Abstract
Chromosomally encoded toxin-antitoxin (TA) systems are abundantly present in bacteria and archaea. They have become a hot topic in recent years, because—after many frustrating years of searching for biological functions—some are now known to play roles in persister formation. Persister cells represent a subset of a bacterial population that enters a dormant state and thus becomes refractory to the action of antibiotics. TA modules come in several different flavors, regarding the nature of their gene products, their molecular mechanisms of regulation, their cellular targets, and probably their role in physiology. This review will primarily focus on the SOS-associated tisB/istR1 system in Escherichia coli and discuss its nuts and bolts as well as its effect in promoting a subpopulation phenotype that likely benefits long-term survival of a stressed population.
Introduction
Toxin-antitoxin (TA) systems were initially discovered in bacterial plasmidsCitation1,Citation2 where they confer stability of maintenance through post-segregational killing (PSK). PSK systems rely on a key property that subsequently was shown to encompass all other such systems: the toxin is stable, and the antitoxin unstable. Hence, loss of the TA locus (i.e., plasmid loss) quickly results in decreased antitoxin levels, whereas the stable toxin protein or the stable toxin mRNA remains present in the absence of its gene, eventually entailing toxin synthesis or activity. Thus, the toxin kills plasmid-free cells, whereas plasmid-containing cells are unaffected due to continuous de novo synthesis of the antitoxin. The best studied such system is hok/sok of plasmid R1 which has been investigated in great detail by the Gerdes group.Citation2,Citation3 Several years after the discovery of hok/sok and other unrelated PSK systems, it was found that TA systems were also encoded by the chromosomes of numerous bacterial and archaeal species, sometimes in staggering numbers. For instance, E. coli harbors more than 15Citation4,Citation5 and Mycobacterium tuberculosis more than 80 TA loci.Citation6-Citation8
Irrespective of their presence on chromosomes or plasmids, TA systems are today broadly classified by the components they use. Type I TA systems—as for instance the prototypical hok/sok module mentioned above—consist of a toxic protein and a small RNA (sRNA) that acts as the antitoxin. The antitoxin RNA prevents translation/ promotes degradation of the toxin mRNA by an antisense mechanism.Citation9,Citation10 Type II loci encode a toxin whose activity is blocked by an antitoxin protein. Typical examples are relB/relE and mazE/mazF.Citation5,Citation8,Citation11 The complex formed by both proteins regulates the operon that encodes the TA protein pair. Proteolytic destabilization of the antitoxin is performed by proteases and activates toxicity.Citation12 A third, recently discovered type III system is remarkable in that a pseudoknot-containing RNA (the antitoxin) directly binds to the toxin to inhibit its activity.Citation13,Citation14 Toxins have quite a diverse array of different cellular targets and affect for instance DNA replication, translation, and membrane integrity.Citation10,Citation11,Citation15-Citation18 Generally, stress-induced toxicity tends to decrease growth rate or arrest growth entirely. However, an alternative view suggests toxin-dependent altruistic suicide (programmed cell death) under certain conditions—such as exposure to some antibiotics—and is, at this point, controversial.Citation19,Citation20 It is important to note that all TA system toxins work from within and affect only the cells that produce them. They do not act on other cells in the population, and hence—in contrast to toxic proteins such as colicins—they are not used in battles between bacteria.
In spite of the ubiquity of chromosomally encoded TA systems in bacteria and archaea, their functions remained enigmatic for many years. Whereas toxicity could easily be assessed by toxin overexpression, single or even multiple deletions of TA loci initially showed no discernable phenotypes (e.g., ref. Citation21). Recently, though, two breakthrough papers clearly showed that both type I and type II TA systems are involved in persister phenotypes.Citation22,Citation23 Many isogenic bacterial populations, growing under the same environmental and nutritional conditions, display heterogeneity in certain traits and acquire different cell fates.Citation24 This applies, for instance, to chemotactic behavior, biofilm formation, competence, and the susceptibility to antibiotics. Persisters represent a small fraction of a population which stochastically has entered a dormant state and thus is insensitive to antibiotics for extended periods of time; most antibiotics kill only growing/dividing bacteria.Citation7,Citation25 Persister cells are not genetically different since they are resuscitated as normally growing, antibiotics-sensitive cells which in turn subsequently can generate persisters at a low frequency. Stochastic switching that generates heterogeneity of this kind underlies what is often referred to as bet hedgingCitation26: isogenic bacteria with two drastically different phenotypes may be better at surviving unanticipated threats (antibiotics or other stresses) which may kill off the majority of the population while the alternative phenotype may be protected and able to reestablish the population.
This review focuses on tisB/istR1 of E. coli, one of the chromosomally encoded type I TA systems. It is particularly interesting since its regulatory properties and mechanism are unusual, it is the first TA system shown to be involved in the SOS response, and it represents the most clearcut case of a single TA system that has a dramatic effect on persister formation. Other systems will be briefly discussed, but are mostly covered elsewhere in this issue.
tisB/istR1 is an SOS-associated toxin-antitoxin locus
In the aftermath of the first genome-wide searches for bacterial small RNAs (sRNA),Citation27,Citation28 several labs embarked on their functional characterization. Two candidate RNAs, IstR1 and IstR2, were shown to be encoded near a very short, apparently bicistronic, operon of unknown function (ysdAB). The two divergently oriented transcription units shared a bidirectional LexA binding site, suggesting an involvement in the SOS response (); LexA is the master regulator of the SOS regulon in the response to DNA damage. The second of the two reading frames, ysdB, encoded an SOS-induced toxic peptide which, when produced, arrested cell growth and decreased plating efficiency.Citation29 Mutations in ysdB abrogated toxicity, and the toxin was consequently renamed to TisB (toxicity induced by SOS). Mutational analyses indicated that the first, poorly conserved ORF (ysdA/tisA) upstream of tisB was not involved in toxicity,Citation29 and later studies showed that it was not translated.Citation30 The ysdAB mRNA will therefore henceforth be denoted tisB mRNA. This work also showed that the oppositely oriented locus encoded two sRNAs that shared terminators but were transcribed from different promoters. The longer RNA, IstR2 (140 nt), was induced by mitomycin C (MMC) treatment (triggering SOS), whereas IstR1 (74 nt) was constitutively expressed. Transformation assays with plasmids containing various segments of tisB and/or istR clearly indicated that the tisB/istR locus encodes a type I TA system: TisB is the toxin, and IstR1 (inhibitor of SOS-induced toxicity by RNA) is the antitoxin. A 22 nucleotide long uninterrupted sequence complementarity between IstR1/IstR2 and a region in the mRNA far upstream of tisB suggested that one or both sRNAs was an antitoxin acting by an antisense mechanism. Surprisingly, Northern analyses showed that only IstR1 bound to tisB mRNA, indicated by characteristic RNase III-dependent cleavages in both RNAs, whereas IstR2, even though it shared the sequence that targets tisB mRNA, neither inhibited toxicity nor bound its target efficientlyCitation29,Citation30(Darfeuille and Wagner, unpublished). IstR1, in contrast to most other trans-encoded sRNAs, does not require the RNA chaperone Hfq for activity in vivo,Citation30,Citation31 nor does it bind Hfq significantly in vitro.Citation32 Whether IstR2 plays a separate role related to SOS conditions is yet unknown.
Figure 1. Schematics of the tisB-istR1 locus with RNAs encoded. A. The locus is drawn schematically, with promoters (angled arrows), terminators (stemloops) and LexA box indicated. The three primary transcripts are shown. B. RNAs observed under SOS-ON and SOS-OFF (LexA-repressed) conditions. The three mRNA variants (blue) are indicated. “X” at the 5′-end indicates a processing step. In SOS-ON, also IstR1 is processed. See text for details.
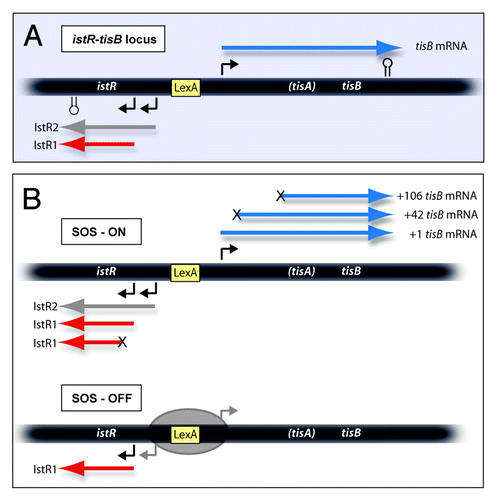
Altogether, this suggested the following model. In normally growing cells, LexA represses transcription of tisB. Constitutive expression of IstR1 prevents inadvertant toxicity that may arise from leaky transcripton of tisB. When SOS conditions are encountered, RecA-induced self-cleavage of LexA derepresses tisB. High tisB mRNA levels then outtitrate the entire IstR1 pool, and subsequent uninhibited toxin synthesis arrests cell growth and permits the cells time to recover.Citation29 Later work suggested another rationale which involves heterogeneity effects in a bacterial population (see below).
Antisense regulation in tisB/istR1 with unexpected features
Conventional regulation by (antisense) sRNAs often involves base-pairing at/near ribosome binding sites (RBS). Binding of the sRNA causes translational inhibition by competition with initiating ribosomes.Citation33,Citation34 How IstR1 inhibited tisB expression was initially enigmatic since the experimentally validated target site was located ≈100 nt upstream of the tisB translation initiation region (TIR). An antisense-target RNA duplex structure so far upstream should be unable to sterically block the formation of 30S initiation complexes. It turned out that tisB/istR1 represents a novel mechanism of translational control with unusual features.
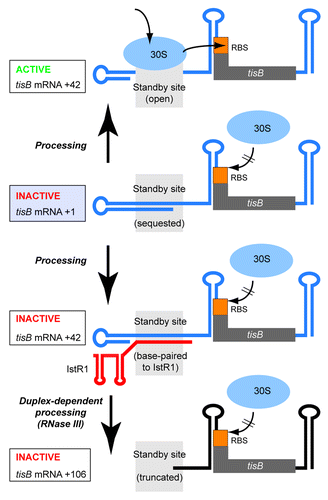
In vivo studiesCitation29 had shown the presence of three distinct variants of tisB mRNA: the primary +1 mRNA transcript (numbers refer to 5′-end position), a low abundance +42 mRNA species derived from a processing event, and +106, generated upon binding of IstR1 followed by RNase III-cleavage of tisB mRNA (). All three mRNAs contain the complete tisB toxin ORF. However, expression of +106 tisB mRNA was known to be non-toxic,Citation29 suggesting this mRNA to be untranslatable in spite of an intact tisB sequence. In vitro experiments surprisingly showed that also +1 mRNA was inactive, and only +42 tisB mRNA was translated.Citation30 Hence, processing of the primary (+1) transcript was required to generate the active (+42) mRNA. Extensive RNA structure mapping and genetic analysis indicated that the three different mRNA species were structurally indistinguishable in their shared 3′-domain, which contained the entire tisB ORF. Moreover, the tisB TIR was located in a stable structure that should be inaccessible to initating ribosomes. Thus, translational activity in tisB had to be determined by sequence/structure differences between the 5′-regions of the +1, +42, and +106 variants. Strikingly, only +42 mRNA displayed a long unstructured segment that also coincided with the IstR1 target site (). Of the two inactive mRNAs, +1 carried instead a stable 5′-structure, and +106 had lost much of the unstructured region through RNase III cleavage. Thus it seemed that the unstructured region in +42 far upstream could “activate” the sequestered tisB TIR and, by inference, binding of IstR1 to this same site should account for preventing this activation event.
Figure 2. Standby requirement for tisB translation and inhibition by IstR1. Inactive +1 tisB mRNA (light blue box) is processed into +42 (up and down arrow). Arrow up: An open standby region (gray field) permits binding of standby 30S subunits, relocation and translation initation at the tisB RBS. Arrows down: Binding of IstR1 blocks the standby site, subsequent RNase III cleavage removes part of it. See text for details.
Inhibition of ribosome standby explains the tisB/istR1 mechanism
The implication of the unstructured 5′-domain of +42 tisB mRNA in both translational activity and regulatibility by IstR1 could not be explained by conventional models such as direct translational repression, induced mRNA decay, translational coupling or others.Citation29,Citation30,Citation35 However, it made perfect sense in light of the “ribosome standby” model of de Smit and van Duin.Citation36,Citation37 Based on studies of the MS2 phage coat protein gene, these authors had shown that translation of a reading frame correlated inversely with the stability of a structure containing the RBS.Citation38 However, they noted that the efficiency of translation of some mRNAs was orders of magniture higher than the calculated structural stability of the TIR region would suggest. De Smit and van Duin solved this paradox by proposing that ribosomes initially can bind sequence-nonspecifically to unstructured regions in an mRNA (“ribosome standby”). Standby binding facilitates initiation at structured RBSs nearby, since a short transient opening of an RNA stemloop (“breathing”) permits sliding of standby ribosomes into the TIR. Without standby, stable structure cannot be overcome in the short time frame of unfolding because free ribosomes need to be recruited from the cytoplasmic pool.Citation36,Citation37 The standby model, which received further support from in vitro analyses,Citation39 provided the framework for how tisB is expressed and regulated. The unstructured region present in +42, but absent from +1 or +106 tisB mRNA, is proposed to be a standby site. Primary transcripts accumulate in the cell as an inactive reservoir. Processing to +42 tisB mRNA enables standby ribosomes to bind to the now unfolded 5′-region, and translation is possible in spite of the stable tisB RBS structure (). This model also explains how IstR1 represses tisB translation in spite of the distance between target site and TIR. IstR1 renders the standby site double-stranded, ribosomes cannot access this RNA segment, and standby-dependent translation of tisB is prevented. Congruent with this, mutations that destabilized the tisB RBS structure of +1 mRNA rendered it partially standby-independent and translation-competent, concomitant with IstR1 losing most of its inhibitory effect.Citation30
The hydrophobic membrane-targeting toxin TisB shuts off macromolecular synthesis
A mechanism by which TisB exerts toxicity was tentatively suggested by its properties. This 29 amino acid peptide was predicted to be hydrophobic and membrane spanning. Indeed, when the expression of FLAG-tagged TisB was induced from a plasmid, it was exclusively detected in the inner membrane fraction.Citation31 Concomitant with membrane localization of TisB, the cells showed a rapid shut-off of macromolecular synthesis; replication, transcription, and translation rates decreased by more than 100-fold within 5 min. This strikingly global effect was suspected to be caused by a shared initial event. Since hydrophopic membrane-penetrating peptides can form pores, this could entail loss of membrane potential and a subsequent drastic drop in intracellular ATP levels. This was observed and tentatively explained the indirect effect on macromolecular synthesis rates.Citation31 These experiments were however conducted under conditions of induced overexpression, and thus cannot inform on the biological effects in normal or SOS-stressed bacteria. Notably, overexpression of tisB is severely toxic and eventually results in cell death in a fraction of the population. Killing was not observed in single-gene dosage, even though tisB expression is 1000-fold induced under SOS conditions.Citation22,Citation31 This is in line with data on heterologous TA systems which failed to promote killing of cells when in single copy.Citation11 We will return to this issue below. Interestingly, an SOS-induced peptide, SidA, of the same size as TisB, has recently been reported in Caulobacter crescentus. Upon sensing DNA damage, it inserts into the cytoplasmic membrane, interacts with FtsW, and causes a delay in cell division.Citation16 Whether this locus also encodes an sRNA is not known but remains an intriguing possibility.
Additional type I TA modules
Some of the properties of TisB are reminiscent of the plasmid-encoded Hok. Hok creates inner membrane pores which cause loss of membrane potential and generate dead cells with so-called “ghost” morphology.Citation2 However, the function of the many hok/sok loci that since have been found on bacterial and archaeal chromomes is still unknown. Their frequently observed mutational corruption may suggest that they have been selectively inactivated.Citation4 Several other chromosomal type I TA systems have properties that in part are similar to those found in tisB/istR1 (). For instance, E. coli K12 encodes four ldr/rdl loci, five ibs/sib loci, and a locus denoted shoB/ohsC.Citation40-Citation42 Bioinformatics suggests that they, and some additional type I systems, are abundantly present in other bacteria.Citation10 Their toxins, in most cases, are small hydrophobic proteins that are known, or suspected, to affect membrane integrity.Citation40 Gene organization is hok/sok- rather than tisB/istR1-like in that the transcription units of toxin and antitoxin overlap, i.e., they represent cis-encoded systems (except: shoB/ohsC). Whole genome expression studies in strains expressing four of these small toxins, IbsC, ShoB, LdrD and TisB, revealed changes in the expression of a shared set of genes, but also indicated individual toxin-specific effects.Citation40 Another interesting type I system is symE/symR. The SOS-induced SymE toxin is 113 amino acids long, not hydrophobic, exhibits structural similarity to the antitoxin MazE (of mazE/mazFCitation43), and may induce RNA degradation.Citation44 The symE/symR system is reviewed elsewhere in this issue.Citation66 In general, whether any of these type I modules contribute to bacterial fitness under any condition, and if so, how, is currently unknown ().
Table 1. Characteristics of some chromosomal type I TA systems in E. coli
What are chromosomally encoded TA systems good for?
The ubiquitous presence of the many different chromosomally encoded TA systems presents a puzzle. Why do bacteria “collect” numerous loci which—when deleted individually or even multiply—show no apparent phenotype? Irrespective of the mechanism of toxicity, and of organization (type I, II, III), it is tempting to look for a common denominator for all systems. Several scenarios have been entertained, such as that they (1) represent merely selfish genes,Citation17 (2) stabilize the genomic regions they inhabit (for instance, superintegrons),Citation45 (3) function in quality control and in protection from nutritional and other stresses,Citation12 (4) induce programmed cell death under adverse conditions such as exposure to antibiotics,Citation19 (5) offer protection against attacking phagesCitation46,Citation64 and (6) alter the frequencies of appearance of certain subpopulation, for instance persister cells.Citation22,Citation23 Many of these proposed roles intuitively make biological sense, but direct experimental evidence has been hard to obtain. For instance, many type II modules encode toxins that are mRNases, i.e., they cleave mRNAs on or off the ribosome.Citation8,Citation11,Citation47-Citation49 Thus, nutrient depletion might induce such TA systems through ppGpp- and Lon-dependent rapid degradation of the antitoxins, followed by enhanced mRNA decay.Citation11,Citation50 This in turn would slow down translation, arrest cell growth, and potentially replenish the aa-tRNA pool.
The Engelberg-Kulka group has argued that induction of the toxin MazF by certain antibiotics promotes altruistic suicide which might benefit the surrounding clonal population.Citation20 This is dependent on cell density via a quorum sensing-like signal (extracellular death factor/EDFCitation51). An interesting recent report demonstrated that MazF cleaves to remove the 3′ end of 16S rRNA containing the anti-Shine-Dalgarno sequence, thus committing a fraction of the ribosomes to the translation of leaderless mRNAs.Citation52 This in turn might reshape the total translational output and reprogram the cell ultimately to promote stress-tolerance. How the latter scenario squares with MazF's role in causing cellular suicide remains to be determined. The frame of this review does not permit us to discuss in depth the controversy between reversible growth arrest models and programmed cell death associated with AT modules.Citation11,Citation17,Citation18,Citation20
TA systems and the persister connection
Regarding the paucity of clearcut clues as to the roles of TA systems in general bacterial physiology, it seemed possible that stochastic expression of toxins, and consequently effects on cells of only a subpopulation, might explain why such modules were present. Many population strategies involve heterogeneity in phenotype. Persisters represent such a heterogeneity phenotype. They are causing major health problems since they are recalcitrant to antibiotics.Citation7,Citation25,Citation53-Citation55 It is well-known that treatment with bacteriocidal antibiotics kills the vast majority of susceptible bacteria. However, dependent on genotype, physiological status, and other less well-defined conditions, some cells stochastically enter a state of very low metabolic activity or even complete dormancy.Citation56 These cells are multi-drug-tolerant (MDT) and persist as viable but nongrowing cells. After often long periods of time, persisters may be resuscitated and resume growth, reestablishing the population of atibiotics-susceptible cells.Citation57 Persistence is not restricted to bacteria since, e.g., it has also been observed in the fungus Candida albicans.Citation58 It is epigenetic in nature, even though many genes by know are known to affect the frequency by which persisters are formed.Citation15,Citation59,Citation60,Citation65 Since persisters are extremely slow growing or totally growth arrested, it has been speculated that expression of toxins might stochastically drive a fraction of a bacterial population into the persister state. In line with this, the first mutant gene that caused a whopping 1000-fold increase in the frequency of persisters in E. coli, hipA7,Citation55 was later recognized to be the toxin of a type II TA gene pair. The HipA toxin is a kinase that phosphorylates EF-Tu, rendering it inactive and causing cell stasis.Citation61 Appearently, the hipA7 mutation weakens interaction with the antitoxin, derepressing the TA locus and increasing persister formation. In general, this suggested that dormancy, and thus persister phenotypes, may be promoted by many TA toxins irrespective of the molecular mechanism by which a dormant state can be achieved. Accordingly, overexpression of the toxins RelE and MazF (both are mRNases) resulted in increased persisters, whereas deletion strains showed no effect.Citation54 Obviously, different physiological cues and stress signals (such as for instance SOS) might work through expression of different TA modules to affect persister levels.
tisB/istR1 as a bona fide persister locus
In 2009, Dörr et al.Citation62 analyzed the effect of the fluoroquinolone ciprofloxacin on the formation of persisters. Fluoroquinolones are antibiotics that target DNA gyrase and DNA topoisomerase, ultimately causing DNA lesions which in turn trigger the SOS response. Pretreatment with low levels of ciprofloxacin induced high levels of ciprofloxacin-tolerant persisters, and this effect was dependent on expression of SOS regulon genes. The data suggested that these persister were not pre-existing but, at least mostly, were induced to enter this state.Citation62 E. coli encodes several SOS-induced TA systems (symE/symR, tisB/istR1, hokE/sokE, yafO/yafN, yafQ/dinJCitation8), and TA genes had previously been found to be among the gene classes that were upregulated in persister cells.Citation56 In a follow-up paper by the Lewis group, it was shown that ciprofloxacin increased persister levels via SOS-dependent induction of TisB.Citation22 A ∆tisB strain showed a 10–100 fold decrease in ciprofloxacin-tolerant persisters, whereas deletion of the antitoxin istR1 instead resulted in a 10–100 fold increase. Remarkably, this was—in contrast to other studies in which persister phenotypes were only observed under overexpression regimes—the first case in which a wild-type TA locus in single copy was associated with so dramatic effects. TisB-dependent persisters were also highly tolerant to multiple antibiotics. This can be reconciled with TisB's effect on membrane integrity, decreasing intracellular ATP levels and thus all macromolecular synthesis ratesCitation31; the state of growth arrest explains the MDT phenotype of the persisters.Citation22
A second landmark paper reinvestigated the contribution of type II TA systems in E. coli to persister formationCitation23; ten TA modules encode mRNase toxins. None of these had previously supported persister phenotypes in single copy. The Gerdes group first showed, by titration of the antitoxin RelB with a binding-competent but non-toxic RelE mutant protein, that endogenous free RelE indeed increased persisters up to 4000-fold; the cells that survived ampicillin treatment showed higher levels of relBE transcription. The authors then created strains carrying from one to ten simultaneous TA deletions. Successive deletions gave an additive effect on persister formation to a level about 100-fold lower than that of the wild-type strain. This showed that TA systems were responsible for persister formation, and suggested that independent stochastic induction of TA systems summed up to give the observed population phenotype. In support of this, overexpression of Lon, the protease that degrades the antitoxins, enhanced toxicity and thereby increased the fraction of persisters, whereas the absence of Lon decreased persister formation.Citation23
Stochastic or deterministic generation of persisters?
TisB-dependent persisters were only observed in exponential growth, when the SOS regulon is maximally expressed upon DNA damage.Citation22 It was argued that the tisB example suggests that persisters are not pre-existing but rather induced, here by SOS. A recent paper showed that bacterial communication, through indole signaling in stationary phase, also induces an increase in persisters.Citation63 This appears to be dependent on activation of the OxyR- and phage shock pathways. By contrast, TA-associated persister formation has, in most publications, suggested to be stochastic, i.e., generating some cells in which growth is arrested. For most systems, this appears to occur at elevated levels in stationary phase.Citation57 It seems to us that stochastic and deterministic interpretations may mostly reflect differences in perspective. The work of Rotem et al.Citation26 presents a general model, based on experimental evidence using the hipA/hipB system. In this model, the levels of the toxin and the antitoxin set a threshold. When the toxin exceeds the threshold, the individual cell will at high probability arrest growth/become dormant. Below the threshold, cells grow normally. Near threshold levels, stochastic bistability in the population will give a majority of normally growing and (usually) few persister cells. Any physiological change, stress signal, and individual factor that affects the toxin/antitoxin balance will alter the frequency of persisters. In this view, changes of the threshold level at which bistability is played out could then be SOS-induction as for TisB,Citation22 signaling with indole,Citation63 activation of Lon, or nutritional or other stresses in stationary phase.Citation23 A difference that likely underlies the different interpretations of stochastic and deterministic effects is the set-up of type I and type II TA systems. The latter are single TA operons. Thus, co-regulation produces both proteins simultaneously, and toxicity requires degradation of the antitoxin. In type I systems such as tisB/istR1 and others, the two genes are individually transcribed. Thus, transcription of the toxin can vary from almost totally repressed to fully induced, creating a different response curve with respect to the threshold model.Citation26 However, even then, stochasticity is evident since only a fraction of the cells is driven into the dormant state.
Concluding remarks
The stunning diversity of the ubiquitous TA systems—representing different induction modes, mechanisms of regulation, different molecular targets, and possibly an interplay of TA modules—makes any simplified general conclusion hazardous. The recently established firm causal connection between some type I and II systems and persisters is a major advance. However, functions for the vast majority of TA modules are yet unknown. Likely, some of these will also affect population phenotypes such as persisters. However, it will also be of interest to find out whether any of the other “biologically plausible” roles that have been proposed for the acquisition of TA systems (see above) will receive experimental support. As for the type I TA family to which the tisB/istR1 locus belongs, there is clearly more work needed to identify bulk or subpopulation phenotypes that might suggest what their roles are. Possibly, more extensive assessment of the conditions under which their toxins are induced might provide clues. From a mechanistic point of view, the tisB/istR1 system has taught us a new way by which sRNAs can inhibit translation of structurally sequestered mRNAs. The mechanism of ribosome standby in tisB/istR1, and its implications, suggests that this mode of translational initiation may be far more prevalent than anticipated.
Acknowledgments
We thank Fredrik Söderbom for critical reading of the manuscript. This work was supported by The Swedish Science Research Council (VR) and the European Commission (EU-STREP BacRNAs).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Ogura T, Hiraga S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci U S A 1983; 80:4784 - 8; http://dx.doi.org/10.1073/pnas.80.15.4784; PMID: 6308648
- Gerdes K, Bech FW, Jørgensen ST, Løbner-Olesen A, Rasmussen PB, Atlung T, et al. Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. EMBO J 1986; 5:2023 - 9; PMID: 3019679
- Gerdes K, Wagner EG. RNA antitoxins. Curr Opin Microbiol 2007; 10:117 - 24; http://dx.doi.org/10.1016/j.mib.2007.03.003; PMID: 17376733
- Pedersen K, Gerdes K. Multiple hok genes on the chromosome of Escherichia coli. Mol Microbiol 1999; 32:1090 - 102; http://dx.doi.org/10.1046/j.1365-2958.1999.01431.x; PMID: 10361310
- Pandey DP, Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res 2005; 33:966 - 76; http://dx.doi.org/10.1093/nar/gki201; PMID: 15718296
- Ramage HR, Connolly LE, Cox JS. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet 2009; 5:e1000767; http://dx.doi.org/10.1371/journal.pgen.1000767; PMID: 20011113
- Lewis K. Persister cells. Annu Rev Microbiol 2010; 64:357 - 72; http://dx.doi.org/10.1146/annurev.micro.112408.134306; PMID: 20528688
- Yamaguchi Y, Inouye M. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat Rev Microbiol 2011; 9:779 - 90; http://dx.doi.org/10.1038/nrmicro2651; PMID: 21927020
- Gerdes K, Wagner EGH. RNA antitoxins. Curr Opin Microbiol 2007; 10:117 - 24; http://dx.doi.org/10.1016/j.mib.2007.03.003; PMID: 17376733
- Fozo EM, Makarova KS, Shabalina SA, Yutin N, Koonin EV, Storz G. Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res 2010; 38:3743 - 59; http://dx.doi.org/10.1093/nar/gkq054; PMID: 20156992
- Gerdes K, Christensen SK, Løbner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol 2005; 3:371 - 82; http://dx.doi.org/10.1038/nrmicro1147; PMID: 15864262
- Christensen SK, Mikkelsen M, Pedersen K, Gerdes K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc Natl Acad Sci U S A 2001; 98:14328 - 33; http://dx.doi.org/10.1073/pnas.251327898; PMID: 11717402
- Blower TR, Pei XY, Short FL, Fineran PC, Humphreys DP, Luisi BF, et al. A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat Struct Mol Biol 2011; 18:185 - 90; http://dx.doi.org/10.1038/nsmb.1981; PMID: 21240270
- Blower TR, Short FL, Rao F, Mizuguchi K, Pei XY, Fineran PC, et al. Identification and classification of bacterial Type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res 2012; 40:6158 - 73; http://dx.doi.org/10.1093/nar/gks231; PMID: 22434880
- Blower TR, Salmond GP, Luisi BF. Balancing at survival’s edge: the structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr Opin Struct Biol 2011; 21:109 - 18; http://dx.doi.org/10.1016/j.sbi.2010.10.009; PMID: 21315267
- Modell JW, Hopkins AC, Laub MT. A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes Dev 2011; 25:1328 - 43; http://dx.doi.org/10.1101/gad.2038911; PMID: 21685367
- Van Melderen L, Saavedra De Bast M. Bacterial toxin-antitoxin systems: more than selfish entities?. PLoS Genet 2009; 5:e1000437; http://dx.doi.org/10.1371/journal.pgen.1000437; PMID: 19325885
- Hayes F. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 2003; 301:1496 - 9; http://dx.doi.org/10.1126/science.1088157; PMID: 12970556
- Amitai S, Yassin Y, Engelberg-Kulka H. MazF-mediated cell death in Escherichia coli: a point of no return. J Bacteriol 2004; 186:8295 - 300; http://dx.doi.org/10.1128/JB.186.24.8295-8300.2004; PMID: 15576778
- Engelberg-Kulka H, Yelin I, Kolodkin-Gal I. Activation of a built-in bacterial programmed cell death system as a novel mechanism of action of some antibiotics. Commun Integr Biol 2009; 2:211 - 2; http://dx.doi.org/10.4161/cib.2.3.7876; PMID: 19641731
- Tsilibaris V, Maenhaut-Michel G, Mine N, Van Melderen L. What is the benefit to Escherichia coli of having multiple toxin-antitoxin systems in its genome?. J Bacteriol 2007; 189:6101 - 8; http://dx.doi.org/10.1128/JB.00527-07; PMID: 17513477
- Dörr T, Vulić M, Lewis K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol 2010; 8:e1000317; http://dx.doi.org/10.1371/journal.pbio.1000317; PMID: 20186264
- Maisonneuve E, Shakespeare LJ, Jørgensen MG, Gerdes K. Bacterial persistence by RNA endonucleases. Proc Natl Acad Sci U S A 2011; 108:13206 - 11; http://dx.doi.org/10.1073/pnas.1100186108; PMID: 21788497
- Losick R, Desplan C. Stochasticity and cell fate. Science 2008; 320:65 - 8; http://dx.doi.org/10.1126/science.1147888; PMID: 18388284
- Lewis K. Persister cells, dormancy and infectious disease. Nat Rev Microbiol 2007; 5:48 - 56; http://dx.doi.org/10.1038/nrmicro1557; PMID: 17143318
- Rotem E, Loinger A, Ronin I, Levin-Reisman I, Gabay C, Shoresh N, et al. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc Natl Acad Sci U S A 2010; 107:12541 - 6; http://dx.doi.org/10.1073/pnas.1004333107; PMID: 20616060
- Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EGH, Margalit H, et al. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol 2001; 11:941 - 50; http://dx.doi.org/10.1016/S0960-9822(01)00270-6; PMID: 11448770
- Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 2001; 15:1637 - 51; http://dx.doi.org/10.1101/gad.901001; PMID: 11445539
- Vogel J, Argaman L, Wagner EGH, Altuvia S. The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr Biol 2004; 14:2271 - 6; http://dx.doi.org/10.1016/j.cub.2004.12.003; PMID: 15620655
- Darfeuille F, Unoson C, Vogel J, Wagner EGH. An antisense RNA inhibits translation by competing with standby ribosomes. Mol Cell 2007; 26:381 - 92; http://dx.doi.org/10.1016/j.molcel.2007.04.003; PMID: 17499044
- Unoson C, Wagner EGH. A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Mol Microbiol 2008; 70:258 - 70; http://dx.doi.org/10.1111/j.1365-2958.2008.06416.x; PMID: 18761622
- Fender A, Elf J, Hampel K, Zimmermann B, Wagner EGH. RNAs actively cycle on the Sm-like protein Hfq. Genes Dev 2010; 24:2621 - 6; http://dx.doi.org/10.1101/gad.591310; PMID: 21123649
- Wagner EGH, Altuvia S, Romby P. Antisense RNAs in bacteria and their genetic elements. Adv Genet 2002; 46:361 - 98; http://dx.doi.org/10.1016/S0065-2660(02)46013-0; PMID: 11931231
- Waters LS, Storz G. Regulatory RNAs in bacteria. Cell 2009; 136:615 - 28; http://dx.doi.org/10.1016/j.cell.2009.01.043; PMID: 19239884
- Unoson C, Wagner EGH. Dealing with stable structures at ribosome binding sites: bacterial translation and ribosome standby. RNA Biol 2007; 4:113 - 7; http://dx.doi.org/10.4161/rna.4.3.5350; PMID: 18094628
- de Smit MH, van Duin J. Control of translation by mRNA secondary structure in Escherichia coli. A quantitative analysis of literature data. J Mol Biol 1994; 244:144 - 50; http://dx.doi.org/10.1006/jmbi.1994.1714; PMID: 7966326
- de Smit MH, van Duin J. Translational standby sites: how ribosomes may deal with the rapid folding kinetics of mRNA. J Mol Biol 2003; 331:737 - 43; http://dx.doi.org/10.1016/S0022-2836(03)00809-X; PMID: 12909006
- de Smit MH, van Duin J. Secondary structure of the ribosome binding site determines translational efficiency: a quantitative analysis. Proc Natl Acad Sci U S A 1990; 87:7668 - 72; http://dx.doi.org/10.1073/pnas.87.19.7668; PMID: 2217199
- Studer SM, Joseph S. Unfolding of mRNA secondary structure by the bacterial translation initiation complex. Mol Cell 2006; 22:105 - 15; http://dx.doi.org/10.1016/j.molcel.2006.02.014; PMID: 16600874
- Fozo EM, Hemm MR, Storz G. Small toxic proteins and the antisense RNAs that repress them. Microbiol Mol Biol Rev 2008; 72:579 - 89; http://dx.doi.org/10.1128/MMBR.00025-08; PMID: 19052321
- Fozo EM, Kawano M, Fontaine F, Kaya Y, Mendieta KS, Jones KL, et al. Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol Microbiol 2008; 70:1076 - 93; http://dx.doi.org/10.1111/j.1365-2958.2008.06394.x; PMID: 18710431
- Kawano M, Oshima T, Kasai H, Mori H. Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli. Mol Microbiol 2002; 45:333 - 49; http://dx.doi.org/10.1046/j.1365-2958.2002.03042.x; PMID: 12123448
- Yamaguchi Y, Inouye M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Prog Mol Biol Transl Sci 2009; 85:467 - 500; http://dx.doi.org/10.1016/S0079-6603(08)00812-X; PMID: 19215780
- Kawano M, Aravind L, Storz G. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol Microbiol 2007; 64:738 - 54; http://dx.doi.org/10.1111/j.1365-2958.2007.05688.x; PMID: 17462020
- Christensen-Dalsgaard M, Gerdes K. Two higBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol Microbiol 2006; 62:397 - 411; http://dx.doi.org/10.1111/j.1365-2958.2006.05385.x; PMID: 17020579
- Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GP. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc Natl Acad Sci U S A 2009; 106:894 - 9; http://dx.doi.org/10.1073/pnas.0808832106; PMID: 19124776
- Zhang Y, Zhang J, Hoeflich KP, Ikura M, Qing G, Inouye M. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol Cell 2003; 12:913 - 23; http://dx.doi.org/10.1016/S1097-2765(03)00402-7; PMID: 14580342
- Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 2003; 112:131 - 40; http://dx.doi.org/10.1016/S0092-8674(02)01248-5; PMID: 12526800
- Christensen SK, Pedersen K, Hansen FG, Gerdes K. Toxin-antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J Mol Biol 2003; 332:809 - 19; http://dx.doi.org/10.1016/S0022-2836(03)00922-7; PMID: 12972253
- Gerdes K. Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. J Bacteriol 2000; 182:561 - 72; http://dx.doi.org/10.1128/JB.182.3.561-572.2000; PMID: 10633087
- Kolodkin-Gal I, Hazan R, Gaathon A, Carmeli S, Engelberg-Kulka H. A linear pentapeptide is a quorum-sensing factor required for mazEF-mediated cell death in Escherichia coli. Science 2007; 318:652 - 5; http://dx.doi.org/10.1126/science.1147248; PMID: 17962566
- Vesper O, Amitai S, Belitsky M, Byrgazov K, Kaberdina AC, Engelberg-Kulka H, et al. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell 2011; 147:147 - 57; http://dx.doi.org/10.1016/j.cell.2011.07.047; PMID: 21944167
- Lafleur MD, Qi Q, Lewis K. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob Agents Chemother 2010; 54:39 - 44; http://dx.doi.org/10.1128/AAC.00860-09; PMID: 19841146
- Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 2004; 186:8172 - 80; http://dx.doi.org/10.1128/JB.186.24.8172-8180.2004; PMID: 15576765
- Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. Persister cells and tolerance to antimicrobials. FEMS Microbiol Lett 2004; 230:13 - 8; http://dx.doi.org/10.1016/S0378-1097(03)00856-5; PMID: 14734160
- Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. Persisters: a distinct physiological state of E. coli. BMC Microbiol 2006; 6:53; http://dx.doi.org/10.1186/1471-2180-6-53; PMID: 16768798
- Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science 2004; 305:1622 - 5; http://dx.doi.org/10.1126/science.1099390; PMID: 15308767
- LaFleur MD, Kumamoto CA, Lewis K. Candida albicans biofilms produce antifungal-tolerant persister cells. Antimicrob Agents Chemother 2006; 50:3839 - 46; http://dx.doi.org/10.1128/AAC.00684-06; PMID: 16923951
- Girgis HS, Harris K, Tavazoie S. Large mutational target size for rapid emergence of bacterial persistence. Proc Natl Acad Sci U S A 2012; 109:12740 - 5; http://dx.doi.org/10.1073/pnas.1205124109; PMID: 22802628
- Lewis K. Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol 2008; 322:107 - 31; http://dx.doi.org/10.1007/978-3-540-75418-3_6; PMID: 18453274
- Schumacher MA, Piro KM, Xu W, Hansen S, Lewis K, Brennan RG. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 2009; 323:396 - 401; http://dx.doi.org/10.1126/science.1163806; PMID: 19150849
- Dörr T, Lewis K, Vulić M. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet 2009; 5:e1000760; http://dx.doi.org/10.1371/journal.pgen.1000760; PMID: 20011100
- Vega NM, Allison KR, Khalil AS, Collins JJ. Signaling-mediated bacterial persister formation. Nat Chem Biol 2012; 8:431 - 3; http://dx.doi.org/10.1038/nchembio.915; PMID: 22426114
- Pecota DC, Wood TK. Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J Bacteriol 1996; 178:2044 - 50; PMID: 8606182
- Kim Y, Wood TK. Toxins Hha and CspD and small RNA regulator Hfq are involved in persister cell formation through MqsR in Escherichia coli. Biochem Biophys Res Commun 2010; 391:209 - 13; http://dx.doi.org/10.1016/j.bbrc.2009.11.033; PMID: 19909729
- Fozo EM. New type I toxin-antitoxin families from “wild” and laboratory strains of E. coli: Ibs-Sib, ShoB-OhsC and Zor-Orz. RNA Biol 2012; 9; In press; http://dx.doi.org/10.4161/rna.22568; PMID: 23182878