda K, Yoshikawa Y, Maeda Y, Suzuki T. A novel therapeutic trial of homogentisic aciduria in a murine model of alkaptonuria. J Hum Genet 1999; 44:79 - 84; http://dx.doi.org/10.1007/s100380050114; PMID: 10083729
- Taylor AM, Preston AJ, Paulk NK, Sutherland H, Keenan CM, Wilson PJ, Wlodarski B, Grompe M, Ranganath LR, Gallagher JA, et al. Ochronosis in a murine model of alkaptonuria is synonymous to that in the human condition. Osteoarthritis Cartilage 2012; 20:880 - 6; http://dx.doi.org/10.1016/j.joca.2012.04.013; PMID: 22542924
- Hayami T, Pickarski M, Wesolowski GA, McLane J, Bone A, Destefano J, Rodan GA, Duong T. The role of subchondral bone remodeling in osteoarthritis: reduction of cartilage degeneration and prevention of osteophyte formation by alendronate in the rat anterior cruciate ligament transection model. Arthritis Rheum 2004; 50:1193 - 206; http://dx.doi.org/10.1002/art.20124; PMID: 15077302
- Castañeda S, Roman-Blas JA, Largo R, Herrero-Beaumont G. Subchondral bone as a key target for osteoarthritis treatment. Biochem Pharmacol 2012; 83:315 - 23; http://dx.doi.org/10.1016/j.bcp.2011.09.018; PMID: 21964345
- Karsdal MA, Leeming DJ, Dam EB, Henriksen K, Alexandersen P, Pastoureau P, Altman RD, Christiansen C. Should subchondral bone turnover be targeted when treating osteoarthritis?. Osteoarthritis Cartilage 2008; 16:638 - 46; http://dx.doi.org/10.1016/j.joca.2008.01.014; PMID: 18362080
- Bálint G, Szebenyi B. Hereditary disorders mimicking and/or causing premature osteoarthritis. Baillieres Best Pract Res Clin Rheumatol 2000; 14:219 - 50; http://dx.doi.org/10.1053/berh.2000.0063; PMID: 10925743
- Lagier R. Ochronotic arthropathy, an approach to osteoarthritis bone remodelling. Rheumatol Int 2006; 26:561 - 4; http://dx.doi.org/10.1007/s00296-005-0087-y; PMID: 16435121
- van Lierop AH, Hamdy NAT, van Bezooijen RL, Lowik CW, Papapoulos SE. The role of sclerostin in the pathophysiology of sclerosing bone dysplasias. Clinical Reviews in Bone and Mineral Metabolism 2012; 10:108; http://dx.doi.org/10.1007/s12018-011-9123-5
- Moester MJC, Papapoulos SE, Löwik CW, van Bezooijen RL. Sclerostin: current knowledge and future perspectives. Calcif Tissue Int 2010; 87:99 - 107; http://dx.doi.org/10.1007/s00223-010-9372-1; PMID: 20473488