Abstract
In the past ten years, long non-protein-coding RNAs (lncRNAs) have been shown to comprise a major part of the mammalian transcriptome and are predicted from their highly specific expression patterns, to play a role in regulating protein-coding gene expression in development and disease. Many lncRNAs particularly those lying in imprinted clusters, are predominantly unspliced “macro” transcripts that can also show a low level of splicing, and both the unspliced and spliced transcript have the potential to be functional. Three known imprinted macro lncRNAs have been shown to silence from three to ten genes in cis in imprinted gene clusters. We review here the potential for functional macro lncRNAs, defined here as “inefficiently-spliced lncRNAs” to play a wider cis-regulatory role in the mammalian genome. This potential has been underestimated by the inability of current RNA-seq technology to annotate unspliced macro lncRNAs.
Introduction
The goal of the Human Genome Project to understand the genome and improve treatment of major human diseases is made more difficult by the subsequent demonstration of the complexity of the transcriptome. This has shown that protein-coding transcripts comprise a minority of the transcriptome, despite the finding that abundant alternative splicingCitation1 and RNA-editingCitation2 increase their diversity. In contrast, non-protein-coding (nc) RNAs whose expression patterns indicate a regulatory role, contribute the majority of the transcriptome.Citation3-Citation6 Currently non-coding RNA nomenclature is inconsistent and ncRNAs are named according to size (e.g., small, short, micro, long) but may also be annotated by their interaction partners (piRNAs), cellular localization (snRNAs) or by their position and orientation relative to the nearest protein-coding gene (asRNAs). However, initiatives are underway to coordinate nomenclatureCitation7 and to combine the growing number of specialized databases to create a common international database “RNAcentral” for all ncRNA sequences.Citation8
How can non-coding RNAs function in gene regulation?
Non-coding RNAs have the potential, because of their highly tissue, developmental and cancer-specific expression to play a role in regulating the expression of protein-coding genes during these processes. Regulatory functions for ncRNAs can be broadly classified as occurring in cis or in trans. Trans-acting ncRNAs could act at many different levels to influence protein-coding gene activity either by regulating transcription itself, or, at a post-transcriptional level by regulating diverse processes including mRNA splicing, ribosomal function, mRNA translation or protein activity. In mammals three major classes of small gene-regulatory ncRNAs are known that mostly appear to regulate gene expression post-transcriptionally: micro (miRNAs ~22 nt) that appear to function only in somatic cells of the post-implantation embryo, endogenous small interfering (endo-siRNAs 21 nt) whose function appears to be restricted to oocytes and ES cells, and Piwi-interacting (piRNAs 26–31 nt) that function in spermatogenesis (reviewed in ref. Citation9) and may also induce epigenetic transcriptional silencing effects.Citation10 The recent “competing endogenous RNA” hypothesis proposes however, that all transcribed RNAs have the potential to titrate the cells miRNA population.Citation11,Citation12 Mammals also have numerous additional regulatory small nuclear ncRNAs that play general roles in gene expression such as: short nucleolar snoRNAs (60–300 nt) that guide rRNA/tRNA chemical modifications,Citation13 small nuclear ribonucleoprotein snrpnRNAs (~150 nt) involved in RNA splicing,Citation14 the 7Sk RNA (272 bp) that negatively regulates the PTEFβ RNA polymerase CTD-kinase,Citation15 and B2 RNA (178 nt) that negatively regulates RNA polymerase in heat-shocked mouse cells.Citation16 Recently, a new and abundant class of noncoding RNA has been distinguished that is generally referred to as lnc (long non-coding) RNAs and defined as being longer than 200nt. One of the most studied subclasses are lincRNAs (large intergenic non-coding) that are classified by their intergenic position. The HOTAIR ncRNA was the prototype lincRNA in humans and was shown to have a role in regulating repressive chromatin at the HOXD gene cluster and in modulating the cancer epigenome,Citation17,Citation18 however this function may not be conserved in mice.Citation19 Many thousands of lincRNAs have now been identified,Citation20-Citation25 that mostly appear to have trans-regulatory functions acting at the level of transcription,Citation26 making this the largest lncRNA subclass in the mouse and human genome.
In contrast to the large number of possible functions for trans-acting lncRNAs, cis-acting lncRNAs whose regulatory functions are restricted to genes on the same chromosome are likely to operate only at the level of transcription. However, this needs to be experimentally verified as trans-acting lncRNA may also affect genes on the same chromosome (see comment Citation26). To date, the known functional cis-acting lncRNAs are all in the (very) long size class. One example is the 19kb long Xist (X-inactive specific transcript) lncRNA that induces epigenetic silencing of one of the two X chromosomes in female mammalian cells.Citation27,Citation28 Other examples that silence small clusters of protein-coding genes on autosomes are the imprinted lncRNAs and the ANRIL lncRNA that silences the p15/INK4B tumor suppressor gene (see below). Imprinted lncRNAs have been identified at several imprinted gene clusters and are unusual as they show inefficient co-transcriptional splicing and polyadenylation and have been termed 'macro' lncRNAs to reflect that their main product is unspliced, in contrast to the fully-spliced Xist lncRNA and all known lincRNAs.Citation21,Citation29,Citation30
In this review, we focus on functional cis-acting macro lncRNAs and their role in disease and review three main areas: (1) imprinted cis-acting macro lncRNAs, their unusual transcript biology and how they differ from spliced lncRNAs, (2) non-imprinted cis-acting macro lncRNAs linked to disease and (3), methods available to test the functions of large data sets of newly annotated but still uncharacterized lncRNAs.
Macro lncRNAs Act as cis-silencers in Genomic Imprinting
Genomic imprinting is a form of epigenetic gene regulation whereby genes are repressed in a parent-of-origin specific manner. This process affects ~140 genes in mice, many of which form clusters of 2–12 genes that with some exceptions, are conserved in humans.Citation31-Citation34 In each of the six well-studied imprinted gene clusters, at least one gene is a macro lncRNA.Citation29 Expression of three imprinted macro lncRNAs: Airn (118 kb), Kcnq1ot1 (91 kb) and Nespas (> 14 kb), has been shown to be required for imprinted silencing of genes respectively in the Igf2r, Kcnq1 and Gnas clusters.Citation35-Citation37 The single imprint control element (ICE) in each of these clusters has been genetically defined by in vivo deletions, to harbor the respective macro lncRNA promoter. On the maternal chromosome, the ICE in these three clusters is modified by a DNA methylation imprint established in the oocyte, that silences the macro lncRNA promoter. On the paternal chromosome, the ICE is unmethylated allowing expression of the macro lncRNA, which represses flanking protein-coding genes to generate imprinted expression (). The methylated ICE on the maternal chromosome prevents macro lncRNA expression thereby allowing expression of the flanking protein-coding genes. This regulatory system is unique as the DNA methylation imprint on the ICE correlates with protein-coding gene expression, thus its role is to repress the expression of the cis-acting silencer, i.e., the macro lncRNA, on the maternal allele.
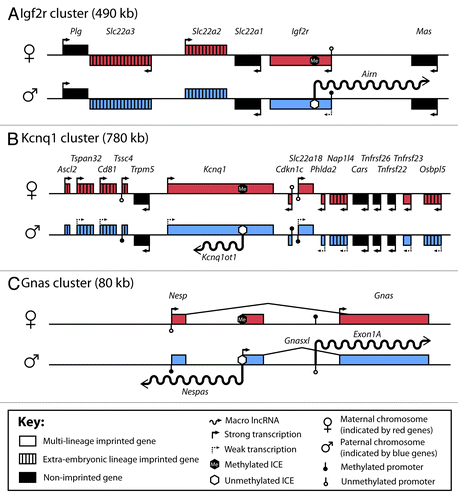
Figure 1. Macro lncRNAs silence imprinted genes in cis. (A-C) Genomic organization of the mouse Igf2r, Kcnq1 and Gnas imprinted gene clusters (not drawn to scale). Red boxes: imprinted expression from the maternal chromosome. Blue boxes: imprinted expression from the paternal chromosome. Genes that are bi-allelically expressed are depicted as black boxes. Open colored boxes: genes with multi-lineage imprinted expression. Colored boxed with black stripes: genes whose imprinted expression is restricted to the extra-embryonic lineage. Genes above the line are expressed from the (+) strand, genes below the line are expressed from the (-) strand. Arrows show transcriptional direction. Solid arrows indicate strong transcription whereas dashed arrows indicate weak transcription. Macro lncRNAs are shown as wavy lines. See key for further details. ICE, imprint control element
The Airn macro lncRNA silences the Igf2r imprinted gene cluster
The Igf2r imprinted gene cluster on mouse chromosome 17 (human chromosome 6) spans a 490kb region that includes six protein-coding genes and the 118kb long Airn macro lncRNA (). Airn is expressed from its unmethylated CpG island promoter contained in the defined ICE on the paternal chromosome and represses in cis three of the six protein-coding genes in the cluster, of which only Igf2r is overlapped in antisense orientation by Airn. The non-overlapped genes Slc22a2 and Slc22a3 in contrast to Igf2r, show restricted imprinted expression only in extra-embryonic tissues such as placenta and visceral yolk sac (VYS).Citation38,Citation39 Deletion of the Airn promoter resulted in a loss of imprinted expression in this cluster and Igf2r, Slc22a2 and Slc22a3 show bi-allelic expression.Citation40 To discriminate whether the promoter or Airn expression leads to repression of these protein-coding genes, a polyadenylation cassette was inserted 3kb downstream from the Airn transcript start site.Citation36 This strategy left the promoter and transcription initiation intact, but shortened the Airn RNA to 4% of its original length and this truncated lncRNA failed to silence Igf2r, Slc22a2 and Slc22a3. This demonstrated that macro lncRNA expression was necessary for silencing but did not discriminate if the lncRNA product or the act of its transcription played the key silencing role (see review Citation41). In humans, IGF2R shows polymorphic imprinted expression in some fetal tissues but biallelic expression in adult tissues.Citation42 The human AIRN promoter has been identified, however, a clear correlation between AIRN expression and IGF2R repression has not yet been established.Citation43
The Kcnq1ot1 macro lncRNA silences the Kcnq1 imprinted gene cluster
The Kcnq1 imprinted gene cluster on mouse chromosome 7 (human chromosome 11) spans a 780kb region that contains 15 protein-coding genes and the ~90 kb Kcnq1ot1 macro lncRNA (). On the paternal chromosome Kcnq1ot1 is expressed in antisense orientation to Kcnq1 but only overlaps internal introns and exons. The Kcnq1ot1 CpG island promoter also serves as the ICE of the cluster that is modified and repressed by a maternal-specific methylation imprint acquired during oocyte development. Kcnq1ot1 expression is necessary to silence 10 protein-coding genes in this cluster.Citation35 Four genes: Kcnq1, Cdkn1c, Phlda2 and Slc22a18 are ubiquitously silenced (i.e, in embryonic and extra-embryonic tissues) on the paternal chromosome from which Kcnq1ot1 is expressed. Six genes: Ascl2, Tspan32, Cd81, Tssc4, Nap1l4 and Osbpl5 only show imprinted expression in placental tissues.Citation42,Citation44 Recent studies however, challenge the imprinted status of Tssc4, Nap1l4 and Osbpl5, since their high expression in the maternal decidua invalidates their analysis in mouse placenta.Citation39,Citation45,Citation46
The Nespas macro lncRNA silences the Gnas imprinted gene cluster
The Gnas imprinted gene cluster on mouse chromosome 2 (human chromosome 20) spans a ~100 kb region that contains the two maternally expressed protein-coding Gnas isoforms Nesp and Gnas. The paternal allele expresses one protein-coding Gnas-xl isoform plus two paternally expressed macro lncRNAs Nespas and Exon1aCitation47 (). On the paternal chromosome, the Nespas macro lncRNA is expressed in antisense orientation and overlaps and represses the Nesp protein-coding isoform.Citation37 The maternal Nespas lncRNA promoter is modified and repressed by a maternal-specific methylation imprint acquired during oocyte development. Whether the other Gnas protein-coding isoforms in the cluster are also regulated by Nespas is not yet known.
Cis-silencing by imprinted macro lncRNAs
Despite the fact that the Airn, Kcnq1ot1 and Nespas imprinted macro lncRNAs are known to be necessary for repression of flanking protein-coding genes, the mechanism by which they act and how their influence is restricted to a small cluster of genes, is unknown. In particular, for lncRNAs such as Airn that produce a majority of unspliced and a minority of spliced products, it is unknown which variant controls its repressor function. One of the key questions not yet settled is whether the macro lncRNA product or the act of its transcription causes repression, or if both features are involved.Citation41 In the Igf2r and Kcnq1 clusters, the Airn and Kcnq1ot1 repressor macro lncRNAs overlap only one of the genes that they silence. In addition, silencing of most of the non-overlapped genes is restricted to extra-embryonic tissues (i.e., the placenta and membranes of the developing embryo).Citation38 This could indicate that different mechanisms act downstream of the macro lncRNA to repress overlapped and non-overlapped genes and that these mechanisms have different levels of activity in embryo vs. extra-embryonic tissues. In placenta, a role for the lncRNA product has been shown for Airn that accumulates at the distant Slc22a3 promoter and recruits the G9A/EHMT2 histone methyltransferase, which is necessary for Slc22a3 repression.Citation48 However, Igf2r repression is not dependent on G9A/EHMT2 in placenta indicating that at least two mechanisms act downstream of Airn in this tissue. Silencing models based on Airn transcription interference in which the activity of promoters or enhancers is reduced by overlapping macro lncRNA transcription have been proposed, but none have yet been demonstrated.Citation29,Citation41 Expression of the Kcnq1ot1 macro lncRNA correlates with accumulation of repressive histone H3K9me3 and H3K27me3 on the silenced flanking genes in placental but not in embryonic tissues.Citation49,Citation50 However despite the demonstration that the responsible G9A/EHMT2 and PRC2 histone methyltransferases physically interact with Kcnq1ot1,Citation51 mice lacking G9A/EHMT2 and PRC2 only show stochastic loss of imprinted expression of some genes in the placenta.Citation52,Citation53 A role for the Kcnq1ot1 macro lncRNA product was recently dismissed based on a siRNA-mediated knockdown experiment that did not change imprinted expression of these genes in undifferentiated embryonic stem cells.Citation54 However, this may not be relevant since RNAi activity is restricted to the cytoplasm and Kcnq1ot1 is nuclear-localized.Citation51,Citation55,Citation56 Thus the key questions of how macro lncRNAs silence flanking genes in cis and if they depend on more than one downstream mechanism, remains to be determined.
The Unusual Transcript Biology of Macro lncRNAs
When the first mammalian imprinted genes Igf2rCitation57 and Igf2Citation58 were discovered in 1991, repression by lncRNAs was not expected despite the simultaneous discovery of the Xist lncRNA responsible for X chromosome inactivation.Citation27,Citation28,Citation59 The respective lncRNAs in these two clusters (Airn and H19), although markedly different in transcript biology and function, served as model lncRNAs for many years,Citation60 see . We have used the term 'macro' ncRNA in several publications describing Airn, to reflect its unique biology that produces mainly unspliced transcripts.Citation29 This term is used to contrast inefficiently processed transcripts with fully-spliced lncRNAs such as H19 and Xist.Citation61 In the past ten years studies have identified many thousands of lncRNAs that are currently defined as 'longer than 200 nt'–a definition that relates to the standard method used to harvest RNA that collects transcripts greater than this size.Citation62 LncRNAs include large intergenic 'lincRNAs' that occupy an intergenic position as well as those with a sense or antisense overlap of a protein-coding gene.Citation6,Citation20 Many lincRNAs have been shown by knockdown of their spliced product, to play a trans-acting gene regulatory role (see review Citation26). New categories of spliced or unspliced lncRNAs with potential cis-regulatory functions have also been identified as divergent transcripts at promoters or enhancers.Citation63-Citation65 However it is not yet clear if these transcripts are a byproduct of open chromatin or if they play a functional cis-acting role in regulating transcription (see discussion in Citation66).
Figure 2. Macro lncRNAs are unusual transcripts that differ from fully-spliced lincRNAs. Left panel: Macro lncRNAs are inefficiently spliced lncRNAs. In contrast to lncRNAs, macro lncRNAs are ultra-long, lack sequence conservation and have a short half-life. They are also inefficiently spliced and imprecisely terminated at their 3′ end. Some macro lncRNAs such as Kcnq1ot1 are only found as unspliced transcripts, others such as Airn produce both unspliced and spliced variants. Only spliced Airn transcripts are exported to the cytoplasm. Macro lncRNAs could repress in two ways: through their lncRNA product or through a form of transcriptional interference particularly if they overlap a silenced gene. Right panel: LncRNAs are ncRNAs of more than 200 nt, which are currently defined only by their spliced product. On average they contain two to three exons and are approximately 1–3 kb in length. LncRNAs are generally considered to act in trans e.g., by binding and recruiting chromatin-modifying enzymes to regulate gene expression. In contrast to macro lncRNAs they are highly spliced, relatively stable and likely to be exported to the cytoplasm. Only transcriptional effects are displayed, for details of post-transcriptional effects see text: How can non-coding RNAs function in gene regulation?
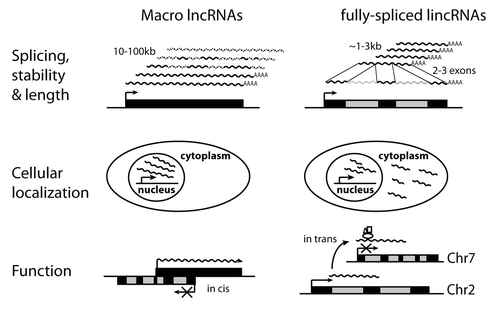
The majority of lncRNAs not associated with enhancers and promoters are only annotated as fully-spliced products estimated to have an average length of ~1 kb with ~2.9 exonsCitation67 or ~3.2 kb with 3.7 exons.Citation21 It is not yet known if these annotated lncRNA genes also produce a significant proportion of unspliced transcripts as seen for imprinted macro lncRNAs. However, strategies based on assembling overlapping cDNAs,Citation68 or analysis of RNA-seq dataCitation69 indicate that macro lncRNAs can be found outside of imprinted clusters. This is an important distinction when considering function, because if the bulk of the lncRNA is unspliced it is possible that the unspliced but not the spliced product is functional. Until this issue is further investigated we provisionally retain the use of 'macro' to describe the subset of lncRNAs that show the inefficient splicing behavior of Airn and Kcnq1ot1. The Airn and Kcnq1ot1 macro lncRNAs are also distinguished by a relatively short half-life, a high interspersed repeat content, and low sequence conservation.Citation51,Citation60,Citation70 One additional feature visible from genome-tiling-array and RNA-seq analysisCitation30,Citation60,Citation71 is that the Airn and Kcnq1ot1 macro lncRNAs show reduced RNA abundance toward the 3′ end of the gene, indicating imprecise termination. Together these features indicate that the macro lncRNA product may be less important than its transcription in regulating imprinted expression. If the Airn and Kcnq1ot1 macro lncRNA products are not important, the alternative is that they exert repressive effects by the act of their transcription. Models have been proposed whereby macro lncRNAs particularly those that overlap silenced genes, may induce a form of transcriptional interference either of the promoter or of essential enhancers.Citation41,Citation72,Citation73 These models do not exclude that the macro lncRNA product may also have a functional role for example, to repress non-overlapping genes.
An explanation of the unusual transcriptional biology of the Airn and Kcnq1ot1 macro lncRNA is lacking. One genetic element that may be relevant is the promoter as it was shown in fission yeast that promoters of some genes specify splicing.Citation74 However, replacement of the Airn promoter with the mouse Pgk1 promoter showed that the endogenous promoter does not control the low splicing capacity of the Airn lncRNA.Citation75 Both the Airn and Kcnq1ot1 macro lncRNAs are unusual in that their CpG island lies immediately downstream of the promoter and although they lack sequence similarities, they each contain a series of tandem direct repeats that have been suggested to guide epigenetic modifications such as the acquisition of the maternal-specific methylation imprint.Citation76 In support of this, the Airn tandem direct repeats have recently been shown to be necessary for the oocyte-specific DNA methylation imprint.Citation71 The Kcnq1ot1 tandem direct repeats have only been partially deleted as two separate overlapping deletions, and although they are necessary to block somatic methylation of the normally unmethylated paternal ICE, no other functional role has so far been identified.Citation35,Citation77
Different strategies are needed to detect macro lncRNAs and fully-spliced lncRNAs
The main differences between macro lincRNAs and fully-spliced lncRNAs are shown in Table 1. Today much attention is centered on RNA-seq strategies to identify lncRNAs and this highlights another difference: their ability to be detected by this technology. LncRNAs are reliably detected due to their spliced nature, while detection of continuous very long unspliced macro lncRNAs is challenging for current bioinformatic approaches. The approach of ab initio reconstruction of the transcriptome based on identifying transcription islands and connecting them by split-mapped reads, has allowed the detection of many thousands of novel spliced lncRNAs.Citation22,Citation25,Citation67 Bioinformatic pipelines have been developed including programs such as Scripture,Citation22 CufflinksCitation78 and the split-mapper TopHat,Citation79 which greatly increased the numbers and reliability of detected lncRNAs. The spliced nature of lncRNAs thus increases confidence in identifying novel transcripts but a bioinformatic pipeline to detect macro lncRNAs in RNA-seq data are lacking. Traditionally, whole-transcriptome analysis to investigate the expression of mRNAs is based on polyA-enrichedCitation25,Citation67 or polyA-amplifiedCitation24 RNA as input. However, genome-tiling-array data sets show that more than 40% of transcripts are non-polyadenylatedCitation80 and although imprinted macro lncRNAs are polyadenylated, their extreme length does not allow capture by polyA-enrichment due to RNA fragmentation before the polyA-selection step. Therefore, an effective RNA-seq strategy to discover novel macro lncRNAs should include an efficient rRNA removal step to allow an unbiased view of the complete non-ribosomal transcriptome.Citation30 In addition, strand-specific RNA-seq is necessary to identify overlapping lncRNAs (e.g., Airn) and those lying inside annotated genes (e.g., Kcnq1ot1). Some studies have shown that macro lncRNAs such as Airn and Kcnq1ot1 are better visible in RNA-seq data sets produced by a pipeline that replaces polyA+ selection with rRNA depletion.Citation30,Citation69 One unsolved problem is how to determine if ultra-long macro lncRNAs are one continuous transcript or composed of transcripts from clusters of smaller genes. For the Airn and Kcnq1ot1 macro lncRNAs continuous transcription was demonstrated by laborious experiments that demonstrated their repressor function,Citation35,Citation36 since insertion of a polyA signal downstream of the promoter removed all downstream transcripts. However, for the large number of novel lncRNAs now being identified, a bioinformatic approach needs to be developed.
Cis-silencing Macro lncRNAs in Human Disease
To date, three cases are known in which the expression of an aberrant RNA or a macro lncRNA is causative for transcriptional silencing of an overlapped gene leading to a disease phenotype in humans. Additionally, several human imprinting disorders such as the Beckwith-Wiedemann syndrome,Citation81 Prader-Willi syndrome,Citation82 Angelman syndromeCitation82 and Russell-Silver syndromeCitation83 are known in which deregulation of an imprinted gene or gene cluster causes severe disease phenotypes, but as involvement of imprinted lncRNAs has not been directly demonstrated, they will not be discussed further here.
A LUC7L-HBA2 fusion transcript silences HBA2 in cis
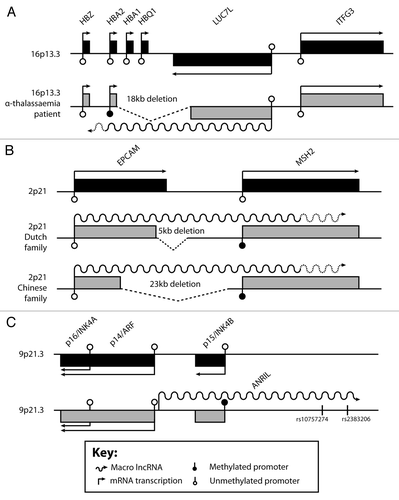
α-Thalassemia is an autosomal recessive blood disease arising from reduced functional α-globin production from the clustered HBA1 and HBA2 genes on chromosome 16. This leads to anemia by unbalancing the mix of α, β and δ globin chains that produce hemoglobin. The promoters of all the genes in the α-globin cluster () are associated with unmethylated CpG islands irrespective of expression.Citation84 A thalassemia patient was described to carry a unique 18kb deletion that includes HBA1, HBQ1 and the 3′ end of LUC7L.Citation85 Although the HBA2 gene remained structurally intact and the deletion did not remove any known cis-activating sequences, HBA2 was silent and its CpG island was densely methylated (). A follow-up study was able to explain this finding by identifying the expression of an aberrant RNA resulting from the deletion, which creates a gene fusion of the widely expressed upstream LUC7L gene with HBA2.Citation86 Transcription of the 3′ truncated LUC7L from the antisense strand overlaps the HBA2 gene on the sense strand in antisense orientation (). The fusion locus was also analyzed in a transgenic mouse model and in differentiating embryonic stem cells that reproduced the correlation between cis-expression of the aberrant antisense transcript, silencing HBA2 and de novo methylation of its CpG island. This fusion antisense transcript shares some but not all features of macro lncRNAs as described here. It is more than 40kb long, originates from the LUC7L promoter but due to the fact that the last 3 exons and transcription termination site are deleted, transcription extends at least into the HBA2 CpG island.Citation86 The majority of fusion transcripts consist of correctly spliced and protein-coding LUC7L mRNA.Citation86 In contrast, the 3′ part antisense to HBA2 may be more like a macro lncRNA, as it acts in cis (as the wild type HBA2 allele on the other parental chromosome is unaffected), and is most probably not spliced and not likely to be protein-coding.
Figure 3. Aberrant and macro lncRNAs associated with human disease. (A-C) Genomic organization of three loci expressing disease-associated aberrant or macro lncRNAs (not drawn to scale). Each locus is depicted in the wild type (black boxes) and at least one disease situation (gray boxes). Genes above the line are expressed from the (-) strand, genes below the line from the (+) strand. Arrows show transcriptional direction. Aberrant or macro lncRNAs are shown as wavy lines, unmapped 5′ ends are indicated by dashed wavy lines. In (C), two disease conferring SNPs are shown. See key for further details.
EPCAM-MSH2 fusion transcripts silence MSH2 in cis
A second example of transcription-mediated promoter methylation and gene silencing was found in patient families suffering from Lynch syndrome. This genetic condition confers high susceptibility to mainly colorectal cancer due to inactivation of mismatch repair genes: MLH1, MSH2, MSH6 or PMS2. Affected Dutch and Chinese families were shown to carry a heterozygous germline deletion of the last two or four exons of EPCAM and as a consequence transcription of EPCAM overlaps the neighboring MSH2 mismatch repair gene in sense orientation ().Citation87 The MSH2 promoter is silent and methylated in cis, but only in EPCAM expressing tissues that express the aberrant fusion transcript. Further studies confirmed the 3′ end EPCAM deletions as a recurrent cause of Lynch syndrome.Citation88,Citation89 Similar to the LUC7L-HBA2 fusion transcript, the EPCAM-MSH2 transcripts are likely to consist mainly of co-transcriptionally spliced EPCAM mRNA and 30kb or 3kb of RNA transcribed from the intergenic region, depending on the size of the deletion (). This gene silencing by transcriptional read-through in sense direction is an additional mechanism to the above-mentioned silencing of HBA2 by antisense read-through. How exactly transcriptional read-through leads to silencing of the overlapped promoter and a gain of DNA methylation, is unknown in both cases. Taken together, these examples indicate that aberrant coding as well as macro lncRNAs are implicated in epigenetic cis-silencing of overlapped genes.
ANRIL regulates a cluster of tumor suppressor genes
Strong support that macro lncRNAs are also involved in epigenetic silencing of tumor suppressor genes comes from a number of studies showing that the ANRIL macro lncRNA regulates the expression of the p16INK4A/p14ARF/p15INK4B tumor suppressor gene cluster in physiology and disease. Initially, ANRIL was discovered as a spliced 3.8kb lncRNA spanning a genomic region of 126kb that was expressed from a bidirectional promoter shared with p14ARF ().Citation90 This study showed coordinated transcriptional regulation of ANRIL and the p16INK4A/p14ARF/p15INK4B tumor suppressor genes, based on correlation analysis of expression levels in 22 normal tissues and 12 breast tumor tissues. Later the p15AS RNA was described as a 35kb unspliced nuclear lncRNA (the whole sequence of which is contained in the defined ANRIL locus) that inversely correlates with p15INK4B expression in acute myeloid and lymphoblastic leukemia.Citation91 Two acute myeloid cell lines with highly-expressed p15AS and lowly-expressed p15INK4B exhibited DNA hypermethylation and heterochromatin formation at the p15INK4B CpG island promoter. However, induction of p15AS expression using an episomal report system shows that heterochromatin formation in cis rather than DNA methylation correlates with gene silencing.Citation91 More recently, ANRIL has been shown to directly interact with the Polycomb repressive complex (PRC1) and thereby target repressive histone modification to all three tumor suppressor genes in cis, resulting in their epigenetic silencing.Citation92 Sequence-specific structural features of ANRIL were suggested to bind PRC1, but whether they arise from spliced or unspliced ANRIL is unknown. A role in disease was suggested as upregulation of ANRIL and PRC1 components in prostate cancer tissues correlates with significant downregulation of p16INK4A. ANRIL has also been shown to bind to PRC2 in vivo and to facilitate the recruitment of H3K27me3Citation93 to silence p15INK4B, but not p16INK4A or p14ARF. Interestingly, recent genome-wide association studies identified the ANRIL gene as a genetic susceptibility locus associated with coronary artery disease, type II diabetes,Citation94 atherosclerosis,Citation95 and multiple cancers such as breast cancer,Citation96 basal cell carcinoma,Citation97 and glioma.Citation98 How exactly ANRIL associates with these quite varying disease phenotypes is not yet clear but may involve anti-proliferative mechanisms such as senescence and apoptosis that are regulated by telomere shortening and the activities of p53 and the p16INK4A/p14ARF/p15INK4B tumor suppressor genes.Citation99 These cancer-protective pathways may also interfere with the replicative function of stem cell as they age.Citation99 The resulting altered regenerative capacity could explain some ANRIL-mediated age-associated diseases such as type II diabetes, atherosclerosis and cancer.Citation100 More than eight ANRIL splice variants are knownCitation101 and it was proposed that SNPs in the ANRIL gene body could regulate alternative splicing of ANRIL and thus the expression of p16INK4A and p15INK4B in diseases that rely on a certain replication potential.Citation99 The SNPs linked to coronary artery disease are located in a 58kb interval on human chromosome 9p21 () and the deletion of the orthologous 70kb region on mouse chromosome 7 showed that expression levels of p16INK4A and p15INK4B are markedly reduced in cis.Citation102 Despite the existence of at least eight non-coding splice variants,Citation101 the functions of unspliced ANRIL in regulating p15INK4B in cis cannot be dismissed.Citation91
Assigning Functions to Large lncRNA Data Sets
A major challenge for the field is that for most newly annotated lncRNAs only a bioinformatic classification is available, and little is known with respect to their RNA biology and cellular distribution and function. Since lncRNAs are also generally lowly expressed relative to protein-coding genes,Citation21,Citation30 it is also not known if some could arise from 'opportunistic' non-specific RNA polymerase activity that reflects an open chromatin state (see discussion in Citation66). Thus, it is currently difficult to devise the correct nomenclature as well as to model possible lncRNA functions. Some features are relatively simple to investigate, for example to test where lncRNAs are sub-localized in the cell, if they are efficiently spliced, if they have a long or short half-life, all of which would allow a more accurate approach to study their function. How to determine the functionality of lncRNAs is not trivial. It could be assumed that all expressed lncRNAs are potentially functional, thus the current lncRNA data sets could be considered to contain RNA genes selected for expression.Citation17,Citation18,Citation20-Citation25 This contrasts with the discovery of imprinted macro lncRNAs from the last decade where despite a negative correlation between expression of imprinted protein-coding genes and the macro lncRNA, functionality was not accepted until demonstrated in mice carrying deleted lncRNA alleles.Citation35-Citation37 This approach is unfeasible for large lncRNA data sets. One possibility for large data sets that is starting to be applied is to test a series of more easily implemented features and use this to predict a possibility of function. This would include the features discussed above i.e., splicing efficiency, length, cellular sub-localization, short or long half-life. Publically available genome-wide chromatin-immuno-precipitation data sets (http://genome.ucsc.edu/ENCODE/) could be mined to predict promoters. In addition, tissue, developmental and cancer-specific expression status can be characterized, and if a sufficient number of different tissues are analyzed it would also be possible to characterize lncRNA expression relative to protein-coding genes to predict possible cis or trans regulatory effects.Citation67 The cumulative score for all these features would be used to generate a 'functionality index' for the lncRNA. For imprinted macro lncRNAs, the functionality index could also determine differential-methylation status of the promoter and SNP analysis of RNA-seq data could determine parental allelic expression. This approach will identify families of lncRNAs sharing standardized parameters and subsequently allows predictions of distinct functions for a particular class of lncRNAs. An obstacle is clearly that in-depth knowledge of the investigated transcripts is needed to define suitable parameters. Recently, a study determined a catalog of > 30 properties including sequence, structural, expression and orthology features, for more than 8000 human lincRNAs and revealed specific subclasses. This study also demonstrated that lincRNAs show remarkable tissue-specific expression compared with coding genes, and are typically co-expressed with neighboring genes to the same extent as seen for pairs of neighboring protein-coding genes.Citation67 These large-scale bioinformatic studies are valuable to predict functions for lncRNA classes but detailed studies on individual lncRNAs are still important to understand mechanisms of action.
How to directly experimentally test for lncRNA function?
The chosen experimental strategy would involve an approach to deplete the lncRNA and assess the effects on gene expression and on cell biology. However, the chosen strategy would clearly depend on knowing some degree of lncRNA biology. For example if the chosen lncRNA is nuclear localized, RNAi cannot be deployed, as its activity is restricted to the cytoplasm.Citation55,Citation56 However, the nuclear localized Xist lncRNAs can be depleted by siRNA micro-injected into the zygote nucleus, but RNAi activity against a nuclear localized lncRNA is thought to be limited to the brief window during nuclear membrane breakdown in the cell cycle.Citation103 Alternative depletion methods based on antisense DNA oligos and endogenous nuclear RNaseH activity have been shown to deplete nuclear lncRNAs.Citation56,Citation104 If the chosen lncRNA is predominantly unspliced and is predicted to be cis-acting, there is a possibility that its transcription and not its lncRNA product is functional as suggested for the Airn macro lncRNA.Citation41 As a consequence lncRNA depletion that acts post-transcriptionally will not reveal a function, and these lncRNAs would require removal by homologous recombination in embryonic stem cells as described above for imprinted macro lncRNAs, or by zinc finger nucleases that can be used in any cell type.Citation105 Many studies have successfully used RNAi to knock-down spliced lncRNAs,Citation17,Citation18,Citation21,Citation24,Citation25,Citation106 however it was not known if these were nuclear localized or exported to the cytoplasm. For example, a recent large-scale RNAi knockdown study which showed that 137 out of 147 lincRNAs had a significant effect on the expression of 100–200 genes in mouse embryonic stem cells, did not determine their nuclear or cytoplasmic localization.Citation21 The method of choice to investigate lncRNA function could depend on knowledge of which class and which mechanism of action would be expected.
Conclusions
The field of lncRNAs is rapidly expanding and will be further accelerated as the depth of RNA-seq transcriptome studies increases.Citation107 However, we argue here that omitting the analysis of unspliced macro lncRNAs leads to a gap in our knowledge about this potential new layer of cis-regulatory information in the mammalian genome. The remarkable tissue-specificity of lncRNAsCitation67 strongly argues that they represent a functional layer of the transcriptome, rather than transcriptional noise (see discussion in Citation66). The recent emphasis to identify non-annotated transcripts will certainly be switched to focus on functional aspects of these transcripts. The fact that many lncRNAs have been found to be highly expressed in cancer, has also led to them being termed 'cancer-specific' lncRNAs.Citation25,Citation106 However, it would be unfavorable for an organism to encode a gene whose expression leads to disease. Therefore, such lncRNAs are more likely to be involved at a time point during development of the organism or differentiation of the respective tissue and only become deregulated in cancer. Therefore, dual studies to determine the physiological function and the disease function of non-coding transcripts will be essential in the future.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Financial Disclosure
PG is a recipient of a DOC Fellowship of the Austrian Academy of Sciences; the laboratory is supported by GEN-AU III Epigenetic Control Of Cell Identity (GZ200.141/1-VI/12009), and by the Austrian Science Fund SFB RNA Regulation Of The Transcriptome (F4302-B09).
Acknowledgments
We thank Aleksandra Kornienko and Florian Pauler for comments on the manuscript.
Related Research Data
References
- Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008; 321:956 - 60; http://dx.doi.org/10.1126/science.1160342; PMID: 18599741
- Bahn JH, Lee JH, Li G, Greer C, Peng G, Xiao X. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res 2012; 22:142 - 50; http://dx.doi.org/10.1101/gr.124107.111; PMID: 21960545
- Pauli A, Rinn JL, Schier AF. Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet 2011; 12:136 - 49; http://dx.doi.org/10.1038/nrg2904; PMID: 21245830
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet 2009; 10:155 - 9; http://dx.doi.org/10.1038/nrg2521; PMID: 19188922
- Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev 2009; 23:1494 - 504; http://dx.doi.org/10.1101/gad.1800909; PMID: 19571179
- Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, et al. The reality of pervasive transcription. PLoS Biol 2011; 9:e1000625; discussion e1001102 PMID: 21765801
- Wright MW, Bruford EA. Naming ‘junk’: human non-protein coding RNA (ncRNA) gene nomenclature. Hum Genomics 2011; 5:90 - 8; PMID: 21296742
- Bateman A, Agrawal S, Birney E, Bruford EA, Bujnicki JM, Cochrane G, et al. RNAcentral: A vision for an international database of RNA sequences. RNA 2011; 17:1941 - 6; http://dx.doi.org/10.1261/rna.2750811; PMID: 21940779
- Suh N, Blelloch R. Small RNAs in early mammalian development: from gametes to gastrulation. Development 2011; 138:1653 - 61; http://dx.doi.org/10.1242/dev.056234; PMID: 21486922
- Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, Hannon GJ. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science 2008; 322:1387 - 92; http://dx.doi.org/10.1126/science.1165171; PMID: 19039138
- Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?. Cell 2011; 146:353 - 8; http://dx.doi.org/10.1016/j.cell.2011.07.014; PMID: 21802130
- Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011; 147:358 - 69; http://dx.doi.org/10.1016/j.cell.2011.09.028; PMID: 22000014
- Bratkovič T, Rogelj B. Biology and applications of small nucleolar RNAs. Cell Mol Life Sci 2011; 68:3843 - 51; http://dx.doi.org/10.1007/s00018-011-0762-y; PMID: 21748470
- Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol 2011; 3; http://dx.doi.org/10.1101/cshperspect.a003707; PMID: 21441581
- Prasanth KV, Camiolo M, Chan G, Tripathi V, Denis L, Nakamura T, et al. Nuclear organization and dynamics of 7SK RNA in regulating gene expression. Mol Biol Cell 2010; 21:4184 - 96; http://dx.doi.org/10.1091/mbc.E10-02-0105; PMID: 20881057
- Goodrich JA, Kugel JF. Dampening DNA binding: a common mechanism of transcriptional repression for both ncRNAs and protein domains. RNA Biol 2010; 7:305 - 9; http://dx.doi.org/10.4161/rna.7.3.11910; PMID: 20436282
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010; 464:1071 - 6; http://dx.doi.org/10.1038/nature08975; PMID: 20393566
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007; 129:1311 - 23; http://dx.doi.org/10.1016/j.cell.2007.05.022; PMID: 17604720
- Schorderet P, Duboule D. Structural and functional differences in the long non-coding RNA hotair in mouse and human. PLoS Genet 2011; 7:e1002071; http://dx.doi.org/10.1371/journal.pgen.1002071; PMID: 21637793
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009; 458:223 - 7; http://dx.doi.org/10.1038/nature07672; PMID: 19182780
- Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011; 477:295 - 300; http://dx.doi.org/10.1038/nature10398; PMID: 21874018
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 2010; 28:503 - 10; http://dx.doi.org/10.1038/nbt.1633; PMID: 20436462
- Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 2010; 42:1113 - 7; http://dx.doi.org/10.1038/ng.710; PMID: 21057500
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA 2009; 106:11667 - 72; http://dx.doi.org/10.1073/pnas.0904715106; PMID: 19571010
- Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 2011; 29:742 - 9; http://dx.doi.org/10.1038/nbt.1914; PMID: 21804560
- Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature 2012; 482:339 - 46; http://dx.doi.org/10.1038/nature10887; PMID: 22337053
- Lee JT. Gracefully ageing at 50, X-chromosome inactivation becomes a paradigm for RNA and chromatin control. Nat Rev Mol Cell Biol 2011; 12:815 - 26; http://dx.doi.org/10.1038/nrm3231; PMID: 22108600
- Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet 2011; 12:542 - 53; http://dx.doi.org/10.1038/nrg3035; PMID: 21765457
- Koerner MV, Pauler FM, Huang R, Barlow DP. The function of non-coding RNAs in genomic imprinting. Development 2009; 136:1771 - 83; http://dx.doi.org/10.1242/dev.030403; PMID: 19429783
- Huang R, Jaritz M, Guenzl P, Vlatkovic I, Sommer A, Tamir IM, et al. An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS ONE 2011; 6:e27288; http://dx.doi.org/10.1371/journal.pone.0027288; PMID: 22102886
- Williamson CM, Blake A, Thomas S, Beechey CV, Hancock J, Cattanach BM, et al. MRC Harwell, Oxfordshire. World Wide Web Site - Mouse Imprinting Data and References. 2012.
- Barlow DP. Genomic imprinting: a mammalian epigenetic discovery model. Annu Rev Genet 2011; 45:379 - 403; http://dx.doi.org/10.1146/annurev-genet-110410-132459; PMID: 21942369
- Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet 2011; 12:565 - 75; http://dx.doi.org/10.1038/nrg3032; PMID: 21765458
- Abramowitz LK, Bartolomei MS. Genomic imprinting: recognition and marking of imprinted loci. Curr Opin Genet Dev 2011; http://dx.doi.org/10.1016/j.gde.2011.12.001; PMID: 22195775
- Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev 2006; 20:1268 - 82; http://dx.doi.org/10.1101/gad.1416906; PMID: 16702402
- Sleutels F, Zwart R, Barlow DP. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002; 415:810 - 3; http://dx.doi.org/10.1038/415810a; PMID: 11845212
- Williamson CM, Ball ST, Dawson C, Mehta S, Beechey CV, Fray M, et al. Uncoupling antisense-mediated silencing and DNA methylation in the imprinted Gnas cluster. PLoS Genet 2011; 7:e1001347; http://dx.doi.org/10.1371/journal.pgen.1001347; PMID: 21455290
- Hudson QJ, Kulinski TM, Huetter SP, Barlow DP. Genomic imprinting mechanisms in embryonic and extraembryonic mouse tissues. Heredity (Edinb) 2010; 105:45 - 56; http://dx.doi.org/10.1038/hdy.2010.23; PMID: 20234385
- Hudson QJ, Seidl CI, Kulinski TM, Huang R, Warczok KE, Bittner R, et al. Extra-embryonic-specific imprinted expression is restricted to defined lineages in the post-implantation embryo. Dev Biol 2011; 353:420 - 31; http://dx.doi.org/10.1016/j.ydbio.2011.02.017; PMID: 21354127
- Wutz A, Smrzka OW, Schweifer N, Schellander K, Wagner EF, Barlow DP. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature 1997; 389:745 - 9; http://dx.doi.org/10.1038/39631; PMID: 9338788
- Pauler FM, Koerner MV, Barlow DP. Silencing by imprinted noncoding RNAs: is transcription the answer?. Trends Genet 2007; 23:284 - 92; http://dx.doi.org/10.1016/j.tig.2007.03.018; PMID: 17445943
- Monk D, Arnaud P, Apostolidou S, Hills FA, Kelsey G, Stanier P, et al. Limited evolutionary conservation of imprinting in the human placenta. Proc Natl Acad Sci USA 2006; 103:6623 - 8; http://dx.doi.org/10.1073/pnas.0511031103; PMID: 16614068
- Yotova IY, Vlatkovic IM, Pauler FM, Warczok KE, Ambros PF, Oshimura M, et al. Identification of the human homolog of the imprinted mouse Air non-coding RNA. Genomics 2008; 92:464 - 73; http://dx.doi.org/10.1016/j.ygeno.2008.08.004; PMID: 18789384
- Engemann S, Strödicke M, Paulsen M, Franck O, Reinhardt R, Lane N, et al. Sequence and functional comparison in the Beckwith-Wiedemann region: implications for a novel imprinting centre and extended imprinting. Hum Mol Genet 2000; 9:2691 - 706; http://dx.doi.org/10.1093/hmg/9.18.2691; PMID: 11063728
- Okae H, Hiura H, Nishida Y, Funayama R, Tanaka S, Chiba H, et al. Re-investigation and RNA sequencing-based identification of genes with placenta-specific imprinted expression. Hum Mol Genet 2012; 21:548 - 58; http://dx.doi.org/10.1093/hmg/ddr488; PMID: 22025075
- Proudhon C, Bourc’his D. Identification and resolution of artifacts in the interpretation of imprinted gene expression. Brief Funct Genomics 2010; 9:374 - 84; http://dx.doi.org/10.1093/bfgp/elq020; PMID: 20829207
- Peters J, Williamson CM. Control of imprinting at the Gnas cluster. Epigenetics 2007; 2:207 - 13; http://dx.doi.org/10.4161/epi.2.4.5380; PMID: 18094621
- Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008; 322:1717 - 20; http://dx.doi.org/10.1126/science.1163802; PMID: 18988810
- Terranova R, Yokobayashi S, Stadler MB, Otte AP, van Lohuizen M, Orkin SH, et al. Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev Cell 2008; 15:668 - 79; http://dx.doi.org/10.1016/j.devcel.2008.08.015; PMID: 18848501
- Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, et al. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 2004; 36:1296 - 300; http://dx.doi.org/10.1038/ng1467; PMID: 15516932
- Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 2008; 32:232 - 46; http://dx.doi.org/10.1016/j.molcel.2008.08.022; PMID: 18951091
- Wagschal A, Sutherland HG, Woodfine K, Henckel A, Chebli K, Schulz R, et al. G9a histone methyltransferase contributes to imprinting in the mouse placenta. Mol Cell Biol 2008; 28:1104 - 13; http://dx.doi.org/10.1128/MCB.01111-07; PMID: 18039842
- Mager J, Montgomery ND, de Villena FP, Magnuson T. Genome imprinting regulated by the mouse Polycomb group protein Eed. Nat Genet 2003; 33:502 - 7; http://dx.doi.org/10.1038/ng1125; PMID: 12627233
- Golding MC, Magri LS, Zhang L, Lalone SA, Higgins MJ, Mann MR. Depletion of Kcnq1ot1 non-coding RNA does not affect imprinting maintenance in stem cells. Development 2011; 138:3667 - 78; http://dx.doi.org/10.1242/dev.057778; PMID: 21775415
- Zeng Y, Cullen BR. RNA interference in human cells is restricted to the cytoplasm. RNA 2002; 8:855 - 60; http://dx.doi.org/10.1017/S1355838202020071; PMID: 12166640
- Ideue T, Hino K, Kitao S, Yokoi T, Hirose T. Efficient oligonucleotide-mediated degradation of nuclear noncoding RNAs in mammalian cultured cells. RNA 2009; 15:1578 - 87; http://dx.doi.org/10.1261/rna.1657609; PMID: 19535462
- Barlow DP, Stöger R, Herrmann BG, Saito K, Schweifer N. The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 1991; 349:84 - 7; http://dx.doi.org/10.1038/349084a0; PMID: 1845916
- DeChiara TM, Robertson EJ, Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 1991; 64:849 - 59; http://dx.doi.org/10.1016/0092-8674(91)90513-X; PMID: 1997210
- Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, et al. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991; 349:38 - 44; http://dx.doi.org/10.1038/349038a0; PMID: 1985261
- Seidl CI, Stricker SH, Barlow DP. The imprinted Air ncRNA is an atypical RNAPII transcript that evades splicing and escapes nuclear export. EMBO J 2006; 25:3565 - 75; http://dx.doi.org/10.1038/sj.emboj.7601245; PMID: 16874305
- Pfeifer K, Tilghman SM. Allele-specific gene expression in mammals: the curious case of the imprinted RNAs. Genes Dev 1994; 8:1867 - 74; http://dx.doi.org/10.1101/gad.8.16.1867; PMID: 7525406
- Gingeras TR. Origin of phenotypes: genes and transcripts. Genome Res 2007; 17:682 - 90; http://dx.doi.org/10.1101/gr.6525007; PMID: 17567989
- Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, et al. Divergent transcription from active promoters. Science 2008; 322:1849 - 51; http://dx.doi.org/10.1126/science.1162253; PMID: 19056940
- Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010; 143:46 - 58; http://dx.doi.org/10.1016/j.cell.2010.09.001; PMID: 20887892
- Kowalczyk MS, Hughes JR, Garrick D, Lynch MD, Sharpe JA, Sloane-Stanley JA, et al. Intragenic enhancers act as alternative promoters. Mol Cell 2012; 45:447 - 58; http://dx.doi.org/10.1016/j.molcel.2011.12.021; PMID: 22264824
- Kowalczyk MS, Higgs DR, Gingeras TR. Molecular biology: RNA discrimination. Nature 2012; 482:310 - 1; http://dx.doi.org/10.1038/482310a; PMID: 22337043
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011; 25:1915 - 27; http://dx.doi.org/10.1101/gad.17446611; PMID: 21890647
- Furuno M, Pang KC, Ninomiya N, Fukuda S, Frith MC, Bult C, et al. Clusters of internally primed transcripts reveal novel long noncoding RNAs. PLoS Genet 2006; 2:e37; http://dx.doi.org/10.1371/journal.pgen.0020037; PMID: 16683026
- Kapranov P, St Laurent G, Raz T, Ozsolak F, Reynolds CP, Sorensen PH, et al. The majority of total nuclear-encoded non-ribosomal RNA in a human cell is ‘dark matter’ un-annotated RNA. BMC Biol 2010; 8:149; http://dx.doi.org/10.1186/1741-7007-8-149; PMID: 21176148
- Lyle R, Watanabe D, te Vruchte D, Lerchner W, Smrzka OW, Wutz A, et al. The imprinted antisense RNA at the Igf2r locus overlaps but does not imprint Mas1. Nat Genet 2000; 25:19 - 21; http://dx.doi.org/10.1038/75546; PMID: 10802648
- Koerner MV, Pauler FM, Hudson QJ, Santoro F, Sawicka A, Guenzl PM, et al. A downstream CpG island controls transcript initiation and elongation and the methylation state of the imprinted Airn macro ncRNA promoter. PLoS Genet 2012; 8:e1002540; http://dx.doi.org/10.1371/journal.pgen.1002540; PMID: 22396659
- Rougeulle C, Heard E. Antisense RNA in imprinting: spreading silence through Air. Trends Genet 2002; 18:434 - 7; http://dx.doi.org/10.1016/S0168-9525(02)02749-X; PMID: 12175797
- Palmer AC, Egan JB, Shearwin KE. Transcriptional interference by RNA polymerase pausing and dislodgement of transcription factors. Transcr 2011; 2:9 - 14; http://dx.doi.org/10.4161/trns.2.1.13511; PMID: 21326903
- Moldón A, Malapeira J, Gabrielli N, Gogol M, Gómez-Escoda B, Ivanova T, et al. Promoter-driven splicing regulation in fission yeast. Nature 2008; 455:997 - 1000; http://dx.doi.org/10.1038/nature07325; PMID: 18815595
- Stricker SH, Steenpass L, Pauler FM, Santoro F, Latos PA, Huang R, et al. Silencing and transcriptional properties of the imprinted Airn ncRNA are independent of the endogenous promoter. EMBO J 2008; 27:3116 - 28; http://dx.doi.org/10.1038/emboj.2008.239; PMID: 19008856
- Walter J, Hutter B, Khare T, Paulsen M. Repetitive elements in imprinted genes. Cytogenet Genome Res 2006; 113:109 - 15; http://dx.doi.org/10.1159/000090821; PMID: 16575169
- Guseva N, Mondal T, Kanduri C. Antisense noncoding RNA promoter regulates the timing of de novo methylation of an imprinting control region. Dev Biol 2012; 361:403 - 11; http://dx.doi.org/10.1016/j.ydbio.2011.11.005; PMID: 22119056
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28:511 - 5; http://dx.doi.org/10.1038/nbt.1621; PMID: 20436464
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009; 25:1105 - 11; http://dx.doi.org/10.1093/bioinformatics/btp120; PMID: 19289445
- Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 2005; 308:1149 - 54; http://dx.doi.org/10.1126/science.1108625; PMID: 15790807
- Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 2010; 154C:343 - 54; http://dx.doi.org/10.1002/ajmg.c.30267; PMID: 20803657
- Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet 2010; 154C:365 - 76; http://dx.doi.org/10.1002/ajmg.c.30273; PMID: 20803659
- Eggermann T. Russell-Silver syndrome. Am J Med Genet C Semin Med Genet 2010; 154C:355 - 64; http://dx.doi.org/10.1002/ajmg.c.30274; PMID: 20803658
- Flint J, Thomas K, Micklem G, Raynham H, Clark K, Doggett NA, et al. The relationship between chromosome structure and function at a human telomeric region. Nat Genet 1997; 15:252 - 7; http://dx.doi.org/10.1038/ng0397-252; PMID: 9054936
- Barbour VM, Tufarelli C, Sharpe JA, Smith ZE, Ayyub H, Heinlein CA, et al. alpha-thalassemia resulting from a negative chromosomal position effect. Blood 2000; 96:800 - 7; PMID: 10910890
- Tufarelli C, Stanley JA, Garrick D, Sharpe JA, Ayyub H, Wood WG, et al. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet 2003; 34:157 - 65; http://dx.doi.org/10.1038/ng1157; PMID: 12730694
- Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet 2009; 41:112 - 7; http://dx.doi.org/10.1038/ng.283; PMID: 19098912
- Kempers MJ, Kuiper RP, Ockeloen CW, Chappuis PO, Hutter P, Rahner N, et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol 2011; 12:49 - 55; http://dx.doi.org/10.1016/S1470-2045(10)70265-5; PMID: 21145788
- Kuiper RP, Vissers LE, Venkatachalam R, Bodmer D, Hoenselaar E, Goossens M, et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum Mutat 2011; 32:407 - 14; http://dx.doi.org/10.1002/humu.21446; PMID: 21309036
- Pasmant E, Laurendeau I, Héron D, Vidaud M, Vidaud D, Bièche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res 2007; 67:3963 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-06-2004; PMID: 17440112
- Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, et al. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 2008; 451:202 - 6; http://dx.doi.org/10.1038/nature06468; PMID: 18185590
- Yap KL, Li S, Muñoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 2010; 38:662 - 74; http://dx.doi.org/10.1016/j.molcel.2010.03.021; PMID: 20541999
- Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011; 30:1956 - 62; http://dx.doi.org/10.1038/onc.2010.568; PMID: 21151178
- Broadbent HM, Peden JF, Lorkowski S, Goel A, Ongen H, Green F, et al, PROCARDIS consortium. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet 2008; 17:806 - 14; http://dx.doi.org/10.1093/hmg/ddm352; PMID: 18048406
- Holdt LM, Beutner F, Scholz M, Gielen S, Gäbel G, Bergert H, et al. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol 2010; 30:620 - 7; http://dx.doi.org/10.1161/ATVBAHA.109.196832; PMID: 20056914
- Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, et al, Breast Cancer Susceptibility Collaboration (UK). Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet 2010; 42:504 - 7; http://dx.doi.org/10.1038/ng.586; PMID: 20453838
- Stacey SN, Sulem P, Masson G, Gudjonsson SA, Thorleifsson G, Jakobsdottir M, et al. New common variants affecting susceptibility to basal cell carcinoma. Nat Genet 2009; 41:909 - 14; http://dx.doi.org/10.1038/ng.412; PMID: 19578363
- Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet 2009; 41:899 - 904; http://dx.doi.org/10.1038/ng.407; PMID: 19578367
- Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol 2007; 8:703 - 13; http://dx.doi.org/10.1038/nrm2241; PMID: 17717515
- Pasmant E, Sabbagh A, Masliah-Planchon J, Ortonne N, Laurendeau I, Melin L, et al. NF France Network. Role of noncoding RNA ANRIL in genesis of plexiform neurofibromas in neurofibromatosis type 1. J Natl Cancer Inst 2011; 103:1713 - 22; http://dx.doi.org/10.1093/jnci/djr416; PMID: 22034633
- Folkersen L, Kyriakou T, Goel A, Peden J, Mälarstig A, Paulsson-Berne G, et al, PROCARDIS consortia. Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS ONE 2009; 4:e7677; http://dx.doi.org/10.1371/journal.pone.0007677; PMID: 19888323
- Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 2010; 464:409 - 12; http://dx.doi.org/10.1038/nature08801; PMID: 20173736
- Matoba S, Inoue K, Kohda T, Sugimoto M, Mizutani E, Ogonuki N, et al. RNAi-mediated knockdown of Xist can rescue the impaired postimplantation development of cloned mouse embryos. Proc Natl Acad Sci USA 2011; 108:20621 - 6; http://dx.doi.org/10.1073/pnas.1112664108; PMID: 22065773
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, et al. Regulating gene expression through RNA nuclear retention. Cell 2005; 123:249 - 63; http://dx.doi.org/10.1016/j.cell.2005.08.033; PMID: 16239143
- Gutschner T, Baas M, Diederichs S. Noncoding RNA gene silencing through genomic integration of RNA destabilizing elements using zinc finger nucleases. Genome Res 2011; 21:1944 - 54; http://dx.doi.org/10.1101/gr.122358.111; PMID: 21844124
- Tsai MC, Spitale RC, Chang HY. Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res 2011; 71:3 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-10-2483; PMID: 21199792
- Mercer TR, Gerhardt DJ, Dinger ME, Crawford J, Trapnell C, Jeddeloh JA, et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol 2012; 30:99 - 104; http://dx.doi.org/10.1038/nbt.2024; PMID: 22081020