Abstract
Intracellular transport, processing and stability of mRNA play critical roles in the functional physiology of the cell and defects in these processes are thought to underlie the pathogenesis in a number of neurodegenerative disorders. One of the cellular sites that regulate the mRNA half-life is the processing bodies, the dynamic cytoplasmic structures that represent the non-translating mRNA and the ribonucleoprotein complex that also control the decapping and translation of mRNA. In the present study we explored the possible role of malin E3 ubiquitin ligase in the mRNA decay pathway via the processing bodies. Defects in malin are associated with Lafora disease (LD)—a neurodegenerative disorder characterized by myoclonus seizures. We show here that malin is recruited to the processing bodies and that malin regulates the recruitment of mRNA decapping enzyme Dcp1a by promoting its degradation via the ubiquitin proteasome system. Depletion of malin results in elevated levels of Dcp1a and an altered microRNA-mediated gene silencing activity. Our study suggests that malin is one of the critical regulators of processing bodies and that defects in the mRNA processing might underlie some of the disease symptoms in LD.
Introduction
An emerging theme in the understanding of various neurodegenerative disorders suggests that malfunctions in the mRNA metabolic pathways could be one of the common underlying mechanisms that lead to the neurodegenerative changes.Citation1 For example, in disorders caused by the triplet repeat expansion, the transcripts bearing the expanded repeat form nuclear foci that recruit several splicing factors leading to abnormal splicing of transcripts.Citation2-Citation6 Besides the mRNA, the protein products of some of these transcripts result in the formation of intracellular inclusions which recruit the transactive response binding protein of 43 kDa (TDP-43) and fused-in-sarcoma (FUS) proteins that normally bind to the RNA and are thought to regulate mRNA processing.Citation7-Citation13 Indeed one of the pathological hallmarks of the TDP-43 and FUS in neurodegenerative conditions is their redistribution from the nucleus to the cytoplasm and studies have shown that this nuclear loss could result in the neurodegenerative changes.Citation14-Citation22 Intriguingly, the wild-type version of the polyglutamine disease associated proteins, the ataxin-2 and the huntingtin protein, are involved in the formation and/or the function of processing bodies and stress granules.Citation23,Citation24 Among these, the processing bodies (PBs) represent the ubiquitous form of cytoplasmic structures that house the RNA decay machinery.Citation25,Citation26 PBs are thought to mediate the mRNA silencing by several distinct mechanisms, and the number and position of PBs could vary depending on the cell type and/or the amount of mRNAs degraded via the silencing pathways.Citation26-Citation28 Emerging studies suggest that PBs might help in regulating several neuronal functions, including the learning and memory, thereby underscoring the role of RNA processing in the neurodegenerative disorders.Citation29
Lafora disease (LD) is a fatal form of progressive myoclonus epilepsy characterized by neurodegeneration and the presence of polyglucosan inclusions in the neurons and other affected tissues.Citation30-Citation32 The symptoms of LD include ataxia, dementia and hallucination besides the epileptic seizures.Citation30-Citation32 LD can be caused due to defects in the EPM2A gene that codes for the laforin protein phosphatase or the NHLRC1 gene that codes for the malin E3 ubiquitin ligase.Citation31 Studies on cellular models have shown that laforin and malin interact with each other, and appear to regulate, as a functional complex, several cellular functions such as glycogen metabolism, cellular stress response, and the proteolytic processes.Citation33-Citation41 Laforin is known to associate with the polyribosomes, therefore a possible role for the two proteins in mRNA metabolism was proposed but not tested.Citation42,Citation43 Here we show that malin is recruited to the PBs, and is one of the critical regulators of the PB resident protein, Dcp1a.
Results
Malin is recruited to processing bodies (PBs)
We have shown earlier that laforin and malin show stress-specific subcellular localization; proteasomal blockade results in their recruitment to aggresomes,Citation41 heat shock induces their translocation to the nucleus,Citation38 and the glucose starvation induced only laforin to translocate to the nucleus.Citation44 We extended our studies to other stressors, and found a significant increase in the frequency of cells exhibiting cytoplasmic granule-like foci for the transiently expressed malin when the cells were treated with zinc or puromycin (). Such cytoplasmic foci were also seen in untreated cells but at a lower frequency as compared with cells treated with either zinc or puromycin (). Such effects were seen in more than one cell line (HeLa, COS7, and Neuro2A) (; Fig. S1) and were independent of the fusion tags used for detecting the overexpressed protein (; Fig. S1). Since zinc and puromycin treatments are known to induce PBsCitation45,Citation46 − the subcellular sites where decapping and degradation of mRNAs takes placeCitation25,Citation26—we wanted to check whether the malin-positive cytoplasmic foci are indeed PBs. For this we coexpressed malin with argonaute2 (Ago2), an established PB marker.Citation47 Interestingly, malin showed complete colocalization with the cytoplasmic foci of Ago2 (). Similar observations were made when malin was transiently expressed with the mRNA-decapping enzyme 1A (Dcp1a) − another well-known PB marker (). The cytoplasmic granules of transiently expressed malin also showed colocalization with the endogenous PB markers GW182 and Xrn1 ().Citation48,Citation25 None of the available antibodies detected the endogenous malin in immunofluorescence staining, therefore we were unable to establish whether the endogenous malin gets recruited to the PBs or not. Transiently expressed laforin however did not co-localize with endogenous PB markers (Fig. S2). Therefore laforin was not considered for the rest of the analyses.
Figure 1. Recruitment of malin to the PBs. (A) Representative images showing subcellular localization of transiently expressed GFP-tagged malin in HeLa cells. Note the punctate cytoplasmic granules of malin (identified with arrowheads). (B) Bar diagram showing the frequency of cells showing the punctate cytoplasmic granules (as shown in (A) for the GFP-tagged malin in HeLa cells upon treatment with puromycin or zinc or without any treatment (control), as indicated (***, p < 0.0005; t-test; n = 3). (C) Representative images showing the colocalization of Myc-tagged malin with the PB marker Ago2 (GFP-tagged) or GFP tagged malin with Dcp1a (RFP-tagged) in HeLa cells when transiently coexpressed. (D) Representative images showing the colocalization pattern of transiently expressed malin (Myc-tagged) with endogenous markers for PBs—the GW182 and Xrn1, as indicated. Scale bar, 10 μM (A, C-D).
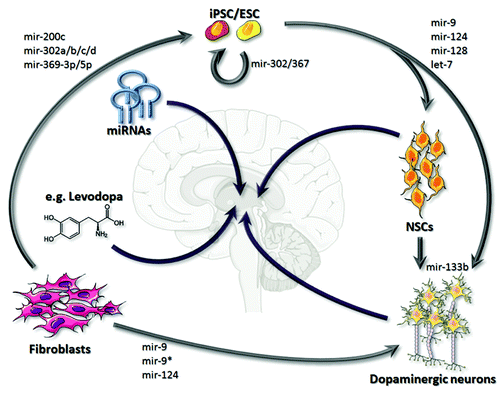
Malin regulates the recruitment of mRNA-decapping enzyme 1A (Dcp1a) to processing bodies
Decapping is a critical step for the degradation of cytoplasmic mRNA, and this process is regulated by a multiprotein complex.Citation49,Citation50 Among them Dcp2 is the primary decapping enzyme whose activity is modulated by a few co-factors including the Dcp1a.Citation49-Citation51 Decapping is necessary for the degradation of mRNAs, both in normal mRNA turnover and in the nonsense- mediated mRNA decay.Citation26,Citation51 Dcp1a, when expressed alone, resulted in the formation of PBs in nearly 100% of the transfected cells (). However, when coexpressed with the wild-type malin there was a significant reduction in the number of cells that showed Dcp1a-positive PBs (). However no such reduction was noted when Dcp1a was co-expressed with catalytically inactive malin mutants (). The malin mutants used are known to cause LD phenotype in recessive condition, and are known to be inactive as ubiquitin ligase.Citation31 It is intriguing to note that cells that strongly expressed wild-type malin showed loss of Dcp1a-postive foci (, panel 1) while no such difference was noted when malin mutants were coexpressed with Dcp1a (, panels 2 and 3). Similar observations were made when the endogenous Dcp1a was visualized using an antibody in the wild-type or the mutant malin expressing cells (Fig. S3). Interestingly, this effect of wild-type malin was restricted to Dcp1a as Ago2 did not show such a difference in localization pattern when co-expressed with the wild-type malin (), suggesting that malin could probably regulate the recruitment of only Dcp1a to the PBs. To investigate this possibility we co-transfected cells with the constructs that code for Dcp1a, Ago2 and either the wild-type malin, or its catalytic inactive mutant (C26S), or an empty vector and the frequency of cells showing PBs positive for both Dcp1a and Ago2 was scored. As shown in , nearly 75% of the transfected cells showed PBs that were positive both for Dcp1a and Ago2 when only these two proteins were transiently co-expressed or when they were co-expressed with the mutant malin. However, coexpression of wild-type malin resulted in a dramatic reduction in number of cells with PBs that were positive both for Dcp1a and Ago2 (), suggesting that malin probably negatively regulates the recruitment of Dcp1a to PBs. To further confirm this observation, we looked at the number of endogenous Dcp1a-positive PBs in cells in which malin was partially knocked down by the RNAi approach. As shown in , there was a significant increase in the number of PBs that were positive for the endogenous Dcp1a when malin was suppressed. Similar observations were made even for the overexpressed Dcp1a but not for overexpressed Ago2 (), suggesting that malin indeed regulates the recruitment of Dcp1a to the PBs. The efficiency of shRNAi knockdown constructs have been established previouslyCitation40,Citation44 and have been validated once again in the present study (Fig. S4A).
Figure 2. Malin regulates the recruitment of Dcp1a to PBs. (A) Bar diagram showing frequency of cells having Dcp1a or Ago2-positive PBs in HeLa cells cotransfected with the expression constructs as indicated (***, p < 0.0005; t-test; n = 3). (B) Representative images showing the colocalization pattern of wild-type malin or its mutant forms—C26S mutation affecting the RING domain, and deletion mutant F216_D233 del (identified as “NHL del”) affecting the carboxyl-terminal NHL domain—when coexpressed with the RFP-tagged Dcp1a in HeLa cells. Both mutants are known to cause LD in recessive condition, and are shown to lack E3 ubiquitin ligase activity.Citation31 Note the absence of Dcp1a positive PBs in the cell that had higher levels of wild-type malin expression and the presence of Dcp1a-positive PBs in the cell that had lower expression of wild-type malin (identified by arrows). No such difference was noted when malin mutants were coexpressed.
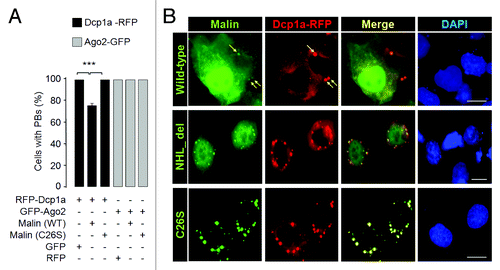
Figure 3. Malin regulates the recruitment of Dcp1a to the PBs. (A) Bar diagram showing frequency of cells having PBs that are positive both for Dcp1a and Ago2 when they were coexpressed with the wild-type malin or its mutant C26S, as indicated (***, p < 0.0005; t-test; n = 3). (B) A bar diagram showing the number of endogenous Dcp1a-positive PBs observed per cell when the cells were transfected with the RNAi construct to knockdown malin, or with the non-silencing RNAi (NS-RNAi) construct, as indicated (***, p < 0.0005; t-test; n = 3). (C) Bar diagram showing number of Dcp1a-, or Ago2-positive PBs observed per cell when Dcp1a or Ago2 was transiently expressed with the knockdown construct for, malin or the non-silencing control RNAi (NS-RNAi) construct, as indicated (***, p < 0.0005; t-test; n = 3). (D) Representative images showing the Dcp1a- or Ago2-positive PBs in HeLa cells transfected with the malin and non-silencing RNAi (NS-RNAi) constructs as indicated. Scale bar, 10 μM.
Malin regulates the cellular levels of Dcp1a by promoting its degradation via the ubiquitin-proteasome system
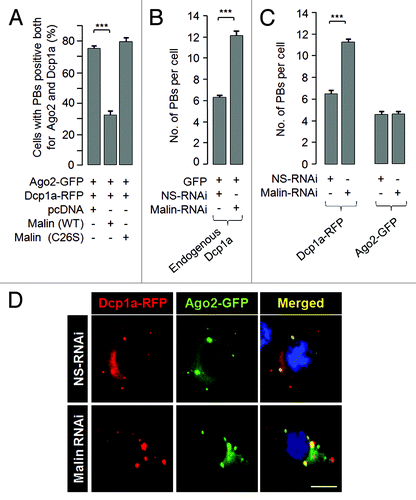
Since malin is an E3 ubiquitin ligase, and since overexpression of malin excludes Dcp1a from the PBs, we speculated that malin might interact with and promote the degradation of Dcp1a. To test this possibility, we looked at the cellular level of endogenous Dcp1a in conditions when either the wild-type or catalytically inactive mutant malin was overexpressed. As shown in , transient expression of wild-type malin (lane 2), but not it’s catalytic inactive mutant (lane 3), was able to decrease the cellular level of endogenous Dcp1a. Further, treating wild-type malin expressing cells with the proteasomal blocker MG132 rescued the loss of endogenous Dcp1a (; compare lane 2 with 4) suggesting that malin probably regulates Dcp1a by promoting its degradation via the proteasome. Similar observations were made with the overexpressed Dcp1a as well; wild-type malin () but not its mutant () was able to decrease the cellular level of overexpressed Dcp1a in a dose-dependent manner. We also found an increase in the cellular level of endogenous Dcp1a when malin was knocked down using the RNAi approach (), suggesting that malin promotes the degradation of Dcp1a. This effect of malin was specific to Dcp1a as the protein levels of two other PB markers GW182, and Ago2 did not alter when malin was partially knocked-down (). Having found that malin regulates the cellular level of Dcp1a, we checked whether Dcp1a levels are altered in the malin knockout mouse. Malin knockout mice develop most of the symptoms/pathology seen in LD.Citation52 As shown in , the level of Dcp1a was significantly higher in the brain tissue of malin knockout mice as compared with the brain tissue of wild-type littermates. These results clearly suggest that loss of malin results in increased levels of Dcp1a both in the cellular and animal models.
Figure 4. Malin promotes the degradation of Dcp1a. (A) Immunoblot showing the cellular level of endogenous Dcp1a in HeLa cells transiently transfected with the indicated expression constructs and probed for indicated proteins. Note the reduced levels of endogenous Dcp1a in the presence of wild-type malin (lane 2) and its rescue when the cells were treated with the proteasomal blocker MG132 (lane 4). The bar diagram shown below depicts the fold difference in the levels of endogenous Dcp1a (as measured by densiometric analysis) upon expressing the wild-type or the mutant form (C26S) of malin and when the cells were treated with MG132 as identified. (***, p < 0.0005; t-test; n = 3). (B and C) Immunoblot analysis for the lysates of the cells transfected with the expression constructs for RFP-tagged Dcp1a, GFP (in lanes 2-to-4) and increasing proportion of constructs (in lanes 3 and 4; identified by the slanting triangle on the top) for the wild-type (WT) (B) or the mutant form (C26S) of malin (C), as indicated. GFP served as the control for transfection efficiency. The bar diagrams shown below depicts the fold difference in the levels of RFP-tagged Dcp1a (as measured by densiometric analysis) when increasing concentration of wild-type (B) or mutant form of malin was coexpressed (C). (**,p < 0.005; ***, p < 0.0005; t-test; n = 3).
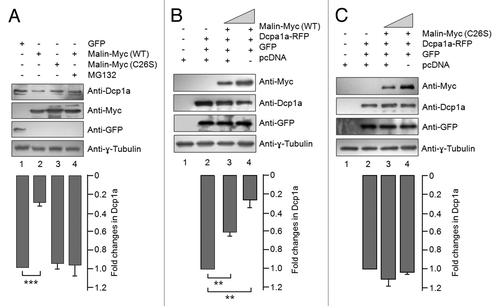
Figure 5. Malin interacts with and ubiquitinates Dcp1a. (A) Immunoblot showing the cellular levels of endogenous Dcp1a, GW182, and the transiently expressed GFP-tagged Ago2 in cells transfected with the RNAi construct for the knockdown of malin or a non-silencing control RNAi construct (NS-RNAi), as indicated. Probing for tubulin served as the loading control. Note that except Dcp1a, no other PB marker showed change in level on malin knockdown. For Ago2, the relative level was evaluated using an expression construct coding for GFP-tagged Ago2 and probed using anti-GFP antibody as indicated. (B) Immunoblot showing the levels of endogenous Dcp1a in two pairs of brain tissues of the malin knockout mice (KO) as compared with their wild-type littermates (WT). (C) Malin physically interacts with endogenous Dcp1a. Myc-tagged malin or an empty vector (pcDNA) was transiently expressed in HeLa cells and the cell lysates were processed for immunoprecipitation using anti-Myc antibody. The immunoprecipitate (IP) or the whole cell lysate (WCL) was probed with the indicated antibodies. The cells were pretreated with MG132 to arrest the degradation of Dcp1a. (D) Malin ubiquitinates Dcp1a. HeLa cells transiently transfected with an expression construct coding for RFP-tagged Dcp1a and wild-type malin or its catalytically inactive mutant form (C26S), treated with the proteasomal blocker MG132 and the cell lysate was immunoprecipitated (IP) using anti-ubiquitin antibody to trap the ubiquitinated proteins. The IP fraction and the whole cell lysates (WCL) were immunoblotted with indicated antibodies. Arrow identifies the expected molecular weight at which the full length Dcp1a-RFP should migrate (105 kDa).
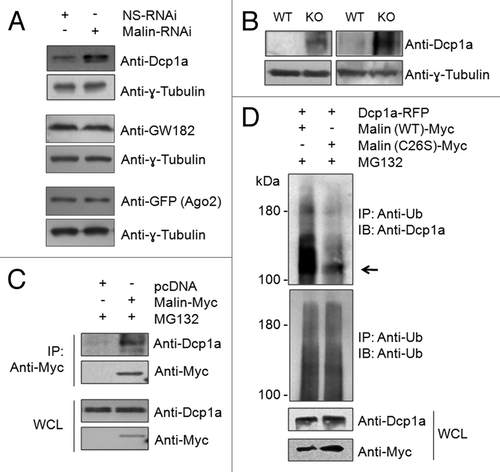
We next checked whether malin physically interacts with Dcp1a to promote its degradation. For this, cells transiently transfected with an expression construct coding for the Myc-tagged malin or an empty vector were treated with proteasome blocker MG132 to prevent Dcp1a degradation, and the cell lysates were processed for immunoprecipitation with anti-Myc antibody and immunoblotting with an antibody against Dcp1a. As shown in , malin was able to pull-down endogenous Dcp1a. We further analyzed whether malin ubiquitinates Dcp1a to promote its degradation. For this, we transiently coexpressed RFP-tagged Dcp1a with either the wild-type malin or its catalytically inactive mutant (C26S). The cells were treated with the proteasomal blocker MG132, the ubiquitinated proteins were pulled down using anti-ubiquitin antibody, and the pulled-down product and the whole cell lysates were immunoblotted to detect the transiently expressed RFP-tagged Dcp1a. As shown in , coexpression of wild-type malin resulted in a marked increase in the high molecular weight ubiquitinated forms for Dcp1a as compared with the set wherein the mutant malin was coexpressed, suggesting that the difference noted for Dcp1a in the ubiquitinated fraction is indeed due to the ubiquitin ligase activity of malin.
Malin regulates mRNA silencing via the microRNA pathway
PBs are multi-protein complexes, and depletion of some of their components is known to compromise the PB-mediated mRNA surveillance. For example, depletion of GW182, Xrn1 or Dcp2 are known to alter the number and size of PBsCitation45,Citation53 and loss of GW182 or the Dcp1-Dcp2 complex affects miRNA-mediated silencing of mRNAs.Citation53 Since malin regulates the cellular level of Dcp1a and its recruitment to the PBs, we wanted to check whether loss of malin would alter the PB function. The PBs are known to be important for the function of the miRNA pathway.Citation47,Citation53 To check for the possible role of malin on miRNA-dependent mRNA silencing, we first knocked down endogenous malin in HeLa cells by RNAi approach and after 24 h, the cells were transfected with a reporter construct coding for an mRNA that has two target sequence sites for the endogenous let-7 miRNA (referred to here as “Let7R-WT”) or with a reporter construct encoding a transcript with a mutant version of the target sequence for let-7 miRNA such that the latter cannot be silenced by the endogenous miRNA (referred to here as “Let7R-MT”). Both constructs had a coding sequence for the Renilla luciferase, hence the activity of this protein was measured to determine the miRNA-mediated mRNA silencing. To normalize the transfection efficiency, a construct that codes for the firefly luciferase was cotransfected. We have used a shRNA construct for the PB marker GW182 (Fig. S4B) and shRNA construct for GFP as the positive and negative controls, respectively. As shown in , the knockdown of GW182 significantly reduced the miRNA-mediated repression of Let7R-WT reporter as noted previously,Citation24,Citation53 while the knockdown of GFP did not show any such effect. Partial knockdown of malin using two independent shRNA constructs resulted in a significant increase in miRNA mediated Let7R-WT reporter repression, suggesting that malin might impair the let-7-dependent mRNA silencing (). There was no change in the activity for the mutant construct Let7R-MT for any of the knockdowns (GW182, malin, GFP) (). We have also checked whether overexpression of malin would inhibit the let-7-mediated mRNA degradation. This we did by co-expressing the wild-type or mutant form of malin with the reporter constructs and measured the luciferase activity. As shown in , overexpression of wild-type malin resulted in a significant reduction in the miRNA- mediated silencing of the Let7R-WT reporter activity as compared with the cells that coexpressed the mutant malin or GFP. No significant change in the activity for the Let7R-MT mutant reporter was noted (). To further establish that the observed difference in the let-7 mediated reporter activity is indeed due to the malin-dependent changes in the Dcp1a level, HeLa cells were transiently transfected with RNAi constructs for malin, Dcp1a, and the Let-7 reporter construct and the luciferase activity was measured. As shown in , the double knockdown significantly rescued the reporter repression seen when only malin was knocked-down. The efficiency of Dcp1a siRNA was established by immunoblotting (Fig. S4C). Taken together our results suggest that malin negatively regulates the let-7 mediated mRNA silencing by modulating the cellular level of Dcp1a.
Figure 6. Malin negatively regulates the let-7 miRNA-mediated gene silencing pathway. (A) Bar diagram showing the let-7 miRNA-mediated repression of a reporter construct. HeLa cells were transfected with shRNAi constructs for GW182, malin, or GFP and at 24 h post-transfection they were split into two sets and transfected again with a construct that code for renilla luciferase and having a target sequence for the endogenous let-7 miRNA (LetR-WT) or a mutated target site for the let-7 miRNA (Let7R-MT) and a construct that code for the firefly luciferase as indicated. The luciferase activity for each set was measured as ratio of firefly to renilla and was normalized with activity of control set (taken as 1 fold) and plotted as fold change in the miRNA-mediated repression of the reporter. Similar assay was done on cells that expressed the wild-type or the mutant form of malin in the place of the knockdown constructs(B), or in conditions of either malin knockdown, or Dcp1a knockdown or in cells were both malin and Dcp1a were knocked down (C) and the activity of the luciferase was measured. Each bar in A, B and C represents the mean of three independent experiments (***, p < 0.0005; t-test). (D) Immunoblot showing the level of Dicer in HeLa cells transfected with shRNAi for malin or a non-silencing shRNAi construct (NS-RNAi), as indicated. (E) Immunoblot showing the level of Dicer in the brain tissue lysates of malin knockout (KO) or their wild-type littermates (WT), as indicated. Two different pairs of animals were used for the immunoblot. The same blot shown in was used to probe with anti-Dicer antibody.
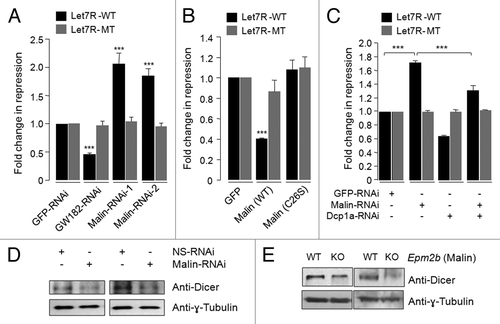
To further probe malin’s role in miRNA-mediated mRNA silencing we looked at the level of one of the known endogenous targets for let-7 miRNA. Dicer, an endoribonuclease of RNase III family,Citation54,Citation55 is known to be regulated by let-7 miRNA.Citation56,Citation57 Studies have shown that overexpression of let-7 reduces the dicer expression significantly both at the protein and mRNA levels and let-7 knockdown has the opposite effect.Citation56 Let-7 target sequences are present in the coding region of the Dicer transcript and therefore, let-7 is thought to have a direct effect on Dicer expression.Citation56,Citation57 If knockdown of malin led to an increase in the let-7 mediated mRNA silencing then dicer levels should also go down. As expected, there was a reduction in the level of Dicer, both at protein () and mRNA levels (Fig. S4D), when malin was transiently knocked down in HeLa cells. A reduction in the level of Dicer protein was also observed in the brain tissue lysates of malin knockout mice (). Taken together these results are strong and compelling enough to suggest that malin negatively regulates the miRNA mediated gene silencing by regulating the level of Dcp1a both at the cellular and organism levels.
Discussion
We show here that transiently expressed malin is recruited to the PBs and this observation has been confirmed by co-staining of malin with the overexpressed as well as endogenous markers for these dynamic structures. The possible role of malin in PBs was further established by using reporter assays and also by evaluating some of the endogenous target genes. We however could not establish the presence of endogenous malin in the PBs since none of the available antibodies against malin work well in the in situ detection.
The Dcp1a is an indispensable member of the mRNA-decapping complex that recruits mRNAs to the PBs for the translational repression and their eventual degradation via the miRNA pathway.Citation58,Citation59 It is known that the individual components of PBs, including the decapping enzymes, shuttle dynamically within the cytoplasmic pool and this dynamics are regulated by some critical cofactors.Citation60-Citation62 For example, phosphorylation regulates the localization of Ago2 and Dcp1a to the PBs.Citation63,Citation64 In addition, Ago2 localization is also regulated by mLin41, an E3 ubiquitin ligase, by modulating the half-life of Ago2.Citation65 Our observations that malin’s cellular level shows an inverse correlation with the Dcp1a-positive PBs (number of cells with PBs or the number of PBs per cell) proposes malin to be one of the regulators of Dcp1a. Since the loss or the overexpression of malin did not alter the level and localization of Ago2 (and other makers tested) to PBs, it could be argued that malin regulates the targeting of specific components of PBs, like the Dcp1a, but not the formation of PBs itself. This notion was supported by our demonstration that malin physically interacts with and promotes the degradation of Dcp1a via the ubiquitin-proteasome pathway. Increased level of Dcp1a was also observed in the brain tissue lysates of malin deficient mice. Thus, malin appears to regulate the PB-specific functions of Dcp1a by modulating its turnover—a mechanism very similar to the one proposed for the mLin41 E3 ligase on the stability and recruitment of Ago2 to PBs; overexpression of mLin41 partially rescued the silencing of let-7 miRNA targets by antagonizing Ago2.Citation65 In this regard it is of interest to note that Trim32, a malin-like ubiquitin ligase,Citation66 interacts with the Argonaute 1 protein and regulates the miRNA-mediated inhibitory mechanisms.Citation67 Since malin and Trim32 are phylogenetically related, and since both share some substrates for ubiquitination, it appears that the RING family ubiquitin ligases have functional roles in the miRNA-mediated gene silencing. Consistent with this model we have shown here that loss of malin resulted in an enhanced repression of a reporter for the let-7 miRNA and decreased levels of Dicer, an endogenous target of let-7, possibly due to the increased level of Dcp1a. The contrasting levels of Dcp1a and Dicer were also observed in the brain tissues of malin knockout mice—an animal model for LD that replicated most of the pathological changes seen in human LD.Citation52 In this regard it is intriguing to note that loss of Dicer in the brain results in neurodegeneration, the hyperphosphorylation of tau protein, and symptoms like seizures and ataxia,Citation68-Citation71 and these defining neuropathological changes are also seen in LD.Citation30-Citation32,Citation72 Therefore it is tempting to speculate that dysregulation in mRNA metabolism may underlie some of the symptoms in LD, and these could be secondary to the increased levels of Dcp1a or the decreased levels of Dicer. It is equally likely that malin may also promote the degradation of an additional PB associated factor not tested in the present study, and loss of which—along with Dcp1a—could result in the deficient miRNA activity and the neurodegeneration in LD. Indeed defects in the dicer-micro RNA network have been implicated in etiology of several neurodegenerative disorders like Parkinson disease, Alzheimer disease and several polyglutamine disorders including Huntington’s disease.Citation68-Citation70
The emerging knowledge on the role of PBs in the neuronal functions suggests that deficits in PBs may affect several functions of the neurons. For example, PBs have been identified in neuronal dendrites and near synapses and they have been implicated in the signal-dependent mRNA trafficking, processing and/or localized protein synthesis.Citation29,Citation73 Given the critical roles of PBs in neuronal processes, and the suspected role of LD protein in the mRNA regulation,Citation42,Citation44 the present set of findings call for further studies on malin in the mRNA processing, and its specific role in the LD neuropathology.
Materials and Methods
Cell culture, transfections and treatments
All experiments were performed in HeLa cells unless otherwise stated. The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 25 mM of glucose and 10% (vol/vol) fetal bovine serum. Cells were transfected using Polyfect reagent (Qiagen) as recommended by the manufacturer. Duration and concentration of various treatments given were as follows unless mentioned otherwise: MG132 (12 h, 20 μM), ZnCl2 (2 h, 100 µM) and puromycin (1 h, 100 μg/ml).
Expression constructs and chemicals
The mammalian expression constructs that code for the wild-type or mutant form of malin are previously described.Citation41,Citation42 The construct that code for red fluorescent protein (RFP)-tagged Dcp1a was generously provided by Ken Fujimura (University of Tokyo, Japan). The shRNA knockdown constructs (RNAi) for malin, GFP and the non-silencing RNAi (NS-RNAi) were purchased from Open Biosystems USA, and have been validated in our previous studiesCitation40,Citation44 and are shown here as well (Fig. S4). The shRNAi construct for GW182 and the siRNA duplex for Dcp1a were obtained from Sigma-Aldrich (India) Pvt Ltd and their efficieny has been confirmed (Fig. S4). The miRNA reporter constructs Let7R-WT and Let7R-MT, and the one that code for green fluorescent protein (GFP)-tagged argonaute-2 (Ago2) were obtained from Addgene (Plasmid ID 11324, 11325 and 11590, respectively). All the chemicals were purchased from Sigma-Aldrich Pvt Ltd unless stated otherwise.
Immunostaining and antibodies
Cells, grown on a gelatin-coated sterile glass coverslip, were processed for immunofluorescence microscopy as described earlier.Citation40,Citation41 Briefly, cells were fixed with 4% paraformaldehyde, permeabilized with TritonX-100 (0.05%) and subsequently incubated with primary and secondary antibodies. For staining the nuclei, cells were treated with 10 µM of 4',6-diamidino-2-phenylindole (DAPI) for 5 min. Immunofluorescence images were captured using an epifluorescence microscope (Nikon Eclipse 80i, Japan, 40× oil objective) and the images were assembled using Adobe Photoshop. The following antibodies were used in the present study: Anti-Myc and anti-GFP (Roche India); anti-Myc (Cell Signaling Technologies); anti-Xrn1, anti-Dcp1a, anti-Dicer and anti-tubulin (Sigma-Aldrich); anti-GW182 (generously gifted by Dr. Marvin J Fritzler); and anti-polyubiquitin (Enzo Life Sciences). Secondary antibodies were obtained from Jackson Immuno Research Inc.
Cell counting
Cell counting was performed in a blinded manner. Each experiment was done in triplicates and 300 cells were counted in each experimental set.
Immunoprecipitation
Immunoprecipitation was performed as detailed earlier.Citation72 Briefly, cell lysates were pre-cleared by incubating with protein A agarose beads (Bangalore Genei, India) for 2 h at 4°C, followed by incubation with anti-Myc antibody (CST) overnight at 4°C. Finally the protein A agarose beads were added and incubated for 2-to-3 h at 4°C. The beads were then washed 3 times and the bound proteins were eluted by boiling the beads with SDS sample buffer.
Malin knockout mouse tissue lysates
Brain tissues of malin-deficient mice and their wild-type littermates,Citation52 obtained in frozen condition from Dr. Berge Minassian (The Hospital for Sick Children, Canada), were homogenized and processed for immunoblotting as reported earlier.Citation72
Immunoblotting
For immunoblotting, protein samples were resolved on SDS-PAGE gel and transferred onto 0.22 µm nitrocellulose membrane (MDI). After blocking with 5% non-fat modified milk (G Biosciences), the membranes were processed for sequential incubations with primary antibody followed by incubation with HRP- tagged secondary antibody, as reported previously.Citation44 Immunoreactive proteins on the filter were visualized using a chemiluminscent substrate (SuperSignal West PICO, Pierce).
Ubiquitination assay
The cells were treated with 10 µM MG132 at 24 h post-transfection for 12 h, and the lysates were then processed for immunoprecipitation with anti-polyubiquitin antibody and for immunoblotting as described.
Micro-RNA reporter assay
The reporter assay as described by Savas et al.Citation24 was followed to measure the miRNA activity. Briefly, the cells were transfected with shRNAi construct to knockdown GW182, malin, GFP (control) or with siRNA for Dcp1a or with an expression construct for the wild-type or mutant form of malin or GFP. At 24 h post-transfection, the cells were split into two sets and transfected again with either the Let7R-WT construct having a target of endogenous let-7 miRNA and coding for renilla luciferase or with the Let7R-MT construct having a mutated target site for let-7 miRNA and coding for renilla luciferase. In each set a construct that code for the firefly luciferase was cotransfected to serve as transfection control. The luciferase activity for each set was measured as ratio of firefly to renilla and was normalized with activity of control set and plotted as fold change in the miRNA-mediated repression of the reporter.
Statistical analysis
Data were analyzed by two-tailed, unpaired Student’s t-test using GraphPad software. Differences were considered significant at p < 0.05 and the same was denoted by *, ** and *** when the p-value is less than 0.05, 0.005 and 0.0005, respectively.
| Abbreviations: | ||
| LD | = | Lafora Disease |
| PBs | = | Processing bodies |
| Ago2 | = | argonaute-2 |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| GFP | = | green fluorescent protein |
| RFP | = | red fluorescent protein |
| TDP-43 | = | transactive response binding protein of 43 kDa |
| FUS | = | fused-in-sarcoma |
| GW182 | = | glycine-tryptophan protein of 182 kDa |
Additional material
Download Zip (676.1 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Ken Fujimura, University of Tokyo, Japan, for providing us the RFP-tagged Dcp1a expression construct, Dr. Marvin J. Fritzler, University of Calgary, Canada, for the gift of GW182 antibody, Dr. Berge Minassian, Hospital for Sick Children, Canada, for the tissue lysates of the malin knockout mice, and Dr Amitabha Bandyopadhyay, IIT Kanpur, for extending his laboratory facilities. We also thank the anonymous reviewers for their comments and suggestions on an earlier version of this article. This work was supported by a sponsored research grant from the Department of Atomic Energy, Government of India, to S.G. S.G. is a Ramanna Fellow and Gill-Joy Chair Professor at IIT Kanpur. S.S. and P.K.S. were supported by a senior research fellowship from the Council of Scientific and Industrial Research, Government of India.
Supplemental Material
Supplemental material may be found here:
http://www.landesbioscience.com/journals/rnabiology/article/22708/
Related Research Data
References
- Renoux AJ, Todd PK. Neurodegeneration the RNA way. Prog Neurobiol 2012; 97:173 - 89; http://dx.doi.org/10.1016/j.pneurobio.2011.10.006; PMID: 22079416
- Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci 2006; 29:259 - 77; http://dx.doi.org/10.1146/annurev.neuro.29.051605.113014; PMID: 16776586
- Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A 1997; 94:7388 - 93; http://dx.doi.org/10.1073/pnas.94.14.7388; PMID: 9207101
- Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet 2004; 41:e43; http://dx.doi.org/10.1136/jmg.2003.012518; PMID: 15060119
- Chen IC, Lin HY, Lee GC, Kao SH, Chen CM, Wu YR, et al. Spinocerebellar ataxia type 8 larger triplet expansion alters histone modification and induces RNA foci. BMC Mol Biol 2009; 10:9; http://dx.doi.org/10.1186/1471-2199-10-9; PMID: 19203395
- Dahm R, Macchi P. Human pathologies associated with defective RNA transport and localization in the nervous system. Biol Cell 2007; 99:649 - 61; http://dx.doi.org/10.1042/BC20070045; PMID: 17939777
- Doi H, Mitsui K, Kurosawa M, Machida Y, Kuroiwa Y, Nukina N. Identification of ubiquitin-interacting proteins in purified polyglutamine aggregates. FEBS Lett 2004; 571:171 - 6; http://dx.doi.org/10.1016/j.febslet.2004.06.077; PMID: 15280037
- Doi H, Koyano S, Suzuki Y, Nukina N, Kuroiwa Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci Res 2010; 66:131 - 3; http://dx.doi.org/10.1016/j.neures.2009.10.004; PMID: 19833157
- Schwab C, Arai T, Hasegawa M, Yu S, McGeer PL. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J Neuropathol Exp Neurol 2008; 67:1159 - 65; http://dx.doi.org/10.1097/NEN.0b013e31818e8951; PMID: 19018245
- Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 2007; 114:221 - 9; http://dx.doi.org/10.1007/s00401-007-0261-2; PMID: 17653732
- Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 2007; 61:435 - 45; http://dx.doi.org/10.1002/ana.21154; PMID: 17469117
- Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 2007; 1184:284 - 94; http://dx.doi.org/10.1016/j.brainres.2007.09.048; PMID: 17963732
- Lin WL, Dickson DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol 2008; 116:205 - 13; http://dx.doi.org/10.1007/s00401-008-0408-9; PMID: 18607609
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130 - 3; http://dx.doi.org/10.1126/science.1134108; PMID: 17023659
- Belzil VV, Valdmanis PN, Dion PA, Daoud H, Kabashi E, Noreau A, et al, S2D team. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 2009; 73:1176 - 9; http://dx.doi.org/10.1212/WNL.0b013e3181bbfeef; PMID: 19741216
- Corrado L, Del Bo R, Castellotti B, Ratti A, Cereda C, Penco S, et al. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J Med Genet 2010; 47:190 - 4; http://dx.doi.org/10.1136/jmg.2009.071027; PMID: 19861302
- Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009; 323:1205 - 8; http://dx.doi.org/10.1126/science.1166066; PMID: 19251627
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323:1208 - 11; http://dx.doi.org/10.1126/science.1165942; PMID: 19251628
- Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, et al. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging 2011; 32:2323 - , e27-40; http://dx.doi.org/10.1016/j.neurobiolaging.2010.06.010; PMID: 20674093
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ Jr., et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 2010; 19:4160 - 75; http://dx.doi.org/10.1093/hmg/ddq335; PMID: 20699327
- Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, Strong MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res 2009; 1249:202 - 11; http://dx.doi.org/10.1016/j.brainres.2008.10.021; PMID: 19046946
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 2010; 5:e13250; http://dx.doi.org/10.1371/journal.pone.0013250; PMID: 20948999
- Nonhoff U, Ralser M, Welzel F, Piccini I, Balzereit D, Yaspo ML, et al. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell 2007; 18:1385 - 96; http://dx.doi.org/10.1091/mbc.E06-12-1120; PMID: 17392519
- Savas JN, Makusky A, Ottosen S, Baillat D, Then F, Krainc D, et al. Huntington’s disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc Natl Acad Sci U S A 2008; 105:10820 - 5; http://dx.doi.org/10.1073/pnas.0800658105; PMID: 18669659
- Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003; 300:805 - 8; http://dx.doi.org/10.1126/science.1082320; PMID: 12730603
- Cougot N, Babajko S, Séraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. J Cell Biol 2004; 165:31 - 40; http://dx.doi.org/10.1083/jcb.200309008; PMID: 15067023
- Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 2007; 8:9 - 22; http://dx.doi.org/10.1038/nrm2080; PMID: 17183357
- Teixeira D, Sheth U, Valencia-Sanchez MA, Brengues M, Parker R. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 2005; 11:371 - 82; http://dx.doi.org/10.1261/rna.7258505; PMID: 15703442
- Zeitelhofer M, Karra D, Macchi P, Tolino M, Thomas S, Schwarz M, et al. Dynamic interaction between P-bodies and transport ribonucleoprotein particles in dendrites of mature hippocampal neurons. J Neurosci 2008; 28:7555 - 62; http://dx.doi.org/10.1523/JNEUROSCI.0104-08.2008; PMID: 18650333
- Ganesh S, Puri R, Singh S, Mittal S, Dubey D. Recent advances in the molecular basis of Lafora’s progressive myoclonus epilepsy. J Hum Genet 2006; 51:1 - 8; http://dx.doi.org/10.1007/s10038-005-0321-1; PMID: 16311711
- Singh S, Ganesh S. Lafora progressive myoclonus epilepsy: a meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum Mutat 2009; 30:715 - 23; http://dx.doi.org/10.1002/humu.20954; PMID: 19267391
- Ramachandran N, Girard JM, Turnbull J, Minassian BA. The autosomal recessively inherited progressive myoclonus epilepsies and their genes. Epilepsia 2009; 50:Suppl 5 29 - 36; http://dx.doi.org/10.1111/j.1528-1167.2009.02117.x; PMID: 19469843
- Fernández-Sánchez ME, Criado-García O, Heath KE, García-Fojeda B, Medraño-Fernández I, Gomez-Garre P, et al. Laforin, the dual-phosphatase responsible for Lafora disease, interacts with R5 (PTG), a regulatory subunit of protein phosphatase-1 that enhances glycogen accumulation. Hum Mol Genet 2003; 12:3161 - 71; http://dx.doi.org/10.1093/hmg/ddg340; PMID: 14532330
- Vilchez D, Ros S, Cifuentes D, Pujadas L, Vallès J, García-Fojeda B, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci 2007; 10:1407 - 13; http://dx.doi.org/10.1038/nn1998; PMID: 17952067
- Vernia S, Solaz-Fuster MC, Gimeno-Alcañiz JV, Rubio T, García-Haro L, Foretz M, et al. AMP-activated protein kinase phosphorylates R5/PTG, the glycogen targeting subunit of the R5/PTG-protein phosphatase 1 holoenzyme, and accelerates its down-regulation by the laforin-malin complex. J Biol Chem 2009; 284:8247 - 55; http://dx.doi.org/10.1074/jbc.M808492200; PMID: 19171932
- Worby CA, Gentry MS, Dixon JE. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J Biol Chem 2008; 283:4069 - 76; http://dx.doi.org/10.1074/jbc.M708712200; PMID: 18070875
- Solaz-Fuster MC, Gimeno-Alcañiz JV, Ros S, Fernandez-Sanchez ME, Garcia-Fojeda B, Criado Garcia O, et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum Mol Genet 2008; 17:667 - 78; http://dx.doi.org/10.1093/hmg/ddm339; PMID: 18029386
- Sengupta S, Badhwar I, Upadhyay M, Singh S, Ganesh S. Malin and laforin are essential components of a protein complex that protects cells from thermal stress. J Cell Sci 2011; 124:2277 - 86; http://dx.doi.org/10.1242/jcs.082800; PMID: 21652633
- Vernia S, Rubio T, Heredia M, Rodríguez de Córdoba S, Sanz P. Increased endoplasmic reticulum stress and decreased proteasomal function in lafora disease models lacking the phosphatase laforin. PLoS One 2009; 4:e5907; http://dx.doi.org/10.1371/journal.pone.0005907; PMID: 19529779
- Garyali P, Siwach P, Singh PK, Puri R, Mittal S, Sengupta S, et al. The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system. Hum Mol Genet 2009; 18:688 - 700; http://dx.doi.org/10.1093/hmg/ddn398; PMID: 19036738
- Mittal S, Dubey D, Yamakawa K, Ganesh S. Lafora disease proteins malin and laforin are recruited to aggresomes in response to proteasomal impairment. Hum Mol Genet 2007; 16:753 - 62; http://dx.doi.org/10.1093/hmg/ddm006; PMID: 17337485
- Ganesh S, Agarwala KL, Ueda K, Akagi T, Shoda K, Usui T, et al. Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum Mol Genet 2000; 9:2251 - 61; http://dx.doi.org/10.1093/oxfordjournals.hmg.a018916; PMID: 11001928
- Minassian BA, Andrade DM, Ianzano L, Young EJ, Chan E, Ackerley CA, et al. Laforin is a cell membrane and endoplasmic reticulum-associated protein tyrosine phosphatase. Ann Neurol 2001; 49:271 - 5; http://dx.doi.org/10.1002/1531-8249(20010201)49:2<271::AID-ANA52>3.0.CO;2-D; PMID: 11220751
- Singh PK, Singh S, Ganesh S. The laforin-malin complex negatively regulates glycogen synthesis by modulating cellular glucose uptake via glucose transporters. Mol Cell Biol 2012; 32:652 - 63; http://dx.doi.org/10.1128/MCB.06353-11; PMID: 22124153
- Eulalio A, Behm-Ansmant I, Schweizer D, Izaurralde E. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol Cell Biol 2007; 27:3970 - 81; http://dx.doi.org/10.1128/MCB.00128-07; PMID: 17403906
- Blumenthal J, Ginzburg I. Zinc as a translation regulator in neurons: implications for P-body aggregation. J Cell Sci 2008; 121:3253 - 60; http://dx.doi.org/10.1242/jcs.033266; PMID: 18799791
- Liu J, Rivas FV, Wohlschlegel J, Yates JR 3rd, Parker R, Hannon GJ. A role for the P-body component GW182 in microRNA function. Nat Cell Biol 2005; 7:1261 - 6; http://dx.doi.org/10.1038/ncb1333; PMID: 16284623
- Eystathioy T, Jakymiw A, Chan EK, Séraphin B, Cougot N, Fritzler MJ. The GW182 protein colocalizes with mRNA degradation associated proteins hDcp1 and hLSm4 in cytoplasmic GW bodies. RNA 2003; 9:1171 - 3; http://dx.doi.org/10.1261/rna.5810203; PMID: 13130130
- Coller J, Parker R. Eukaryotic mRNA decapping. Annu Rev Biochem 2004; 73:861 - 90; http://dx.doi.org/10.1146/annurev.biochem.73.011303.074032; PMID: 15189161
- Franks TM, Lykke-Andersen J. The control of mRNA decapping and P-body formation. Mol Cell 2008; 32:605 - 15; http://dx.doi.org/10.1016/j.molcel.2008.11.001; PMID: 19061636
- Lykke-Andersen J. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol Cell Biol 2002; 22:8114 - 21; http://dx.doi.org/10.1128/MCB.22.23.8114-8121.2002; PMID: 12417715
- Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, Draginov AG, et al. Glycogen hyperphosphorylation underlies lafora body formation. Ann Neurol 2010; 68:925 - 33; http://dx.doi.org/10.1002/ana.22156; PMID: 21077101
- Rehwinkel J, Behm-Ansmant I, Gatfield D, Izaurralde E. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA 2005; 11:1640 - 7; http://dx.doi.org/10.1261/rna.2191905; PMID: 16177138
- Fierro-Monti I, Mathews MB. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem Sci 2000; 25:241 - 6; http://dx.doi.org/10.1016/S0968-0004(00)01580-2; PMID: 10782096
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001; 409:363 - 6; http://dx.doi.org/10.1038/35053110; PMID: 11201747
- Tokumaru S, Suzuki M, Yamada H, Nagino M, Takahashi T. let-7 regulates Dicer expression and constitutes a negative feedback loop. Carcinogenesis 2008; 29:2073 - 7; http://dx.doi.org/10.1093/carcin/bgn187; PMID: 18700235
- Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci U S A 2008; 105:14879 - 84; http://dx.doi.org/10.1073/pnas.0803230105; PMID: 18812516
- Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol 2005; 7:719 - 23; http://dx.doi.org/10.1038/ncb1274; PMID: 15937477
- Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev 2006; 20:1885 - 98; http://dx.doi.org/10.1101/gad.1424106; PMID: 16815998
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, et al. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 2005; 169:871 - 84; http://dx.doi.org/10.1083/jcb.200502088; PMID: 15967811
- Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol 2007; 431:61 - 81; http://dx.doi.org/10.1016/S0076-6879(07)31005-7; PMID: 17923231
- Franks TM, Lykke-Andersen J. The control of mRNA decapping and P-body formation. Mol Cell 2008; 32:605 - 15; http://dx.doi.org/10.1016/j.molcel.2008.11.001; PMID: 19061636
- Zeng Y, Sankala H, Zhang X, Graves PR. Phosphorylation of Argonaute 2 at serine-387 facilitates its localization to processing bodies. Biochem J 2008; 413:429 - 36; http://dx.doi.org/10.1042/BJ20080599; PMID: 18476811
- Rzeczkowski K, Beuerlein K, Müller H, Dittrich-Breiholz O, Schneider H, Kettner-Buhrow D, et al. c-Jun N-terminal kinase phosphorylates DCP1a to control formation of P bodies. J Cell Biol 2011; 194:581 - 96; http://dx.doi.org/10.1083/jcb.201006089; PMID: 21859862
- Rybak A, Fuchs H, Hadian K, Smirnova L, Wulczyn EA, Michel G, et al. The let-7 target gene mouse lin-41 is a stem cell specific E3 ubiquitin ligase for the miRNA pathway protein Ago2. Nat Cell Biol 2009; 11:1411 - 20; http://dx.doi.org/10.1038/ncb1987; PMID: 19898466
- Romá-Mateo C, Moreno D, Vernia S, Rubio T, Bridges TM, Gentry MS, et al. Lafora disease E3-ubiquitin ligase malin is related to TRIM32 at both the phylogenetic and functional level. BMC Evol Biol 2011; 11:225; http://dx.doi.org/10.1186/1471-2148-11-225; PMID: 21798009
- Schwamborn JC, Berezikov E, Knoblich JA. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 2009; 136:913 - 25; http://dx.doi.org/10.1016/j.cell.2008.12.024; PMID: 19269368
- Schaefer A, O’Carroll D, Tan CL, Hillman D, Sugimori M, Llinas R, et al. Cerebellar neurodegeneration in the absence of microRNAs. J Exp Med 2007; 204:1553 - 8; http://dx.doi.org/10.1084/jem.20070823; PMID: 17606634
- Hébert SS, Papadopoulou AS, Smith P, Galas MC, Planel E, Silahtaroglu AN, et al. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum Mol Genet 2010; 19:3959 - 69; http://dx.doi.org/10.1093/hmg/ddq311; PMID: 20660113
- Tao J, Wu H, Lin Q, Wei W, Lu XH, Cantle JP, et al. Deletion of astroglial Dicer causes non-cell-autonomous neuronal dysfunction and degeneration. J Neurosci 2011; 31:8306 - 19; http://dx.doi.org/10.1523/JNEUROSCI.0567-11.2011; PMID: 21632951
- Cuellar TL, Davis TH, Nelson PT, Loeb GB, Harfe BD, Ullian E, et al. Dicer loss in striatal neurons produces behavioral and neuroanatomical phenotypes in the absence of neurodegeneration. Proc Natl Acad Sci U S A 2008; 105:5614 - 9; http://dx.doi.org/10.1073/pnas.0801689105; PMID: 18385371
- Puri R, Suzuki T, Yamakawa K, Ganesh S. Hyperphosphorylation and aggregation of Tau in laforin-deficient mice, an animal model for Lafora disease. J Biol Chem 2009; 284:22657 - 63; http://dx.doi.org/10.1074/jbc.M109.009688; PMID: 19542233
- Cougot N, Bhattacharyya SN, Tapia-Arancibia L, Bordonné R, Filipowicz W, Bertrand E, et al. Dendrites of mammalian neurons contain specialized P-body-like structures that respond to neuronal activation. J Neurosci 2008; 28:13793 - 804; http://dx.doi.org/10.1523/JNEUROSCI.4155-08.2008; PMID: 19091970