Abstract
Methyl-6-adenosine (m6A) has been hypothesized to exist since the 1970s,Citation1 but little has been known about the specific RNAs, or sites within them, that are affected by this RNA modification. Here, we report that recent work has shown RNA modifications like m6A, collectively called the “epitranscriptome,” are a pervasive feature of mammalian cells and likely play a role in development and disease. An enrichment of m6A near the last CDS of thousands of genes has implicated m6A in transcript processing, translational regulation and potentially a mechanism for regulating miRNA maturation. Also, because the sites of m6A show strong evolutionary conservation and have been replicated in nearly identical sites between mouse and human, strong evolutionary pressures are likely being maintained for this mark.Citation2,Citation3 Finally, we note that m6A is one of over 100 modifications of RNA that have been reported,Citation4 and with the combination of high-throughput, next-generation sequencing (NGS) techniques, immunoprecipitation with appropriate antibodies and splicing-aware peak-finding, the dynamics of the epitranscriptome can now be mapped and characterized to discern their specific cellular roles.
Background
Dynamic regulation of DNA bases in epigenetics has been established since 1925, and it is known to play a critical role in embryogenesis, imprinting, development and cancer.Citation5 Indeed, incomplete reprogramming of epigenetic marks from cells transformed during somatic cell nuclear transfer (SCNT) is a factor in the inability of these cells to be completely pluripotent.Citation6 The role of epigenetics is so important to biology that some have referred to these bases as the extra fifth and sixth genetic bases of the genetic code.Citation7,Citation8 However, RNA modifications, collectively referred to as “epitranscritpomes,” are a much less-studied area. This is partially due to the fact that RNA degrades quickly and that there are no easy methods to find modified RNA bases, whereas de-amination methods, such as bi-sulfite treatment, work very well in DNA.
Early estimates of m6A frequency were in the range of 0.1–0.4%,Citation9-Citation11 but they required significant effort and work. For example, one of the first studies of m6A used the adenovirus type 2 (Ad-2) system,Citation12 in which viral mRNAs are generated from a large common nuclear precursor RNA in the nucleus. The abundance of viral RNAs transcribed from multiple copies of the virons made it possible to investigate the timing of N-6-methylation on adenosine and the flow of m6A from nuclear precursor RNA to mature cytoplasmic mRNA by kinetic studies. But, this required 3H-methyl-labeled methionine and 14C-labeled uridine, which then were pulsed in intervals and RNA was measured by quantitative RNA hybridization. A comparison of the nuclear dwell time of precursor RNA compared with the mRNA levels in the cytoplasm showed that the ratio of 3H to 14C was significantly higher in cytoplasmic RNA than in nuclear RNA, indicating that m6A was added to transcripts before splicing. These preliminary results implicated a role for m6A in RNA processing or stabilization, or both, during the biogenesis of mRNA.
Even though early papers suggested m6A as a fundamental aspect of RNA biology, little work was done in the field for several decades. This is partly because the known methods to identify m6A sites were limited to a pre-ordained focus on a single transcript, required radio-labeled bases and it was thought that RNA modifications were primarily in rRNACitation13 and tRNA.Citation14 However, in 2011, the FTO gene (associated with fat mass and obesity) was discovered as the enzyme responsible for the oxidative demethylation of m6A.Citation15 The FTO gene is a member of the 2-oxoglutarate (2OG)-dependent oxygenases that can catalyze the de-methylation of RNA and DNA.Citation16,Citation17 Related to FTO is METTL3, a 70-kDA subunit that forms part of the N6-adenosine methyltransferase, and it is thought that METTL3 is responsible for methylating regular adenosines into m6A.Citation18 Notably, additional METTL3 homologs exist in the human and mouse genomes and could also function as RNA adenosine methyltransferases, or in concert with other RNA-binding proteins. For example, the ALKBH5 gene was recently found to demethylate m6A in vitro and in vivo, and knockdown of ALKBH5 not only resulted in higher m6A levels but also altered mRNA export levels and reduced fertility in mice.Citation19 The characterization and discovery of the enzymes responsible for m6A sparked a renewed interest in this particular RNA modification.
Global Methods for Epitranscriptomics
The emergence of a new m6A-specific antibody in 2011 enabled researchers to perform ChIP-seq-like experiments to identify potential m6A sites throughout the transcriptome, also called the “RNA methylome,” which offers a glimpse into the full epitranscriptome.
In 2012, two global methods were independently developed by Meyer et al. (MeRIP-Seq)Citation2 and by Dominissini et al. (m6A-Seq)Citation3 demonstrating that immunoprecipitation and next-generation sequencing (NGS) can be used to efficiently and robustly localize m6A sites in RNA.
In MeRIP-Seq and m6A-Seq, fragmented RNA is immunoprecipitated using an m6A-specific antibody. There are currently two available antibodies, from Synaptic Systems and New England Biolabs (NEB), and both appear to have equal performance.Citation20 The IP RNA fragments are then sequenced, along with the non-IP sample (control), to be analyzed using peak-detection software. One publically available tool, MeRIPPeR (www.sourceforge.net/projects/meripper/), maps reads to windows across the genome and uses Fisher’s exact test to find statistically significant enrichment of reads in the IP vs. the control samples. Windows are then merged into possible peak locations for m6A sites. Both data sets found an enrichment of sites near the stop codon and the beginning of the 3′ UTR, and some evidence was reported of an enrichment of m6A near transcription start site (TSS).Citation2,Citation3,Citation20
While IP-based methods have been successful so far, they are limited insofar as they cannot directly reveal the precise adenosine within a peak, and other methods are now being developed to advance the field. In particular, there are two proof-of-principle studies that have shown that m6A can also be directly detected on the native molecules. The first uses a single-molecule sequencing approach to observe a HIV-based reverse transcriptase as it makes cDNA.Citation20 As bases are incorporated to form the complementary strand, a kinetic signature of each base context can be observed. It has been shown that the steric change of the single methyl group in m6A is large enough to observe a significantly different kinetic signature of base incorporation between m6A vs. A. Second, this same principle can be applied to any single-molecule monitoring technology, and direct RNA sequencing for RNA within a protein nanopore has also been reported.Citation21 As these technologies improve, global methods for m6A detection will likely all become accurate to the exact base, which will help in the analysis and contextualization of these data.
Distribution of m6A across the genome
While MeRIP-Seq and m6A-Seq have mapped putative m6A sites across the transcriptome, their precise biological function and the mechanism of m6A site-specificity is not yet known. The enrichment of m6A sites near the stop codon and in the 3′ UTR suggest that it may be involved in transcription or translation regulation, but experimental evidence of this interaction has not yet been generated. Similarly, the discovery of the FTO, ALKBH5 and METTL3 genes provide insight into how m6A sites are directly manipulated, but these proteins are likely part of a larger network that facilitates dynamic changes in m6A sites in response to environmental changes and cellular stimuli. For example, m6A levels differ between tissues (as demonstrated by immunoblot experiments) with higher levels in the brain, liver and kidney cells, while lower in the heart and lung.Citation2 Also, m6A levels have been shown to increase with neuronal development in mouse tissues, which implies that m6A sites and levels have a defined spatio-temporal trajectory,Citation2 and current evidence shows that m6A levels are more important as an animal ages.
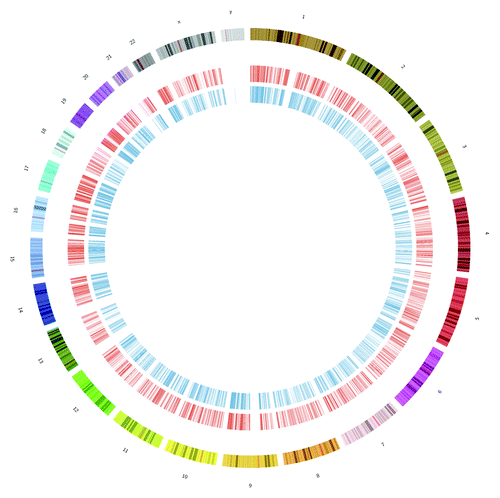
All existing data show that m6A sites cover the majority of the genome (), but there is strong evidence that the sites are not randomly distributed. The enrichment of the peaks near the stop codon and 5′ end of the 3′ UTR has been replicated in both studies,Citation2,Citation3 and the discovered sites have exact-matching orthologous regions found in both human and mouse genes. Also, an analysis of the specific sequences that underlie the m6A sites show very high PhyloP conservation scores,Citation2 which indicates strong evolutionary pressures at these sites. Taken together, these data indicate that m6A sites likely play a key role in the RNA biology and cellular functions of these transcripts, and that their functions are constrained over millions of years of evolution.
Figure 1. Sites of m6A are pervasive across the genome. This shows a circos plot of all m6A peaks for humans, with HEK293T cell peaks from Meyer et al. (in red, inner circle) and HepG2 from Dominissini et al. (in blue, inner-most circle). Peaks were found in over 10,000 genes and in all chromosomes (outer sections, varying colors).
Six Putative Cellular Functions of m6A
Many new questions about RNA biology have been raised by the discovery and validation of this epitranscriptomic modification. While m6A has been localized in thousands of transcripts, its biological function and role are still largely a mystery. Thus far, studies have shown that m6A sites can be found in both mature and immature transcripts, and older data indicate that the m6A may be placed on transcripts before splicing or other RNA processing steps. It is also notable many peaks in the MeRIP papersCitation2,Citation3 were found in introns, non poly-adenylated transcripts and non-coding RNAs, providing further evidence that at least some of the m6A machinery is working in the early stages of RNA processing.
RNA stability
Accordingly, one likely major role for m6A could be in the stabilization of transcripts before they are processed. This hypothesis is supported by work examining methylation patterns in plant rRNA, which showed a stabilizing effect for ribosomal function in response to environmental changes such as temperature.Citation22 Thus, m6A could have a similar function in stabilizing mRNA transcripts and ncRNAs. Studies of m6A in plant mRNAs have further demonstrated the necessity of this modification and implicated its role in genes involved in transport and stress and stimulus response.Citation23
Splicing regulator
Another role for m6A could serve to regulate alternative splicing. For example, the mRNA adenosine methylate (MTA) homolog in plants is proposed to interact with FIP37, which is implicated in RNA splicing.Citation24 Moreover, m6A has previously been shown to be involved in splicing, particularly in alternatively spliced (AS) exons, implicating m6A in the regulation of AS events.Citation3 Interestingly, previous work has shown that AS exons evolve faster than constitutively spliced (CS) exons, and they enable an “evolutionary playground” for genes to modify their minor transcript isoforms while keeping the major isoform present in cells.Citation25 However, since m6A sites show overall strong conservation,Citation2 it is likely that m6A conservation may be contingent upon its genomic location, such that m6A site conservation could be relaxed in fast-evolving genes with complex splicing patterns. More tissues, and more time points, will be needed to examine the distribution of different spliced forms of m6A-enriched transcripts.
miRNA Attenuator
Given the proximity to the stop codon and the 5′ end of the 3′UTR, m6A may also interact with miRNA sites in the 3′UTR.Citation2 It is notable that 67% of UTRs with an m6A peak have a predicted miRNA-binding site, yet only 30% of genes in the genome have predicted miRNA-binding sites,Citation2 representing a 2.2-fold enrichment. Moreover, preliminary data have shown an inverse spatial relationship of miRNA-binding sites and m6A sites,Citation2 even though the m6A-containing transcripts have higher expression of their cognate miRNA targets. Interestingly, some reports have shown that methylated RNA can inhibit miRNA maturation, giving evidence for a direct connection of the methylation of miRNA and miRNA function,Citation26 whereby methylation serves as a negative regulator of the ability of the miRNA to function on the transcript. However, it is not yet known if the methylation of the mRNA is related to the methylation of the miRNA, or if the same enzymes are responsible for controlling methylation on both small and large RNAs. To explain these nascent data, further work is needed to (1) characterize the myriad RNA binding proteins that are predicted and their targets, (2) discern which of the 107 known RNA modifications are present on a target,Citation20 including m6A and (3) examine the molecular dynamics and binding rate changes associated with these ribonucleotide modifications. Depending on these data and where modifications occur in transcripts, both antagonistic and synergistic models of epitranscriptomic control of miRNA function are possible.
RNA editing prevention
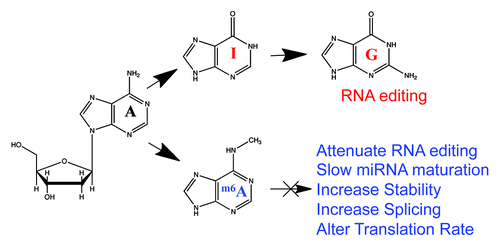
Another potential regulatory role for m6A is in the post-transcriptional modification of A→I editing, which results from the deamination of the N6 in adenosine by the ADAR family of adenosine deaminases. Intriguingly, m6A is known to block the activity of the ADAR enzymes that perform A→I conversion in mRNA.Citation27 Inosine is treated as guanosine in translation and when forming RNA secondary structures,Citation28 so m6A may serve as a mechanism to maintain adenosines in transcripts and hinder RNA editing. However, the challenge is both in finding existing A→I editing sites and seeing a dynamic change induced by an introduction of an m6A site. So far, there is no m6A peak that overlaps with any known RNA editing site, which gives some support for this hypothesis ().
Figure 2. Effects of m6A on RNA function. We hypothesize that the sites of m6A (blue) will prevent the N-6 de-amination that occurs in RNA editing (red), where adenosine (A) gets converted into inosine (I) then guanosine (G). These sites may then have many roles in the function of RNA, from splicing to translation changes (blue).
Disease accelerators
Variants of the FTO allele have been associated with obesity and increased body fat,Citation29,Citation30 where as other mutations in the FTO gene have resulted in lower frontal lobe volumes and are also associated with Alzheimer disease.Citation31-Citation33 Rhein (cassic acid) was recently discovered to be an inhibitor for FTO, binding to its active site in vitro and preventing demethylation of m6A, which may help to study FTO and m6A levels and their association with diseases.Citation34 Also, m6A levels have been observed with substantial variation in different cancer cell linesCitation2 and affect TP53 function,Citation19 meaning that they may play a role in cancer as well. These works potentially implicate m6A in a variety of diseases, but work remains to establish whether m6A is the causative mechanism that results in the higher risk of disease or if it is a secondary effect simply correlated with disease states.
Epigenome control
Lastly, one additional role for modifications of RNA such as m6A could be to serve as a regulator of epigenetic reprogramming and chromatin modification. Specifically, recent work on Alkbh5 (RNA demethylation) has shown that apopotosis- and spermatogenesis-related genes are differentially expressed in Alkbh5-deficient testicular cells when compared with wild-type testicular cells.Citation19 Moreover, several key epigenetic regulatory genes are downregulated in the Alkbh5-deficient cells, including DNA-methyltransferases (Dmnt1), RNA polymerase II elongation factor (Ell3) and Bromodomain-containing protein Brd4. Given the importance of epigenetic reprogramming in development, and with the additional evidence that sperm motility and testicular function is impaired without proper RNA methylation control,Citation19 it is possible that an “epigenome-epitranscriptome” regulatory network may exist in development that is then perturbed in diseases as well.
Conclusion
In summary, despite its known molecular dynamics and the knowledge thousands of its targets, the functions of m6A are still being discovered. Tremendous strides have recently been made toward clarifying key questions related to m6A biology, but much work remains to detail the real impact of m6A in other tissues and organisms. However, for the first time, the regulation, abundance, dynamics and localization of m6A can now be tracked. These epitranscriptomic data will allow us to test key hypotheses related to the function of m6A, providing the foundation for understanding the role of this critical RNA modification and reveal new aspects of RNA biology.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Desrosiers RC, Friderici KH, Rottman FM. Characterization of Novikoff hepatoma mRNA methylation and heterogeneity in the methylated 5′ terminus. Biochemistry 1975; 14:4367 - 74; http://dx.doi.org/10.1021/bi00691a004; PMID: 169893
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012; 149:1635 - 46; http://dx.doi.org/10.1016/j.cell.2012.05.003; PMID: 22608085
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012; 485:201 - 6; http://dx.doi.org/10.1038/nature11112; PMID: 22575960
- Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, et al. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res 2011; 39:Database issue D195 - 201; http://dx.doi.org/10.1093/nar/gkq1028; PMID: 21071406
- Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet 2012; 8:e1002781; http://dx.doi.org/10.1371/journal.pgen.1002781; PMID: 22737091
- Carey BW, Markoulaki S, Hanna JH, Faddah DA, Buganim Y, Kim J, et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell 2011; 9:588 - 98; http://dx.doi.org/10.1016/j.stem.2011.11.003; PMID: 22136932
- Lister R, Ecker JR. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res 2009; 19:959 - 66; http://dx.doi.org/10.1101/gr.083451.108; PMID: 19273618
- Münzel M, Globisch D, Carell T. 5-Hydroxymethylcytosine, the sixth base of the genome. Angew Chem Int Ed Engl 2011; 50:6460 - 8; http://dx.doi.org/10.1002/anie.201101547; PMID: 21688365
- Dubin DT, Taylor RH. The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res 1975; 2:1653 - 68; http://dx.doi.org/10.1093/nar/2.10.1653; PMID: 1187339
- Perry RP, Scherrer K. The methylated constituents of globin mRNA. FEBS Lett 1975; 57:73 - 8; http://dx.doi.org/10.1016/0014-5793(75)80155-4; PMID: 1182024
- Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 1975; 4:379 - 86; http://dx.doi.org/10.1016/0092-8674(75)90158-0; PMID: 164293
- Chen-Kiang S, Nevins JR, Darnell JE Jr.. N-6-methyl-adenosine in adenovirus type 2 nuclear RNA is conserved in the formation of messenger RNA. J Mol Biol 1979; 135:733 - 52; http://dx.doi.org/10.1016/0022-2836(79)90174-8; PMID: 537090
- Saneyoshi M, Harada F, Nishimura S. Isolation and characterization of N6-methyladenosine from Escherichia coli valine transfer RNA. Biochim Biophys Acta 1969; 190:264 - 73; http://dx.doi.org/10.1016/0005-2787(69)90078-1; PMID: 4900574
- Iwanami Y, Brown GM. Methylated bases of ribosomal ribonucleic acid from HeLa cells. Arch Biochem Biophys 1968; 126:8 - 15; http://dx.doi.org/10.1016/0003-9861(68)90553-5; PMID: 5671075
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 2011; 7:885 - 7; http://dx.doi.org/10.1038/nchembio.687; PMID: 22002720
- Falnes PØ, Klungland A, Alseth I. Repair of methyl lesions in DNA and RNA by oxidative demethylation. Neuroscience 2007; 145:1222 - 32; http://dx.doi.org/10.1016/j.neuroscience.2006.11.018; PMID: 17175108
- Gerken T, Girard CA, Tung Y-CL, Webby CJ, Saudek V, Hewitson KS, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 2007; 318:1469 - 72; http://dx.doi.org/10.1126/science.1151710; PMID: 17991826
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997; 3:1233 - 47; PMID: 9409616
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol Cell 2013; 49:18 - 29; PMID: 23177736
- Saletore Y, Meyer K, Korlach J, Vilfan ID, Jaffrey S, Mason CE. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol 2012; 13:175; http://dx.doi.org/10.1186/gb-2012-13-10-175; PMID: 23113984
- Bayley H. Individual RNA Base Recognition in Immobilized Oligonucleotides using a Protein Nanopore. Nano Lett 2012; 2013 In press
- Brown JW, Echeverria M, Qu L-H. Plant snoRNAs: functional evolution and new modes of gene expression. Trends Plant Sci 2003; 8:42 - 9; http://dx.doi.org/10.1016/S1360-1385(02)00007-9; PMID: 12523999
- Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, et al. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008; 20:1278 - 88; http://dx.doi.org/10.1105/tpc.108.058883; PMID: 18505803
- Bodi Z, Zhong S, Mehra S, Song J, Graham N, Li H, et al. Adenosine Methylation in Arabidopsis mRNA is Associated with the 3′ End and Reduced Levels Cause Developmental Defects. Front Plant Sci 2012; 3:48; http://dx.doi.org/10.3389/fpls.2012.00048; PMID: 22639649
- Modrek B, Lee CJ. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat Genet 2003; 34:177 - 80; http://dx.doi.org/10.1038/ng1159; PMID: 12730695
- Xhemalce B, Robson SC, Kouzarides T. Human RNA methyltransferase BCDIN3D regulates microRNA processing. Cell 2012; 151:278 - 88; http://dx.doi.org/10.1016/j.cell.2012.08.041; PMID: 23063121
- Véliz EA, Easterwood LM, Beal PA. Substrate analogues for an RNA-editing adenosine deaminase: mechanistic investigation and inhibitor design. J Am Chem Soc 2003; 125:10867 - 76; http://dx.doi.org/10.1021/ja029742d; PMID: 12952466
- Garrett S, Rosenthal JJC. RNA editing underlies temperature adaptation in K+ channels from polar octopuses. Science 2012; 335:848 - 51; http://dx.doi.org/10.1126/science.1212795; PMID: 22223739
- Yeo GSH. FTO and obesity: a problem for a billion people. J Neuroendocrinol 2012; 24:393 - 4; http://dx.doi.org/10.1111/j.1365-2826.2011.02254.x; PMID: 22248259
- Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007; 316:889 - 94; http://dx.doi.org/10.1126/science.1141634; PMID: 17434869
- Ho AJ, Stein JL, Hua X, Lee S, Hibar DP, Leow AD, et al, Alzheimer’s Disease Neuroimaging Initiative. A commonly carried allele of the obesity-related FTO gene is associated with reduced brain volume in the healthy elderly. Proc Natl Acad Sci USA 2010; 107:8404 - 9; http://dx.doi.org/10.1073/pnas.0910878107; PMID: 20404173
- Benedict C, Jacobsson JA, Rönnemaa E, Sällman-Almén M, Brooks S, Schultes B, et al. The fat mass and obesity gene is linked to reduced verbal fluency in overweight and obese elderly men. Neurobiol Aging 2011; 32:1159 - , e1-5; http://dx.doi.org/10.1016/j.neurobiolaging.2011.02.006; PMID: 21458110
- Keller L, Xu W, Wang H-X, Winblad B, Fratiglioni L, Graff C. The obesity related gene, FTO, interacts with APOE, and is associated with Alzheimer’s disease risk: a prospective cohort study. J Alzheimers Dis 2011; 23:461 - 9; PMID: 21098976
- Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc 2012; 134:17963 - 71; http://dx.doi.org/10.1021/ja3064149; PMID: 23045983