Abstract
Understanding alternative splicing is crucial to elucidate the mechanisms behind several biological phenomena, including diseases. The huge amount of expressed sequences available nowadays represents an opportunity and a challenge to catalog and display alternative splicing events (ASEs). Although several groups have faced this challenge with relative success, we still lack a computational tool that uses a simple and straightforward method to retrieve, name and present ASEs. Here we present SPLOOCE, a portal for the analysis of human splicing variants. SPLOOCE uses a method based on regular expressions for retrieval of ASEs. We propose a simple syntax that is able to capture the complexity of ASEs.
Introduction
Alternative splicing events (ASEs) are present in almost all multi-exonic human genesCitation1,Citation2 and are believed to be one of the most significant components behind the complexity of multi-cellular organisms.Citation1,Citation3,Citation4 Furthermore, ASEs are clearly involved in the etiology of a wide variety of diseases, including cancer,Citation5-Citation7 ischemiaCitation8 and other common human disorders.Citation9 Recently, several studies have shown that constitutive and alternative splicing are regulated by a complex network of cellular elements, which include a set of trans-acting factors and cis-acting sequences found in the primary RNAs.Citation10-Citation18
The complex regulation of splicing and the high frequency of ASEs explain the appearance of Complex Alternative Splicing Events (CASEs), which are composed by a regulated combination of two or more single ASEs in transcripts from the same gene, or even in the same transcript. The most striking example of CASE is the Dscam in Drosophila, a gene containing a cluster of 48 mutually exclusive exons that, in principle, can generate thousands of splicing variants.Citation19
In humans, some ASEs and CASEs occurring in oncogenes and tumor suppressor genes have already been associated to cancer.Citation6,Citation20-Citation23 For example, the gene NTRK1 (nerve growth factor) has a sequence variant, TrkAIII, which is common in certain tumors and lacks three exons that affect a regulatory immunoglobulin-like domain.Citation24 Further CASE examples include the gene CD44, which is a known marker of malignancy and invasiveness and has about ten ASEs that can occur in different combinations in its region coding for the extra-cellular portion of the protein.Citation20,Citation21,Citation23
In spite of the efforts of other groups,Citation25-Citation27 a simple and efficient nomenclature to take into account all the variability generated by alternative splicing, especially for CASEs, is still missing. Here, we present a web portal, SPLOOCE, which uses a method based on regular expressions with an associated syntax. SPLOOCE provides a series of tools that allow users to profile splicing variants and analyze their functional impacts.
Results and Discussion
For the sake of space and clarity, we opted to describe the method used in this report as supplementary material, although an overview is present in . In this section we present an implementation of the method in a computational tool, SPLOOCE and illustrate the use of SPLOOCE discussing a few examples.
Figure 1. General strategy to process and analyze CASEs in a set of expressed sequences.
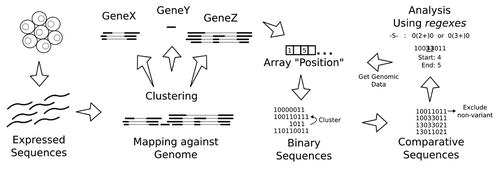
Implementation
To make the method described in the supplementary material available to the community in an easy way, a web tool called SPLOOCE was implemented. SPLOOCE is available at http://www.bioinformatics-brazil.org/splooce. Data sources include RefSeqs, mRNAs, ESTs and data from NGS.
To help the users, SPLOOCE provides in the query box a quick reference table explaining the syntax and showing some illustrative examples. All ASEs and CASEs identified by SPLOOCE can also be displayed for a specific gene by simply typing the gene name between quotes in the query box (). SPLOOCE also provides advanced options for querying. Filters for chromosome, strand, gene name and sequence type are provided. Users can also evaluate the specificity of ASEs and CASEs expression regarding both tissue and pathology. A score for expression specificity is provided, which is a simple ΧCitation2 distribution analysis done among the expressed sequences supporting the corresponding variant. The analysis is based on the annotation provided by eVOCCitation33 for ESTs and manual curation for NGS sequences.
Figure 2. (A) SPLOOCE query form also showing a tab for advanced parameters. (B) Example of the table of results for a query. (C) Some results that can be found in the section “Details” provided by SPLOOCE. In this example, a double skipping (syntax: -e-s-s-e-) that codifies a protein domain is shown.
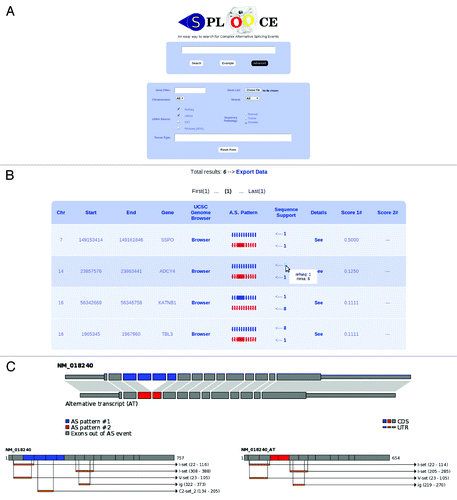
Results provided by SPLOOCE can be downloaded in a GFF file format. By default, results are shown in a table containing chromosome, genomic position, gene name and a pictorial view of the respective ASE or CASE, followed by the amount of their respective supporting sequences. SPLOOCE also provides a link to the UCSC Genome Browser (with tracks), and a local link for additional information.
When a Reference Sequence (RefSeq) is involved in an ASE, it is used as a template for creating a new mRNA sequence containing the specified event. For each of these new sequences, SPLOOCE predicts its open reading frame (ORF), which is then translated to a protein. Moreover, aiming to infer additional biological significance, SPLOOCE analyzes the protein domains using Pfam data and HMMER 3.0 program.Citation34 All these data are shown graphically ().
Analysis
To illustrate the use of SPLOOCE, some basic questions about the frequency and mode of alternative splicing events were addressed. shows the frequency of all types of ASEs in our data set. As expected, exon skipping (ES) is the most frequent type of alternative splicing. One interesting aspect that our method allowed us to explore is the distribution of dual-specific splicing (DSS) events. This type of event was found in 53 (0,27%) human genes (Table S2). In a less restrictive analysis, without considering the number of supporting sequences, the number of genes showing this type of event increased to 577. DSSs events were found to occur frequently in genes such as DIABLO, IRF3 and MAG, and they can occur together with other events as shown in .
Table 1. Frequency of the major types of ASEs
Table 2. Frequency of DSS events coupled to other types of ASEs
Another interesting feature that our method allows us to evaluate is the combination of events occurring in the same mRNA molecule. Each pair-wise comparative sequence in our pipeline contains on average 1.6 events and 46.87% and 12.21% of these pairwise comparisons report more than one or two events, respectively.
To better understand what influences the frequency of ASEs and CASEs, some patterns were further explored. For example, the combination of two adjacent ES events is significantly more frequent among all sets of CASEs (). This excess is absent in situations when both events are not adjacent, like in the patterns –s-E-s- and –s-E-E-s- . Do these adjacent events tend to maintain the phase of an ORF? When adjacent, 60.78% of -E-E-s-s-E-E- maintains the ORF. This is significantly higher than what one would expect by chance based on all pairs of exons in the human genome that maintain an ORF (p < 0.001). This strongly suggests that adjacent ES events are under selection to maintain the ORF. The same pattern is not observed for other types of CASEs (data not shown).
Table 3. Number of genes affected by different types of ASEs and CASEs
Material and Methods
Public data
The human genome reference sequence (NCBI36/hg18) was downloaded from UCSC Genome Bioinformatics portal (http://genome.ucsc.edu). RefSeq sequences were downloaded from the Reference Sequence database (release 25; http://www.ncbi.nlm.nih.gov/RefSeq/). A total of 203,649 mRNAs sequences were downloaded from UCSC Genome Bioinformatics portal (file mrna.fa, for Homo sapiens). ESTs sequences were downloaded from dbEST (http://www.ncbi.nlm.nih.gov/dbEST/). RNA-seq reads were downloaded from SRA database (http://www.ncbi.nlm.nih.gov/sra; IDs: SRX003935, SRX003934, SRX003933, SRX003932, SRX003931, SRX003930, SRX003929, SRX003928, SRX003927, SRX003926).
Sequence alignment
All RefSeqs, mRNAs and ESTs sequences were aligned to the human genome using the protocol described previously.Citation28 In brief, first all long sequences (RefSeq, mRNAs and ESTs) were mapped against the human genome using the BLAT alignment toolCitation29 and only the best alignment for each sequence was selected. Next, those transcripts showing alignment identity greater than 95% and a covering more than 90% of its sequence length were remapped using SIM4Citation30 and stored in a relational database. RNA-seq data were mapped to human genome using Tophat-based pipeline,Citation31 and all mapped reads were submitted to Cufflinks.Citation32 Default parameters were used in both algorithms.
Sequence clustering
All mapped sequences sharing the same genomic region were grouped together using a gene-oriented strategy as described previously.Citation4,Citation28 First, all RefSeq sequences were annotated based on the corresponding “official gene name” from NCBI-Gene (http://www.ncbi.nlm.nih.gov/gene/). Second, all non-RefSeq transcripts (mRNAs, ESTs and RNA-seq) presenting multiple exons and sharing one or more exon-intron boundaries (splice junctions) with a RefSeq sequence were merged together. Third, the remaining non-RefSeq transcripts showing only one exon and overlapping greater than 30 nt with a RefSeq transcript were grouped together. All clustering information was stored in a relational database.
Alternative splicing
Analyses of ASEs and CASEs were done using regular expressions, as detailed in the supplementary material. In the analysis of ASEs, only cDNA clusters containing RefSeqs and/or mRNAs or more than 10 ESTs/RNA-seq were used. The redundancy of ASEs, such as exon skipping and intron retention, was eliminated by comparing all events of the same type from each gene. Afterwards, all events showing position overlap were clustered together and counted as one event. All other events, such as 3′/5′ alternative splice sites and dual-specific splice sites, were counted by verifying the position of the alternative splice site. The number of genes affected by specific CASEs was defined through the analysis of the full list of comparative matrices, not considering the number of supporting sequences.
Conclusion
Although previous methods and interfaces have been proposedCitation25-Citation27 for the study of alternative splicing, all of them present limitations as discussed before.Citation27 SPLOOCE, described here, is an efficient and complementary alternative for the analysis of alternative splicing events due to its high flexibility in the querying patterns and variety of applications.
Like ASTALAVISTA,Citation27 the method used by SPLOOCE is based in a comparison of all transcripts for a given locus. SPLOOCE, however, uses a notation system based on regular expressions to provide a simple and straightforward syntax for splicing events. The design of the syntax was developed to provide a set of simple and intuitive characters, and is actually capable of representing any CASE pattern, including those that have rare DSS events. The proposed syntax was successfully implemented and it may become a standard way for representing ASEs and CASEs.
| Abbreviations: | ||
| ASE | = | alternative splicing event |
| CASE | = | complex alternative splicing event |
| RNA | = | ribonucleic acid |
| mRNA | = | messenger ribonucleic acid |
| UCSC | = | University of California, Santa Cruz |
| NCBI | = | National Center for Biotechnology Information |
| EST | = | expressed sequence tag |
| RNA-seq | = | whole transcriptome shotgun sequencing |
| SRA | = | sequence read archive |
| RefSeq | = | reference sequence |
| BLAT | = | BLAST-like alignment tool |
| cDNA | = | complementary deoxyribonucleic acid |
| NGS | = | next-generation sequencing |
| eVOC | = | expressed sequence annotation for humans |
| ORF | = | open reading frame |
| Pfam | = | protein families |
| ES | = | exon skipping |
| DSS | = | dual-specific splicing |
| Regex | = | regular expression |
| SIM4 | = | Computer Program for Aligning a cDNA Sequence with a Genomic DNA Sequence |
| HMMER | = | Computer Program for Biosequence Analysis Using Profile Hidden Markov |
Additional material
Download Zip (780.4 KB)Acknowledgments
Part of this work was supported by grants from the Fogarty International Center, National Institute of Health (D43TW007015 to L.O.M.); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES); and from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (2007/55790–5 to S.J.S. and 2009/53853–5 to S.J.S.). Funding for open access charge: Fundação de Amparo à Pesquisa do Estado de São Paulo.
Supplemental Material
Supplemental material may be found here: www.landesbioscience.com/journals/rna/article/22181
Related Research Data
References
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456:470 - 6; http://dx.doi.org/10.1038/nature07509; PMID: 18978772
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008; 40:1413 - 5; http://dx.doi.org/10.1038/ng.259; PMID: 18978789
- Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet 2002; 30:13 - 9; http://dx.doi.org/10.1038/ng0102-13; PMID: 11753382
- Galante PA, Sakabe NJ, Kirschbaum-Slager N, de Souza SJ. Detection and evaluation of intron retention events in the human transcriptome. RNA 2004; 10:757 - 65; http://dx.doi.org/10.1261/rna.5123504; PMID: 15100430
- Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 2007; 8:749 - 61; http://dx.doi.org/10.1038/nrg2164; PMID: 17726481
- Venables JP. Aberrant and alternative splicing in cancer. Cancer Res 2004; 64:7647 - 54; http://dx.doi.org/10.1158/0008-5472.CAN-04-1910; PMID: 15520162
- Kirschbaum-Slager N, Parmigiani RB, Camargo AA, de Souza SJ. Identification of human exons overexpressed in tumors through the use of genome and expressed sequence data. Physiol Genomics 2005; 21:423 - 32; http://dx.doi.org/10.1152/physiolgenomics.00237.2004; PMID: 15784694
- Daoud R, Mies G, Smialowska A, Oláh L, Hossmann KA, Stamm S. Ischemia induces a translocation of the splicing factor tra2-beta 1 and changes alternative splicing patterns in the brain. J Neurosci 2002; 22:5889 - 99; PMID: 12122051
- Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nat Biotechnol 2004; 22:535 - 46; http://dx.doi.org/10.1038/nbt964; PMID: 15122293
- Alló M, Buggiano V, Fededa JP, Petrillo E, Schor I, de la Mata M, et al. Control of alternative splicing through siRNA-mediated transcriptional gene silencing. Nat Struct Mol Biol 2009; 16:717 - 24; http://dx.doi.org/10.1038/nsmb.1620; PMID: 19543290
- Schor IE, Rascovan N, Pelisch F, Alló M, Kornblihtt AR. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc Natl Acad Sci USA 2009; 106:4325 - 30; http://dx.doi.org/10.1073/pnas.0810666106; PMID: 19251664
- Muñoz MJ, Pérez Santangelo MS, Paronetto MP, de la Mata M, Pelisch F, Boireau S, et al. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 2009; 137:708 - 20; http://dx.doi.org/10.1016/j.cell.2009.03.010; PMID: 19450518
- Ip JY, Schmidt D, Pan Q, Ramani AK, Fraser AG, Odom DT, et al. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res 2011; 21:390 - 401; http://dx.doi.org/10.1101/gr.111070.110; PMID: 21163941
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science 2010; 327:996 - 1000; http://dx.doi.org/10.1126/science.1184208; PMID: 20133523
- Batsché E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol 2006; 13:22 - 9; http://dx.doi.org/10.1038/nsmb1030; PMID: 16341228
- Saint-André V, Batsché E, Rachez C, Muchardt C. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat Struct Mol Biol 2011; 18:337 - 44; http://dx.doi.org/10.1038/nsmb.1995; PMID: 21358630
- Sakabe NJ, de Souza SJ. Sequence features responsible for intron retention in human. BMC Genomics 2007; 8:59; http://dx.doi.org/10.1186/1471-2164-8-59; PMID: 17324281
- de Souza JES, Ramalho RF, Galante PAF, Meyer D, de Souza SJ. Alternative splicing and genetic diversity: silencers are more frequently modified by SNVs associated with alternative exon/intron borders. Nucleic Acids Res 2011; 39:4942 - 8; http://dx.doi.org/10.1093/nar/gkr081; PMID: 21398627
- Schmucker D, Clemens JC, Shu H, Worby CA, Xiao J, Muda M, et al. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 2000; 101:671 - 84; http://dx.doi.org/10.1016/S0092-8674(00)80878-8; PMID: 10892653
- Hayes GM, Dougherty ST, Davis PD, Dougherty GJ. Molecular mechanisms regulating the tumor-targeting potential of splice-activated gene expression. Cancer Gene Ther 2004; 11:797 - 807; http://dx.doi.org/10.1038/sj.cgt.7700759; PMID: 15359288
- Galiana-Arnoux D, Del Gatto-Konczak F, Gesnel MC, Breathnach R. Intronic UGG repeats coordinate splicing of CD44 alternative exons v8 and v9. Biochem Biophys Res Commun 2005; 336:667 - 73; http://dx.doi.org/10.1016/j.bbrc.2005.08.153; PMID: 16137657
- Tanko Q, Franklin B, Lynch H, Knezetic J. A hMLH1 genomic mutation and associated novel mRNA defects in a hereditary non-polyposis colorectal cancer family. Mutat Res 2002; 503:37 - 42; http://dx.doi.org/10.1016/S0027-5107(02)00031-3; PMID: 12052501
- Venables JP. Unbalanced alternative splicing and its significance in cancer. Bioessays 2006; 28:378 - 86; http://dx.doi.org/10.1002/bies.20390; PMID: 16547952
- Tacconelli A, Farina AR, Cappabianca L, Desantis G, Tessitore A, Vetuschi A, et al. TrkA alternative splicing: a regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 2004; 6:347 - 60; http://dx.doi.org/10.1016/j.ccr.2004.09.011; PMID: 15488758
- Nagasaki H, Arita M, Nishizawa T, Suwa M, Gotoh O. Automated classification of alternative splicing and transcriptional initiation and construction of visual database of classified patterns. Bioinformatics 2006; 22:1211 - 6; http://dx.doi.org/10.1093/bioinformatics/btl067; PMID: 16500940
- Malko DB, Makeev VJ, Mironov AA, Gelfand MS. Evolution of exon-intron structure and alternative splicing in fruit flies and malarial mosquito genomes. Genome Res 2006; 16:505 - 9; http://dx.doi.org/10.1101/gr.4236606; PMID: 16520458
- Sammeth M, Foissac S, Guigó R. A general definition and nomenclature for alternative splicing events. PLoS Comput Biol 2008; 4:e1000147; http://dx.doi.org/10.1371/journal.pcbi.1000147; PMID: 18688268
- Galante PAF, Vidal DO, de Souza JE, Camargo AA, de Souza SJ. Sense-antisense pairs in mammals: functional and evolutionary considerations. Genome Biol 2007; 8:R40; http://dx.doi.org/10.1186/gb-2007-8-3-r40; PMID: 17371592
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res 2002; 12:656 - 64; PMID: 11932250
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res 1998; 8:967 - 74; PMID: 9750195
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009; 25:1105 - 11; http://dx.doi.org/10.1093/bioinformatics/btp120; PMID: 19289445
- Trapnell C, Williams BA, Pertea G, Mortazavi AM, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28:511 - 5; http://dx.doi.org/10.1038/nbt.1621; PMID: 20436464
- Kelso J, Visagie J, Theiler G, Christoffels A, Bardien S, Smedley D, et al. eVOC: a controlled vocabulary for unifying gene expression data. Genome Res 2003; 13:6A 1222 - 30; http://dx.doi.org/10.1101/gr.985203; PMID: 12799354
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, et al. The Pfam protein families database. Nucleic Acids Res 2010; 38:Database issue D211 - 22; http://dx.doi.org/10.1093/nar/gkp985; PMID: 19920124