
Abstract
Senataxin (SETX) is a putative RNA:DNA helicase that is mutated in two distinct juvenile neurological disorders, AOA2 and ALS4. SETX is involved in the response to oxidative stress and is suggested to resolve R loops formed at transcription termination sites or at sites of collisions between the transcription and replication machineries. R loops are hybrids between RNA and DNA that are believed to lead to DNA damage and genomic instability. We discovered that Rrp45, a core component of the exosome, is a SETX-interacting protein and that the interaction depends on modification of SETX by sumoylation. Importantly, we showed that AOA2 but not ALS4 mutations prevented both SETX sumoylation and the Rrp45 interaction. We also found that upon replication stress induction, SETX and Rrp45 co-localize in nuclear foci that constitute sites of R-loop formation generated by transcription and replication machinery collisions. We suggest that SETX links transcription, DNA damage and RNA surveillance, and discuss here how this link can be relevant to AOA2 disease.
Senataxin (SETX) is a large protein of more than 300 kDa that is considered to be the counterpart of the yeast DNA/RNA helicase Sen1. Indeed SETX carries a conserved helicase motif in its C terminus, but so far helicase activity has only been shown for the S. Pombe protein.Citation1 Sen1 (tRNA splicing endonuclease) was originally discovered as an endonuclease that processes tRNACitation2 but has been further characterized as an important factor in non-coding RNA processing and, for some genes, transcription termination.Citation3,Citation4 For example, SETX has been suggested to be involved in transcription termination at G-rich pause sites, which appear to be R-loop hot spots.Citation5 (R loops are hybrids between nascent transcripts and the DNA template.Citation6) Additionally, Sen1 and SETX have both been implicated in R-loop resolution at DNA damage sites.Citation7-Citation10 For example, it is now believed that SETX plays a role in resolving R loops that form when the transcription and replication machineries collide. Defects in R-loop resolution are known to lead to accumulation of DNA double-strand breaks (DSBs), resulting in DNA rearrangements and genome instability.Citation6,Citation11
Interest in SETX increased in 2004 when it was found that mutations in SETX can lead to two distinct neurological disorders, a form of autosomal recessive ataxia named AOA2 (Ataxia with Oculomotor Apraxia 2)Citation12 and a juvenile form of ALS (Amyotrophic Lateral Sclerosis or Lou Gehrig disease), ALS 4.Citation13 Mutations leading to AOA2 are recessive, can be homozygous or compound heterozygous and are believed to result in SETX loss of function, while mutations responsible for ALS4 are dominant and most likely gain of function. In both cases, disease onset occurs generally at adolescence and patient life span is rarely affected. To date, many mutations including missense, nonsense and deletion mutations, scattered throughout the ORF, are linked to AOA2,Citation14 and eight mutations associated with ALS4 have been described.Citation13,Citation15-Citation17 It is important to note that a growing number of SETX mutations have been found in patients that display atypical and/or intermediate phenotypes distinct from AOA2 or ALS4Citation15,Citation16,Citation18-Citation21
While a growing number of AOA2 cases are diagnosed and new SETX mutations are revealed, there has been limited information about how those mutations affect SETX function and how they are related to the disease. In an effort to address this issue, we studied three previously described AOA2 mutations (E65KCitation22 W305C and P413LCitation12) and two ALS4 mutations (T3I and L389SCitation13), all lying within the putative protein interaction domain of SETX located at its N terminus.Citation23 Our approach was to first identify proteins expressed in brain that interact with this region of SETX (Nter-SETX, residues 1–665) using a yeast two-hybrid (Y2H) screen with Nter-SETX and a human brain cDNA library.Citation24 We found that N-ter SETX interacted with a relatively small number of proteins in this assay, and one that was detected repeatedly was Rrp45, a core component of the multisubunit exosome. The exosome is a large complex that processes and/or degrades RNAs in the nucleus, nucleolus as well as the cytoplasm.Citation25-Citation27 It consists of a core of 9 subunits (including Rrp45) and is associated with two 3′-5′ exoribonucleases, Rrp6 and Dis3, that can have distinct RNA specificities.Citation28 This novel interaction, which importantly we also confirmed by co-immunoprecipitation (coIP) in human cellsCitation24 suggested a possible connection between RNA transcription and/or termination and RNA degradation and/or surveillance.Citation29,Citation30 Indeed, while the exosome has a large panel of substrates, Rrp45 has been suggested to confer a specific role in AU-rich mRNAs turnover.Citation31,Citation32 In an effort to elucidate the possible function of the SETX-Rrp45 interaction, we tested whether SETX could be implicated in regulation of AU-rich mRNAs. However, RNA analysis by RT-qPCR after siRNA-mediated SETX knockdown did not show any significant accumulation or degradation of AU-rich mRNAs, and other attempts to implicate SETX in known exosome functions were likewise unsuccessful (unpublished data). Only later were we able to gain insight into the functional significance of this novel interaction.
We next asked whether any of the AOA2 or ALS4 mutations might affect the Rrp45 interaction, again using the Y2H assay. Remarkably, we found that the association between SETX and Rrp45 was abolished or greatly reduced when SETX carried any of the three AOA2 mutations, but unaffected by the ALS4 mutations. How do mutations that are as close as P413L and L389S lead to such different effects while mutations separated by more than 300 aa trigger a similar response? It is possible that the residues affected by the AOA2 mutations are brought together in the 3D structure of SETX and that the mutations affect the same protein interaction domain of the protein. Unfortunately the structure of SETX or even the N-terminal domain is not available yet.
Unexpectedly, the SETX-Rrp45 association also depends on the sumoylation status of SETX. We discovered this after failing to detect interaction in vitro with recombinant proteins, which led us to consider the possible involvement of a posttranslational modification. Indeed, each of the AOA2 mutations tested severely decreased the sumoylated pattern of Nter-SETX in human cells, as detected in several assays, including western blot with an anti-SUMO antibody after immunoprecipitation (IP) of transiently expressed Nter-SETX. Furthermore, in the coIP of endogenous proteins, only sumoylated SETX IPed with Rrp45. Those data indicate that the interaction between SETX and Rrp45 was in fact mediated by SUMO. Additionally, we showed, using the Y2H assay, that the interaction required a previously characterized SUMO interacting motif (SIM) in Rrp45.Citation24
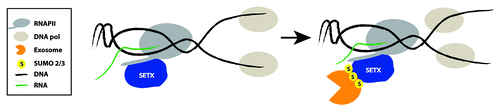
We then wondered what might be the meaning of this interaction and how could we make a connection between RNA surveillance deficiency and AOA2. A clue came from recent studies in yeast and in human cells implicating SETX in resolution of R loops that arise when transcription and replication machineries collide, and which showed the existence of SETX/Sen1 foci that increased after DNA damage induction.Citation8,Citation9 Inspired by these findings, we showed that, following stress replication induction in HeLa cells by aphidicolin (APH), which blocks replication fork progression and enhances collisions, SETX and Rrp45 co-localize in nuclear foci. Importantly, we also showed that the newly defined Rrp45 replication stress-induced foci were R loop-dependent since overexpression of RNase H1, which degrades the RNA moiety of RNA-DNA hybrids, eliminated these foci in APH-treated cells.Citation24 This data led us to the conclusion that SETX and the exosome might collaborate at sites of R-loop formation arising for example when the transcription and DNA replication machineries collide. During transcription, SETX, which has been shown to interact with RNAPII,Citation33 likely unwinds the RNA-DNA hybrid, and then, we propose, the exosome degrades the RNA moiety (). If the exosome is not recruited to the collision sites, the released RNA might itself have deleterious consequences to the cell, or perhaps rehybridize to the DNA, regenerating the R loop and leading to DSBs. Of course, many questions remain regarding this proposed mechanism, not to mention its relevance to AOA2, but perhaps not ALS4.
Figure 1. SETX, SUMO, and the exosome: Working together to fight transcription-related DNA damage. SETX is shown already associated with RNAPII, to resolve any R loops that may form naturally during transcription. Upon transcription and/or replication stress, created for example when RNAPII and DNA polymerase collide, SETX becomes sumoylated. Sumoylated SETX then interacts and/or recruits the exosome through its interaction with Rrp45. SETX resolves the R loop, perhaps inducing RNAPII release from DNA template, while the exosome then degrades the released RNA, to prevent possible reformation of the R loop and/or deleterious effects of the prematurely terminated RNA.
Sumoylation is a dynamic process that has been shown to control a variety of cellular processes. It regulates for example protein-protein interactions, protein stability, localization and transcriptional activity.Citation34-Citation36 Small ubiquitin-like modifiers (SUMO) are peptides of ≈11 kDa that are covalently conjugated to target proteins through the amino side chains of lysine residues. Four SUMO paralogues exist in mammals (SUMO1–4). While SUMO2 and SUMO3 are very similar, SUMO1 shares only 50% sequence identity with SUMO2/3,Citation37 and SUMO4 expression might be restricted to a few cell types, e.g., immune cells.Citation38 Conjugation of SUMO to a target protein is similar to conjugation of ubiquitin, requiring first activation of SUMO by an E1 activating enzyme. This is followed by transfer of SUMO to an E2 conjugating enzyme, Ubc9 (the only known E2), which in turn transfers SUMO to the target protein, facilitated by an E3 ligase. The target lysine is frequently found within the consensus sequence ΨKxE/D (Ψ represents hydrophobic amino acids).Citation39 Interestingly, SUMO has the ability to form poly-SUMO conjugates. In fact, SETX had already been detected as a sumoylated proteinCitation40,Citation41 and had been shown to be poly-sumoylated, by SUMO2, after heat shock.Citation41 Poly-sumoylated SETX shows an apparent MW of ≈600 KDa, which suggests that SETX actually carries about 25 SUMO peptides. Our Y2H screen also revealed interaction of SETX with two other components of the sumoylation machinery, Ubc9 and the E3 ligase PIAS1. Notably, the E65K AOA2 mutant showed the most complete loss of interaction with Rrp45, and sumoylation, and also lost interaction with Ubc9, implying that this mutation prevents sumoylation by disrupting Ubc9 binding. How the other two AOA2 mutations disrupt sumoylation remains to be determined.
Sumoylated SETX in normal cells appears to be present at very low levels as it is barely detectable by western blotting after IP and, as has already been shown with heat shock, is probably increased by stress induction. Indeed, it is well known that various cellular stresses increase sumoylation.Citation37 It is then very likely that the stress induced by APH treatment leads to a hyper-sumoylation of SETX, which in turn is necessary for its association with Rrp45. In the brain, managing various stresses is thought to be more important than in other cell types, and sumoylation is known to play crucial roles.Citation42,Citation43 It is perhaps then not surprising that loss of SETX sumoylation has a direct impact on AOA2 cells. In fact, SETX sumoylation appears to be an efficient and powerful factor in the rapid response to DNA damage.Citation44,Citation45 Indeed, proteins from the sumoylation machinery, including SUMO2/3, PIAS1 and Ubc9, accumulate at sites of DSBs, suggesting that SUMO conjugation takes place at DNA damage sites.Citation46,Citation47 Very interestingly, BLM, another helicase from the RecQ family involved in DSB repair, and which is defective in Bloom syndrome, is also sumoylated, and mutations that impaired its sumoylation led to greater DNA damage.Citation48 There is now more and more evidence that sumoylation is important for accumulation of DNA repair factors at sites of damage.Citation44,Citation49
Many proteins that have the potential to be sumoylated have already been shown to be involved in a variety of diseases. Neurodegenerative disorders seem to be particularly linked to sumoylation.Citation50-Citation52 SETX should now be added to the growing list of disease-associated proteins that are sumoylated. Sumoylation of many such proteins has been linked to their capacity to aggregate in the cell. For example, spinocerebellar ataxia type 1 (SCA1) is caused by an expansion of polyglutamine (polyQ) repeats in ataxin-1. Sumoylation of Ataxin-1 can be exacerbated by oxidative stress and lead to an increase in protein aggregation.Citation53,Citation54 However, Ataxin-1 sumoylation is decreased in a polyQ expansion mutant.Citation55 Some proteins involved in ALS are also known to aggregate in patient cells and animal models. For example, SOD1 (Superoxide Dismutase 1), which is mutated in about 20% of familial cases of ALS, has been shown to be sumoylated in mutant and WT proteins, in both cases possibly leading to an increase and stabilization of aggregates.Citation56 There is so far no evidence of SETX aggregates in AOA2 patient cells.
Neurological diseases have been linked for some time with defects in DNA damage repair pathways.Citation57-Citation59 Patients suffering from Ataxia telangiectasia (AT), another autosomal recessive ataxia, have mutations in ATM (AT mutated), leading to deregulation of many pathways involved in genomic instability including DSB repair.Citation60 AOA2 patient cells have also been shown to be sensitive to oxidative stress, leading to DSBs,Citation61,Citation62 and a number of studies have now established a role for Sen1/SETX in the DNA damage response, most likely by resolving R loops.Citation7-Citation10,Citation24 As already suggested by Yuce and West, SETX might then be considered to be a DNA damage response factor. While it is not known where the DNA damage occurs, SETX stress-induced foci have been suggested to localize at chromosome fragile sites (CFS) or repeated sequences.Citation9,Citation63,Citation64 CFS are large genomic regions that are particularly difficult to replicate and DSBs can be induced at such sites by APH treatment.Citation65 Replication fork stalling at CFS leads to chromosome breakage, accumulation of mutations and genomic aberrations creating genomic instability. Large genes located at some CFS require such a long time until transcription is terminated that the probability for the transcription and replication machineries to meet is high. Such collisions result in the formation and accumulation of R loops that are most likely responsible for chromosome breaks.Citation66 SETX ChIP-seq experiments after APH treatment should confirm whether SETX is indeed recruited to CFS.
If SETX can be classified as a DNA damage response protein, then our data imply that the exosome, or at least Rrp45, can also be considered to be a DNA damage factor. While some evidence of the RNA surveillance machinery being involved in the DNA damage response has been found in yeast,Citation67,Citation68 data in humans was lacking before our study. While knockdown of exosome subunits appears to increase DSBs in human cells (unpublished data), further experiments will be needed to confirm whether the exosome is indeed a DNA damage response factor.
SETX appears to have a specific and crucial role in preventing DSBs created by oxidative stress.Citation61 One of the main features of AOA2 is an atrophy of the cerebellum, and it is known that cerebellar cells are very vulnerable to oxidative stress (OS). Indeed cerebellar granule cells appear significantly more sensitive to OS than do cortical neurons.Citation69 This selective OS neuronal vulnerability could explain why SETX dysfunction seems to be deleterious exclusively in the brain and more prominent in the cerebellum of AOA2 patients. OS is involved in many pathogenic cellular processes and is responsible for a large fraction of DNA damage events.Citation70 In many cases, this might be linked to R-loop formation. Indeed, AOA2 cells have also been shown to be sensitive to camptothecin (CPT),Citation61 which can also generate OS.Citation71 CPT is a topoisomerase I (Top 1) inhibitor that blocks transcription and causes DSBs in an R loop-dependent manner.Citation72,Citation73 Top1 is believed to prevent collisions between the transcription and replication machineries, and thus also prevent formation of R loops.Citation74
An important question raised by our study is how replicative defects, such as transcription-replication fork collisions, can be relevant in a neurological disorder since neurons are mostly post-mitotic cells. We suggest two possibilities. First, AOA2 arises at adolescence when some neurogenesis is still ongoing and replication defects, especially in the cerebellum, might be deleterious enough to lead to disease. Consistent with this, brain cells are known to be highly vulnerable to neurotoxicity during adolescence.Citation75 Second, increasing evidence points to the possibility that neurological diseases are not solely the result of neuronal dysfunction. Glial cells, which “feed” and “support” neurons, appear to have considerable responsibilities in disease pathology,Citation52,Citation76,Citation77 and defects in the response to replicative stress in such cells might then be deleterious to neurons. It is also important to mention that R loops can be generated by various mechanisms,Citation6 and SETX might play a role in resolving them not only when transcription and replication meet but for example also at G-rich sequences such as termination sitesCitation5 or downstream of unmethylated CpG island promoters.Citation78
Our work has highlighted a connection between SETX, RNA surveillance and DNA damage that is disrupted in AOA2. But of course many questions remain. First, we tested only three AOA2 mutations and it is very possible, even likely, that all the reported mutations do not interfere with SETX sumoylation and/or interaction with Rrp45. It would be interesting to test other mutations, especially some outside the N-terminal domain of SETX. Second, SETX is a very large protein that probably performs many functions and it is not difficult to imagine that mutations affect different functions. This would explain the variability observed in patients’ symptoms with SETX mutations.Citation15,Citation18-Citation21 Are all these mutationally-sensitive functions linked to DNA repair? Or can other functions of SETX, e.g., in transcription termination, be relevant to AOA2, or ALS4? Finally, how does disruption of sumoylation and/or exosome interaction caused by the AOA2 mutations we studied affect specifically the brain during the first two decades of life? If SETX/exosome are involved in DNA damage repair and prevent genomic instability, why do AOA2 and ALS4 patients not display a heightened incidence of cancer? The use of iPS cells obtained from AOA2 patients and studies of additional mutations in SETX might shed light on these and other questions.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
This work was supported by the Muscular Dystrophy Association (MDA), the Columbia University Motor Neuron Center (MNC), and an EMBO fellowship to P.R., and NIH grant RO1 GM28983 to J.L.M.
References
- KimHD, ChoeJ, SeoYS. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry1999; 38:14697 - 710; http://dx.doi.org/10.1021/bi991470c; PMID: 10545196
- WineyM, CulbertsonMR. Mutations affecting the tRNA-splicing endonuclease activity of Saccharomyces cerevisiae. Genetics1988; 118:609 - 17; PMID: 3284787
- UrsicD, HimmelKL, GurleyKA, WebbF, CulbertsonMR. The yeast SEN1 gene is required for the processing of diverse RNA classes. Nucleic Acids Res1997; 25:4778 - 85; http://dx.doi.org/10.1093/nar/25.23.4778; PMID: 9365256
- SteinmetzEJ, WarrenCL, KuehnerJN, PanbehiB, AnsariAZ, BrowDA. Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol Cell2006; 24:735 - 46; http://dx.doi.org/10.1016/j.molcel.2006.10.023; PMID: 17157256
- Skourti-StathakiK, ProudfootNJ, GromakN. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell2011; 42:794 - 805; http://dx.doi.org/10.1016/j.molcel.2011.04.026; PMID: 21700224
- AguileraA, García-MuseT. R loops: from transcription byproducts to threats to genome stability. Mol Cell2012; 46:115 - 24; http://dx.doi.org/10.1016/j.molcel.2012.04.009; PMID: 22541554
- MischoHE, Gómez-GonzálezB, GrzechnikP, RondónAG, WeiW, SteinmetzL, AguileraA, ProudfootNJ. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol Cell2011; 41:21 - 32; http://dx.doi.org/10.1016/j.molcel.2010.12.007; PMID: 21211720
- AlzuA, BermejoR, BegnisM, LuccaC, PicciniD, CarotenutoW, SaponaroM, BrambatiA, CocitoA, FoianiM, et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell2012; 151:835 - 46; http://dx.doi.org/10.1016/j.cell.2012.09.041; PMID: 23141540
- YüceÖ, WestSC. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol2013; 33:406 - 17; http://dx.doi.org/10.1128/MCB.01195-12; PMID: 23149945
- BecherelOJ, YeoAJ, StellatiA, HengEY, LuffJ, SuraweeraAM, WoodsR, FlemingJ, CarrieD, McKinneyK, et al. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet2013; 9:e1003435; http://dx.doi.org/10.1371/journal.pgen.1003435; PMID: 23593030
- LiX, ManleyJL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell2005; 122:365 - 78; http://dx.doi.org/10.1016/j.cell.2005.06.008; PMID: 16096057
- MoreiraMC, KlurS, WatanabeM, NémethAH, Le BerI, MonizJC, TranchantC, AubourgP, TazirM, SchölsL, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet2004; 36:225 - 7; http://dx.doi.org/10.1038/ng1303; PMID: 14770181
- ChenYZ, BennettCL, HuynhHM, BlairIP, PulsI, IrobiJ, DierickI, AbelA, KennersonML, RabinBA, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet2004; 74:1128 - 35; http://dx.doi.org/10.1086/421054; PMID: 15106121
- AnheimM, MongaB, FleuryM, CharlesP, BarbotC, SalihM, DelaunoyJP, FritschM, ArningL, SynofzikM, et al. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain2009; 132:2688 - 98; http://dx.doi.org/10.1093/brain/awp211; PMID: 19696032
- HiranoM, QuinziiCM, MitsumotoH, HaysAP, RobertsJK, RichardP, RowlandLP. Senataxin mutations and amyotrophic lateral sclerosis. Amyotroph Lateral Scler2011; 12:223 - 7; http://dx.doi.org/10.3109/17482968.2010.545952; PMID: 21190393
- ZhaoZH, ChenWZ, WuZY, WangN, ZhaoGX, ChenWJ, MurongSX. A novel mutation in the senataxin gene identified in a Chinese patient with sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler2009; 10:118 - 22; http://dx.doi.org/10.1080/17482960802572673; PMID: 19058054
- SaigaT, TateishiT, ToriiT, KawamuraN, NagaraY, ShigetoH, HashiguchiA, TakashimaH, HondaH, OhyagiY, et al. Inflammatory radiculoneuropathy in an ALS4 patient with a novel SETX mutation. J Neurol Neurosurg Psychiatry2012; 83:763 - 4; http://dx.doi.org/10.1136/jnnp-2012-302281; PMID: 22577233
- SchölsL, ArningL, SchüleR, EpplenJT, TimmannD. “Pseudodominant inheritance” of ataxia with ocular apraxia type 2 (AOA2). J Neurol2008; 255:495 - 501; http://dx.doi.org/10.1007/s00415-008-0707-z; PMID: 18350359
- Rudnik-SchönebornS, ArningL, EpplenJT, ZerresK. SETX gene mutation in a family diagnosed autosomal dominant proximal spinal muscular atrophy. Neuromuscul Disord2012; 22:258 - 62; http://dx.doi.org/10.1016/j.nmd.2011.09.006; PMID: 22088787
- BassukAG, ChenYZ, BatishSD, NaganN, OpalP, ChancePF, BennettCL. In cis autosomal dominant mutation of Senataxin associated with tremor/ataxia syndrome. Neurogenetics2007; 8:45 - 9; http://dx.doi.org/10.1007/s10048-006-0067-8; PMID: 17096168
- VantaggiatoC, CantoniO, GuidarelliA, RomanielloR, CitterioA, ArrigoniF, DonedaC, CastelliM, AiroldiG, BresolinN, et al. Novel SETX variants in a patient with ataxia, neuropathy, and oculomotor apraxia are associated with normal sensitivity to oxidative DNA damaging agents. Brain Dev2013; http://dx.doi.org/10.1016/j.braindev.2013.10.003; PMID: 24183476
- DuquetteA, RoddierK, McNabb-BaltarJ, GosselinI, St-DenisA, DicaireMJ, LoiselL, LabudaD, MarchandL, MathieuJ, et al. Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann Neurol2005; 57:408 - 14; http://dx.doi.org/10.1002/ana.20408; PMID: 15732101
- UrsicD, ChinchillaK, FinkelJS, CulbertsonMR. Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res2004; 32:2441 - 52; http://dx.doi.org/10.1093/nar/gkh561; PMID: 15121901
- RichardP, FengS, ManleyJL. A SUMO-dependent interaction between Senataxin and the exosome, disrupted in the neurodegenerative disease AOA2, targets the exosome to sites of transcription-induced DNA damage. Genes Dev2013; 27:2227 - 32; http://dx.doi.org/10.1101/gad.224923.113; PMID: 24105744
- HouseleyJ, LaCavaJ, TollerveyD. RNA-quality control by the exosome. Nat Rev Mol Cell Biol2006; 7:529 - 39; http://dx.doi.org/10.1038/nrm1964; PMID: 16829983
- Lykke-AndersenS, TomeckiR, JensenTH, DziembowskiA. The eukaryotic RNA exosome: same scaffold but variable catalytic subunits. RNA Biol2011; 8:61 - 6; http://dx.doi.org/10.4161/rna.8.1.14237; PMID: 21289487
- AllmangC, MitchellP, PetfalskiE, TollerveyD. Degradation of ribosomal RNA precursors by the exosome. Nucleic Acids Res2000; 28:1684 - 91; http://dx.doi.org/10.1093/nar/28.8.1684; PMID: 10734186
- SchneiderC, KudlaG, WlotzkaW, TuckA, TollerveyD. Transcriptome-wide analysis of exosome targets. Mol Cell2012; 48:422 - 33; http://dx.doi.org/10.1016/j.molcel.2012.08.013; PMID: 23000172
- HillerenP, McCarthyT, RosbashM, ParkerR, JensenTH. Quality control of mRNA 3′-end processing is linked to the nuclear exosome. Nature2001; 413:538 - 42; http://dx.doi.org/10.1038/35097110; PMID: 11586364
- de AlmeidaSF, García-SacristánA, CustódioN, Carmo-FonsecaM. A link between nuclear RNA surveillance, the human exosome and RNA polymerase II transcriptional termination. Nucleic Acids Res2010; 38:8015 - 26; http://dx.doi.org/10.1093/nar/gkq703; PMID: 20699273
- MukherjeeD, GaoM, O’ConnorJP, RaijmakersR, PruijnG, LutzCS, WiluszJ. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J2002; 21:165 - 74; http://dx.doi.org/10.1093/emboj/21.1.165; PMID: 11782436
- van DijkEL, SchildersG, PruijnGJ. Human cell growth requires a functional cytoplasmic exosome, which is involved in various mRNA decay pathways. RNA2007; 13:1027 - 35; http://dx.doi.org/10.1261/rna.575107; PMID: 17545563
- SuraweeraA, LimY, WoodsR, BirrellGW, NasimT, BecherelOJ, LavinMF. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet2009; 18:3384 - 96; http://dx.doi.org/10.1093/hmg/ddp278; PMID: 19515850
- Geiss-FriedlanderR, MelchiorF. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol2007; 8:947 - 56; http://dx.doi.org/10.1038/nrm2293; PMID: 18000527
- SongJ, DurrinLK, WilkinsonTA, KrontirisTG, ChenY. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc Natl Acad Sci U S A2004; 101:14373 - 8; http://dx.doi.org/10.1073/pnas.0403498101; PMID: 15388847
- HayRT. SUMO: a history of modification. Mol Cell2005; 18:1 - 12; http://dx.doi.org/10.1016/j.molcel.2005.03.012; PMID: 15808504
- SaitohH, HincheyJ. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem2000; 275:6252 - 8; http://dx.doi.org/10.1074/jbc.275.9.6252; PMID: 10692421
- WangCY, SheJX. SUMO4 and its role in type 1 diabetes pathogenesis. Diabetes Metab Res Rev2008; 24:93 - 102; http://dx.doi.org/10.1002/dmrr.797; PMID: 17990297
- RodriguezMS, DargemontC, HayRT. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J Biol Chem2001; 276:12654 - 9; http://dx.doi.org/10.1074/jbc.M009476200; PMID: 11124955
- GolebiowskiF, MaticI, TathamMH, ColeC, YinY, NakamuraA, CoxJ, BartonGJ, MannM, HayRT. System-wide changes to SUMO modifications in response to heat shock. Sci Signal2009; 2:ra24; http://dx.doi.org/10.1126/scisignal.2000282; PMID: 19471022
- BrudererR, TathamMH, PlechanovovaA, MaticI, GargAK, HayRT. Purification and identification of endogenous polySUMO conjugates. EMBO Rep2011; 12:142 - 8; http://dx.doi.org/10.1038/embor.2010.206; PMID: 21252943
- WilkinsonKA, NakamuraY, HenleyJM. Targets and consequences of protein SUMOylation in neurons. Brain Res Rev2010; 64:195 - 212; http://dx.doi.org/10.1016/j.brainresrev.2010.04.002; PMID: 20382182
- GwizdekC, CasséF, MartinS. Protein sumoylation in brain development, neuronal morphology and spinogenesis. Neuromolecular Med2013; 15:677 - 91; http://dx.doi.org/10.1007/s12017-013-8252-z; PMID: 23907729
- ShimaH, SuzukiH, SunJ, KonoK, ShiL, KinomuraA, HorikoshiY, IkuraT, IkuraM, KanaarR, et al. Activation of the SUMO modification system is required for the accumulation of RAD51 at sites of DNA damage. J Cell Sci2013; 126:5284 - 92; http://dx.doi.org/10.1242/jcs.133744; PMID: 24046452
- JacksonSP, DurocherD. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell2013; 49:795 - 807; http://dx.doi.org/10.1016/j.molcel.2013.01.017; PMID: 23416108
- GalantyY, BelotserkovskayaR, CoatesJ, PoloS, MillerKM, JacksonSP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature2009; 462:935 - 9; http://dx.doi.org/10.1038/nature08657; PMID: 20016603
- MorrisJR, BoutellC, KepplerM, DenshamR, WeekesD, AlamshahA, ButlerL, GalantyY, PangonL, KiuchiT, et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature2009; 462:886 - 90; http://dx.doi.org/10.1038/nature08593; PMID: 20016594
- OuyangKJ, WooLL, ZhuJ, HuoD, MatunisMJ, EllisNA. SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol2009; 7:e1000252; http://dx.doi.org/10.1371/journal.pbio.1000252; PMID: 19956565
- PinderJB, AttwoodKM, DellaireG. Reading, writing, and repair: the role of ubiquitin and the ubiquitin-like proteins in DNA damage signaling and repair. Front Genet2013; 4:45; http://dx.doi.org/10.3389/fgene.2013.00045; PMID: 23554604
- SargeKD, Park-SargeOK. SUMO and its role in human diseases. Int Rev Cell Mol Biol2011; 288:167 - 83; http://dx.doi.org/10.1016/B978-0-12-386041-5.00004-2; PMID: 21482412
- DorvalV, FraserPE. SUMO on the road to neurodegeneration. Biochim Biophys Acta2007; 1773:694 - 706; http://dx.doi.org/10.1016/j.bbamcr.2007.03.017; PMID: 17475350
- ForanE, BogushA, GoffredoM, RoncagliaP, GustincichS, PasinelliP, TrottiD. Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia2011; 59:1719 - 31; http://dx.doi.org/10.1002/glia.21218; PMID: 21769946
- TerashimaT, KawaiH, FujitaniM, MaedaK, YasudaH. SUMO-1 co-localized with mutant atrophin-1 with expanded polyglutamines accelerates intranuclear aggregation and cell death. Neuroreport2002; 13:2359 - 64; http://dx.doi.org/10.1097/00001756-200212030-00038; PMID: 12488827
- RyuJ, ChoS, ParkBC, LeeH. Oxidative stress-enhanced SUMOylation and aggregation of ataxin-1: Implication of JNK pathway. Biochem Biophys Res Commun2010; 393:280 - 5; http://dx.doi.org/10.1016/j.bbrc.2010.01.122; PMID: 20132795
- RileyBE, ZoghbiHY, OrrHT. SUMOylation of the polyglutamine repeat protein, ataxin-1, is dependent on a functional nuclear localization signal. J Biol Chem2005; 280:21942 - 8; http://dx.doi.org/10.1074/jbc.M501677200; PMID: 15824120
- FeiE, JiaN, YanM, YingZ, SunQ, WangH, ZhangT, MaX, DingH, YaoX, et al. SUMO-1 modification increases human SOD1 stability and aggregation. Biochem Biophys Res Commun2006; 347:406 - 12; http://dx.doi.org/10.1016/j.bbrc.2006.06.092; PMID: 16828461
- JeppesenDK, BohrVA, StevnsnerT. DNA repair deficiency in neurodegeneration. Prog Neurobiol2011; 94:166 - 200; http://dx.doi.org/10.1016/j.pneurobio.2011.04.013; PMID: 21550379
- RassU, AhelI, WestSC. Defective DNA repair and neurodegenerative disease. Cell2007; 130:991 - 1004; http://dx.doi.org/10.1016/j.cell.2007.08.043; PMID: 17889645
- McKinnonPJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci2009; 10:100 - 12; http://dx.doi.org/10.1038/nrn2559; PMID: 19145234
- McKinnonPJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol2012; 7:303 - 21; http://dx.doi.org/10.1146/annurev-pathol-011811-132509; PMID: 22035194
- SuraweeraA, BecherelOJ, ChenP, RundleN, WoodsR, NakamuraJ, GateiM, CriscuoloC, FillaA, ChessaL, et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol2007; 177:969 - 79; http://dx.doi.org/10.1083/jcb.200701042; PMID: 17562789
- AiroldiG, GuidarelliA, CantoniO, PanzeriC, VantaggiatoC, BonatoS, Grazia D’AngeloM, FalconeS, De PalmaC, TonelliA, et al. Characterization of two novel SETX mutations in AOA2 patients reveals aspects of the pathophysiological role of senataxin. Neurogenetics2010; 11:91 - 100; http://dx.doi.org/10.1007/s10048-009-0206-0; PMID: 19593598
- FranchittoA. Genome instability at common fragile sites: searching for the cause of their instability. Biomed Res Int2013; 2013:730714; http://dx.doi.org/10.1155/2013/730714; PMID: 24083238
- DurkinSG, GloverTW. Chromosome fragile sites. Annu Rev Genet2007; 41:169 - 92; http://dx.doi.org/10.1146/annurev.genet.41.042007.165900; PMID: 17608616
- GloverTW, BergerC, CoyleJ, EchoB. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet1984; 67:136 - 42; http://dx.doi.org/10.1007/BF00272988; PMID: 6430783
- HelmrichA, BallarinoM, ToraL. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell2011; 44:966 - 77; http://dx.doi.org/10.1016/j.molcel.2011.10.013; PMID: 22195969
- LunaR, JimenoS, MarínM, HuertasP, García-RubioM, AguileraA. Interdependence between transcription and mRNP processing and export, and its impact on genetic stability. Mol Cell2005; 18:711 - 22; http://dx.doi.org/10.1016/j.molcel.2005.05.001; PMID: 15949445
- GavaldáS, GallardoM, LunaR, AguileraA. R-loop mediated transcription-associated recombination in trf4Δ mutants reveals new links between RNA surveillance and genome integrity. PLoS One2013; 8:e65541; http://dx.doi.org/10.1371/journal.pone.0065541; PMID: 23762389
- WangX, ZaidiA, PalR, GarrettAS, BracerasR, ChenXW, MichaelisML, MichaelisEK. Genomic and biochemical approaches in the discovery of mechanisms for selective neuronal vulnerability to oxidative stress. BMC Neurosci2009; 10:12; http://dx.doi.org/10.1186/1471-2202-10-12; PMID: 19228403
- CookeMS, EvansMD, DizdarogluM, LunecJ. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J2003; 17:1195 - 214; http://dx.doi.org/10.1096/fj.02-0752rev; PMID: 12832285
- PaganoG, ZatteraleA, DeganP, d’IschiaM, KellyFJ, PallardóFV, KodamaS. Multiple involvement of oxidative stress in Werner syndrome phenotype. Biogerontology2005; 6:233 - 43; http://dx.doi.org/10.1007/s10522-005-2624-1; PMID: 16333757
- SordetO, RedonCE, Guirouilh-BarbatJ, SmithS, SolierS, DouarreC, ContiC, NakamuraAJ, DasBB, NicolasE, et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep2009; 10:887 - 93; http://dx.doi.org/10.1038/embor.2009.97; PMID: 19557000
- MarinelloJ, ChillemiG, BuenoS, ManzoSG, CapranicoG. Antisense transcripts enhanced by camptothecin at divergent CpG-island promoters associated with bursts of topoisomerase I-DNA cleavage complex and R-loop formation. Nucleic Acids Res2013; 41:10110 - 23; http://dx.doi.org/10.1093/nar/gkt778; PMID: 23999093
- TuduriS, CrabbéL, ContiC, TourrièreH, Holtgreve-GrezH, JauchA, PantescoV, De VosJ, ThomasA, TheilletC, et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol2009; 11:1315 - 24; http://dx.doi.org/10.1038/ncb1984; PMID: 19838172
- SpearLP. Assessment of adolescent neurotoxicity: rationale and methodological considerations. Neurotoxicol Teratol2007; 29:1 - 9; http://dx.doi.org/10.1016/j.ntt.2006.11.006; PMID: 17222532
- ParkLC, ZhangH, GibsonGE. Co-culture with astrocytes or microglia protects metabolically impaired neurons. Mech Ageing Dev2001; 123:21 - 7; http://dx.doi.org/10.1016/S0047-6374(01)00336-0; PMID: 11640948
- GentlemanSM. Review: microglia in protein aggregation disorders: friend or foe?. Neuropathol Appl Neurobiol2013; 39:45 - 50; http://dx.doi.org/10.1111/nan.12017; PMID: 23339288
- GinnoPA, LottPL, ChristensenHC, KorfI, ChédinF. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell2012; 45:814 - 25; http://dx.doi.org/10.1016/j.molcel.2012.01.017; PMID: 22387027