 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
RNA molecules have highly versatile structures that can fold into myriad conformations, providing many potential pockets for binding small molecules. The increasing number of available RNA structures, in complex with proteins, small ligands and in free form, enables the design of new therapeutically useful RNA-binding ligands. Here we studied RNA ligand complexes from 10 RNA groups extracted from the protein data bank (PDB), including adaptive and non-adaptive complexes. We analyzed the chemical, physical, structural and conformational properties of binding pockets around the ligand. Comparing the properties of ligand-binding pockets to the properties of computed pockets extracted from all available RNA structures and RNA-protein interfaces, revealed that ligand-binding pockets, mainly the adaptive pockets, are characterized by unique properties, specifically enriched in rare conformations of the nucleobase and the sugar pucker. Further, we demonstrate that nucleotides possessing the rare conformations are preferentially involved in direct interactions with the ligand. Overall, based on our comprehensive analysis of RNA-ligand complexes, we suggest that the unique conformations adopted by RNA nucleotides play an important role in RNA recognition by small ligands. We term the recognition of a binding site by a ligand via the unique RNA conformations “RNA conformational readout.” We propose that “conformational readout” is a general way by which RNA binding pockets are recognized and selected from an ensemble of different RNA states.
Introduction
Recent discoveries in RNA biology underscores the importance of RNA in normal and aberrant cellular functions and highlights the potential of RNA as a drug target for many diseases. There are several advantages in targeting RNA over traditional protein targets. For example, RNA targets have the potential for slower development of drug resistance against small molecules.Citation1 More sites are accessible at the RNA level, whereas the “active site” is often the only target on the protein.Citation2 Moreover, unlike DNA, RNA produces unique three-dimensional (3D) pockets suitable for specific binding of small ligands.Citation3 Overall, RNA molecules have highly versatile structures that can fold into different conformations,Citation4 providing specific recognition sites for diverse drugs.
With the increasing number of available RNA structures in the protein structure data base (PDB),Citation5 the design of new therapeutically useful RNA-binding ligands is now technically feasible. Since the majority of drugs are ligands for proteins, RNA provides a unique niche for pharmacological development. Thus far, small molecule ligands for RNA have been developed toward three major classes of targets: antibacterial targets (such as bacterial ribosome); antiviral targets [such as trans-activating response RNA (TAR) in HIV]; and human mRNA targets (such as protein-mediated translation control).Citation1,Citation3,Citation6
RNA ligand interactions are generally divided into three types. The first type includes nonspecific electrostatic interactions between the positively charged ligand and the negatively charged RNA phosphate backboneCitation7,Citation8 that account for at least one-half of the total binding energy in certain aminoglycoside-RNA interactions.Citation3 The second type includes specific interactions generally involving direct hydrogen bonding or van der Waals (VDW) interactions with nucleic acid bases in the deep major groove or the wide shallow minor groove of the RNA helix.Citation7 In a recent study of RNA-ligand complexes, interactions between the ligands and the Watson-Crick edge of the RNA were frequently observed. These interactions were proposed to play a key role in ligand selectivity. In addition, RNA-ligand interactions via the Hoogsteen and/or sugar edge were also observed, though the latter were found to be much less frequent.Citation9 A third type of interaction includes stacking interactions between RNA bases and aromatic ligands.Citation7 In a study by Kondo et al., it was observed that pseudo base pairs using the Watson-Crick edge in combination with stacking interactions were frequently involved in specific ligand recognitions. They showed that binding in the deep major groove was the most preferred recognition mode for bulky sugar ligands such as aminoglycosides.Citation9
In a previous study conducted on the ribosome, we demonstrated that known antibiotic-binding sites, mainly in the large ribosomal subunit, share several unique properties that we defined as the “RNA signature.”Citation10 The most noticeable features found in the majority of known antibiotic-binding sites on the ribosome were the prevalence of non-paired bases, a high frequency of unusual syn conformation and an unusual ribose sugar pucker. The prevalence of syn conformation was also shown in active sites of functional RNAs.Citation11 We previously proposed that the unique nucleotide conformation and the sugar pucker may reflect higher flexibility of rRNA bases in antibiotic-binding sites, which may contribute to antibiotic selectivity and action.Citation10 In an attempt to ask whether unique RNA conformations are a general phenomenon, here we studied ligand-binding sites from diverse (non-ribosomal) RNA targets. Consistent with previous results, we found that in general ligand-binding pockets in RNA, mainly the adaptive pockets, are characterized by the abundance of unique properties, specifically rare sugar pucker and nucleobase conformations. Interestingly, these features were also enriched in the majority of ligand-free (apo) structures, available in the database. Furthermore, we show that nucleotides which adopt rare conformations are indeed involved in direct interactions with the ligand. Our results reinforce the hypothesis that the unique conformations adopted by nucleotides in ligand-binding pockets in RNA contribute to the specific recognition of the binding pockets via an “RNA conformational readout” mode.
Results and Discussion
The unique properties of the ligand binding pockets on RNA
To study the unique properties of ligand binding pocket on RNA we extracted a set of RNA-ligand complexes from the PDB solved by X-ray crystallography or NMR, as described in detail in the Materials and Methods section. Among the complexes, we included “rigid” targets and “adaptive” binding targets (aptamers, riboswitches)Citation12-Citation14 (Table S1). From the extended list of complexes we selected a set of 33 representative pockets from 10 RNA target groups (highlighted in gray in Table S1), on which the statistical analyses were conducted (see Materials and Methods). Subsequent to the data extraction, we computed the physical, chemical and structural features of the ligand-binding pockets in RNA compared with the same features in a large set of predicted pockets (extracted from all RNA structures in PDB). The predicted pockets were calculated using two different pocket extraction methods: Solvent and Fpocket (for details see Materials and Methods). Based on the higher overlap between the pockets predicted by the Solvent program and the known binding pockets in comparison to the overlap obtained between the pockets predicted by the Fpocket program and the real binding pockets (Tables S2 and S3), we chose the pockets predicted by the Solvent program as the preferred background set. Overall, the two different approaches for predicting pockets produced very similar results.
Physicochemical properties
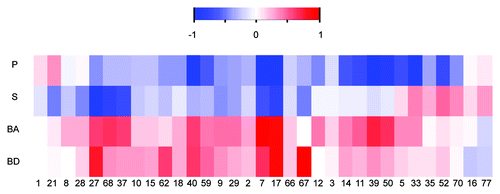
When analyzing the physiochemical properties of the ligand-binding pockets on RNA in comparison to the background, we noticed an overall increased representation of atoms belonging to the nitrogen bases rather than atoms of the sugar-phosphate backbone in the known binding pockets [ (representative pockets) and Fig. S1 (full set)]. The significant enrichment of nucleobases in the known binding pockets relative to the computed pockets was a global phenomenon and was not unique to a specific target group. This phenomenon was also observed for the antibiotic binding sites on the ribosome.Citation10
Figure 1. Enrichment of physicochemical properties in 33 representative structures of ligand binding pockets on RNA. Heatmap demonstrates over- and under- representations of the different physicochemical properties in the binding pockets relative to background. BD denotes base donors, BA base acceptors, S sugar and P phosphate atoms. Numbers represent the index of the complex listed in Table S1. The color scheme refers to the standardized score calculated against a background of 70,912 computed pockets (calculated by the Solvent program). Scores were scaled to range from -1 to 1. Significant biases relative to the background average are colored red and blue for over- and under- representation, respectively.
Structural properties
To characterize the structural properties of RNA within the binding pockets, we used the MC-annotate program.Citation15 We calculated the structural properties of the nucleotides in the known ligand-binding pockets on RNA compared with properties in the predicted pockets. Notably, since the complexes in our data set were solved by different techniques (X-ray crystallography and NMR) to ensure that the properties of the pockets are not biased by the technique used to solved the structure, we analyzed the structural properties of the pockets independently, i.e., pockets solved by X-ray crystallography and pockets solved by NMR were analyzed separately. As demonstrated in (representative pockets) and Figure S2 (all pockets), we noticed an overall increased representation of unique properties such as non-paired nucleotides, and a decreased representation of other properties such as standard Watson-Crick pairing (WWcisCitation15). Interestingly, the overrepresentation of unique properties in the pockets was more pronounced in the subset of pockets belonging to aptamers and riboswitches ().Citation12-Citation14 Importantly, while conformation differences in ligand binding pockets that were solved by different techniques have been previously observed (as for example, in the PreQ1 riboswitchCitation16), our comprehensive analysis shows that, in general, the abundance of unique RNA conformations is common to both X-ray and NMR structures of RNA and does not seem to be influenced by the technique used to solve the structure.
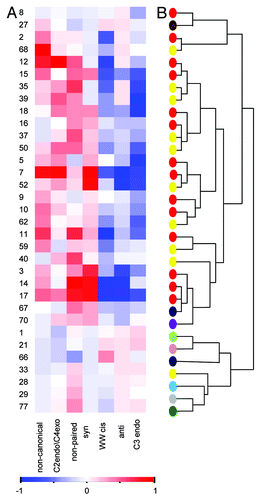
Figure 2. (A) Enrichment of structural properties in 33 representative structures of ligand binding pockets on RNA. Heatmap demonstrates over- and under- representations of the different structural properties in the binding pockets relative to background. The color scheme refers to the standardized score calculated against a background of computed pockets (calculated by the Solvent program). Scores were scaled to range from -1 to 1. Significant preferences of properties relative to the background of all RNA pockets are colored red (1) while blue denotes under-representation (-1). Numbers represent the index of the complex listed in Table S1. (B) Hierarchical clustering of the 33 representative structures of RNA ligand complexes based on their structural properties. The colors of the dots represent different RNA groups (7SK SNRNA – bright green, Aptamer – red, DIS-HIV1 – pink, Duplex – brown, HIV1 Helix – azure, HCV IRES Domain IIa – gray, Riboswitch – yellow, Ribozyme – blue, TAR-purple, Splicing Regulatory – dark green).
Furthermore, we observed a preference for syn over anti conformation of RNA bases in the majority of the known ligand-binding pockets in RNA (). As depicted in and Figure S2 while the preference for the syn over the anti conformation was not detected in all complexes the majority of pockets showed a clear preference for the syn conformation. Similar results were previously observed in antibiotic binding sites on the ribosomeCitation10 and in the active sites of functional RNAs.Citation11 Syn and anti conformations are defined by glycosidic torsion angle χ. The anti conformation is usually more stable, while the syn conformation requires an external stabilizing force. Rotation about the glycosidic bond is hindered, with purines being usually less hindered than pyrimidines.Citation17 As shown in , we also observed an overabundance of the unusual sugar pucker conformation (both C2 endo and C4 exo) in the ligand-binding pockets on RNA over the more common C3 endo form found typically in RNA structures. Notably, while as can be expected from the weak correlation between the torsion angels (χ, δ),Citation4 several of the nucleotide in the syn conformation adopted a C2 endo sugar pucker, in the majority of pockets we did not observe a strong dependency between the two properties (Table S4). Overall, the C2 endo conformation provides less steric hindrance compared with the C3 endo, meaning that the C2 endo conformer is inherently more flexible, accommodating a wider range of allowed χ values and involving a lower energy cost for the aforementioned syn conformation.Citation17-Citation19 While the C2 endo conformation is relatively rare, it has been shown to play functionally important roles in RNA. For example, C2 endo nucleotides have been previously suggested to function as molecular timers in RNA folding and ligand recognition reactions.Citation20 Moreover, deletion of one C2 endo nucleotide at RNase P was shown to accelerate RNA folding by an order of magnitude.Citation21 C2 endo nucleotides were also shown to play a functional role in the spliceosome and in the ribosome.Citation22,Citation23 In the latter, the flexibility of glycosidic bonds of bases in the antibiotic-binding sites of the large ribosomal subunit (specifically at the PTC and the tunnel region) was suggested to explain antibiotic selectivity and action.Citation24,Citation25
As demonstrated in , in addition to the high frequency of rare RNA conformations in the ligand binding pockets, we also noticed that non-paired bases were overrepresented in most of the ligand-binding pockets on RNA. This could likely be due to the greater variation in chemical groups available for structure-specific interactions compared with the nitrogen bases, which are engaged in inter-nucleotide interactions. In addition, we observed a bias for non-canonical WH pairing in the known ligand-binding pockets on RNA. Formation of non-canonical base pairs in RNA is essential because, in contrast to Watson-Crick base pairs in RNA, non-canonical base pairs often play an important functional role, for example, in catalytic RNA. Structural roles for non-canonical pairing was exemplified in the stabilization and formation of the RNA tertiary structure.Citation26
Overall, our results show that the ligand-binding pockets in RNA are characterized by rare structural properties of the RNA. Further, when clustering the ligand-binding pockets on RNA according to their structural properties, we noticed that, in general, the artificial RNA aptamers and the different subgroups of the natural RNA riboswitches were clustered together (), showing a similar pattern of enrichment of the rare properties, specifically the nucleotides in syn conformation, the C2 endo/C4 exo sugar pucker and the non-paired and non-canonical nucleotides
Unique RNA properties are also found in apo structures
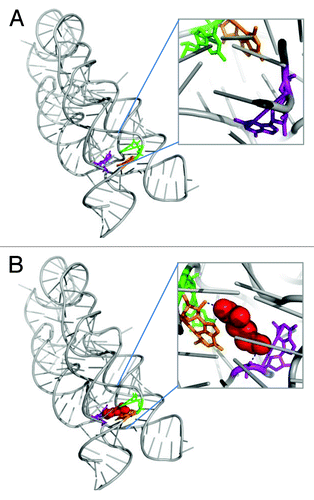
To examine whether the rare RNA conformations are found in the bound (holo) state only we extracted a set of 10 representative RNAs, which their structures were available both in the apo and holo states (see “Material and Methods” section for details). The distributions of the structural properties were calculated independently for the pockets of the bound states and their corresponding nucleotides in the unbound states. As can be noticed from , overall, the properties which were significantly high in the bound pockets (highlighted in bold in ) were usually enriched in the unbound state. For example, as demonstrated in , the three nucleotides in the structures of the lysine riboswitch (3D0U/3D0X, for the bound and unbound, respectively) were found to adopt the syn conformations in both the free and bound form (, ). These results are consistent with Garst et al., observations that the lysine confers only limited local changes upon binding to the RNA pocket.Citation27 Nevertheless, in the case of preQ1 riboswitch, which was shown to be unstructured in its free formCitation28 we indeed noticed a significant difference in the frequencies of the unique RNA properties (). Interestingly, the relative frequencies of the unique RNA conformations were usually higher in the unbound state.
Table 1. Unique RNA conformations in the bound and unbound structures
Figure 3. Demonstration of the unique conformations found in the holo and apo states of the lysine riboswitch. (A) Lysine riboswitch in the unbound state (apo), (PDB ID 3D0X). Nucleotides which adopt the syn conformation are highlighted in color: Nucleotide A8 (orange), Nucleotide A9 (green), Nucleotide A77 (purple). (B) Lysine riboswitch in the bound state (holo), (# 52 PDB ID 3D0U). Nucleotide in syn conformation are highlighted, coloring scheme is as in A. The ligand is shown as red spheres and the hydrogen bonds are represented as blue dashed lines.
Overall, in the majority of the representative pockets non-paired nucleotides were frequent in both the bound and unbound state. One exception was the SAMI riboswitch in which the frequency of non-paired nucleotides was only significant in the bound state. Interestingly, in the latter example the free-state structure revealed that the nucleotide A46 occupies the binding site in place of the SAM ligand.Citation29
Recent dynamic studies of riboswitches suggest that in the absence of ligands, the riboswitch may adopt an ensemble of different states from which the ligand selects a ligand-binding competent conformation, namely a “conformational selection.”Citation29,Citation30 Our results which show that the unique and rare RNA conformations tend to be found in unbound states of different binding pockets, support the “conformational selection” hypothesis, further suggesting that the unique RNA conformations are recognized and selected by the ligand.
RNA-ligand pockets vs. RNA-protein interfaces and all RNA
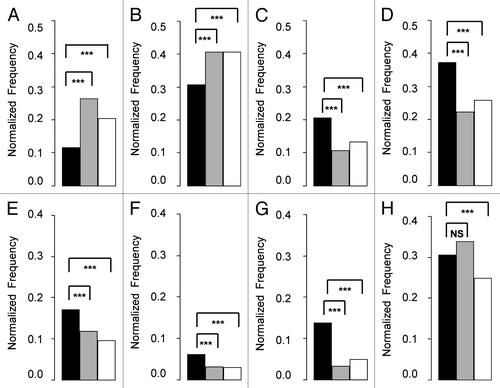
We further compared the properties we observed for the binding pockets on RNA to the properties of RNA-protein interfaces and all RNA extracted from all RNA structures obtained from the PDB (for details see Materials and Methods). Here, again, we noticed an increased representation of atoms belonging to nitrogen bases rather than atoms of the phosphate backbone (). Overall, we observed that nucleobase-specific interactions (involving hydrogen bonds between the ligand and the nucleobases) and non-specific interactions (involving direct contacts between the ligand and the RNA backbone) make up 65% and 35% of RNA-ligand hydrogen bonds, respectively. Thus, the majority of RNA-ligand hydrogen bonds are nucleobase-specific. Interestingly, in recent comprehensive studies analyzing RNA-protein interfaces, it has also been shown that the majority of interactions are rather non-specific.Citation31-Citation33 The different ratio of specific vs non-specific interactions between ligand-RNA and protein-RNA may explain the overall difference in the representation of rare RNA properties, specifically C2 endo/C4exo, syn and non-canonical base pairings within the ligand-binding pockets on RNA compared with protein-binding interfaces in RNA (). Notably, rare conformations (such as syn) have been shown to play an important role in binding specificity of specific RNA-binding proteins (as for example in ref. Citation34). However, while nucleotides with unique RNA conformations may play an important role in conferring protein-RNA specificity, our results suggest that they do not play a general role in protein-RNA recognition. Nonetheless, in both ligand-binding and protein-binding interfaces, we found over-representation of the non-paired conformation over the canonical WW cis base-pairings compared with all RNA (). This phenomenon of favoring non-paired conformation over canonical Watson-Crick base-paring was also observed by Gupta and Gribskov in a recent comprehensive study comparing RNA-protein interfaces to all RNA.Citation31 Taken together, we found that ligand-binding sites in RNA have unique properties that usually differ from RNA-protein interfaces.
Figure 4. Comparison of the different physicochemical and structural property frequencies between known ligand-binding pockets on RNA to RNA-protein interfaces and to overall RNA. The known ligand-binding pockets in RNA are presented as black bars. RNA-protein interfaces are presented as gray bars. Overall RNA is presented as white bars. Bar height represents the normalized frequency of the different properties in each group (known ligand binding pockets on RNA, RNA-protein interfaces, overall RNA). Groups were compared using Fisher exact tests. The stars denote statistical significance (P value < 0.001), NS denotes insignificance. Each panel represents different properties: (A) phosphate, (B) sugar, (C) base donors, (D) base acceptors, (E) C2 endo + C4 exo, (F) syn, (G) non-canonical, (H) non-paired.
Interactions between nucleotides with unique RNA conformations and ligand atoms
Many structural studies of RNA ligand complexes have pointed out that RNA ligands tend to bind non-paired nucleotides or nucleotides in non-canonical pairing conformations (as, for example, in aptamer ligand structuresCitation35). In order to examine whether in general nucleotides with unique RNA structural properties (i.e., C2 endo/C4 exo, syn, non-paired, non-canonical base pairing) are involved preferentially in direct interactions with the ligand, we concentrated on four types of contacts: hydrogen bond, VDW, hydrophobic and aromatic. As shown in , our analysis reinforces that non-paired nucleotides are involved preferentially (statistically significant) in hydrogen bonds with the ligands. Interestingly, among the other contacts (VDW, hydrophobic and aromatic), we did not notice a significant enrichment of contacts toward non-paired nucleotides (; Table S5). This seems reasonable considering that non-paired nucleotides are not engaged in inter-nucleotide hydrogen bond interactions and, thus, offer more conformational flexibility compared with paired nucleotides. On the contrary, the ability of nucleotides to interact with the ligand via hydrophobic contacts is not expected to be affected by its pairing status.
Figure 5. Unique RNA conformations are significantly involved in RNA ligand interactions. (A) The figure demonstrates the RNA properties which are preferentially involved in direct interactions with the ligand, focusing on four types of interactions: hydrogen bonds (HB), van der Waals (VDW), hydrophobic (HD) and aromatic (AR). The rare properties: C2 endo/C4 exo, syn, non-paired and non-canonical pairing are presented as black, light gray, dark gray and white bars, respectively. Bar height represents the –log10 of the P-value of the Fisher exact test. The dashed line marks the level of statistical significance (p = 0.05). (B) Graphical representation of the preferred interactions observed in the RNA ligand complex of arginine aptamer (PDB ID 1KOC). The RNA molecular surface is presented in gray, the ligand as red sticks and the hydrogen bonds as green lines. Highlighted are nucleotide G12 (blue) that interacts with the ligand via VDW contacts and possesses unique properties of C4 exo, non-canonical base pair; nucleotide G35 (yellow) that interacts with the ligand via VDW and hydrophobic contacts and possesses the properties of C2 endo, syn, non-canonical base pairing; nucleotide A33 (green) that interacts with the ligand via hydrogen bonds, VDW and hydrophobic contacts and possesses the rare property of non-paired. (C) nucleotide C13 (purple) that interacts with the ligand via VDW and hydrogen bonds; G31 (pink) that interacts with the ligand via VDW and hydrophobic contacts and is non-paired; nucleotide G30 (orange) that interacts with the ligand via hydrogen bonds and VDW contacts and has the combination of rare properties C4 exo, syn and non-canonical pairing.
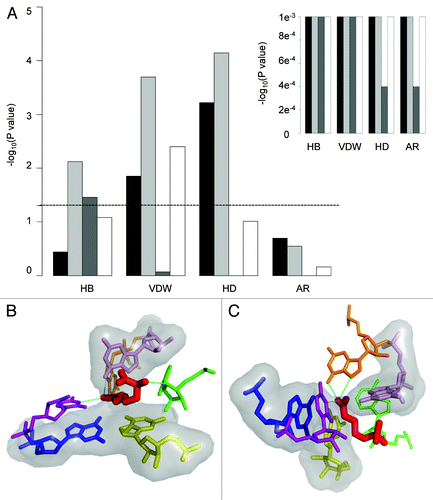
Furthermore, we observed that nucleotides with the unique property syn were preferentially (statistically significant) involved in interactions with the ligand via hydrogen bonds, VDW, and hydrophobic interactions while nucleotides in either C2 endo or C4 exo conformations were significantly more involved in VDW contact in general and in hydrophobic interactions specifically (; Table S5). Notably, while nucleotides possessing both syn and c2 endo or c4 exo conformations were preferentially (statistically significant) involved in interactions with the ligand, in many cases the nucleotides involved in binding had only one of the unique conformations (Table S5). Moreover, nucleotides involved in non-canonical interactions within the RNA were found to interact preferentially (statistically significant) with ligands via VDW contacts (; Table S5). An example of a typical RNA-ligand complex exhibiting preferred interactions between nucleotides having unique RNA features and the ligand is shown in . As shown, nucleotides possessing rare structural properties are involved in direct interactions with the ligand.
Materials and Methods
Data extraction
RNA-ligand complexes
RNA-ligand structures in the holo state were obtained from the PDB (January 2011 Release). Selected structures were solved either by X-ray crystallography (< 3Å) or NMR. Only structures with > 12 nucleotides were included, removing structures with a high structural similarity (RMSD ≤ 1Å) that bind the same ligand. In addition RNA-ligand complexes including proteins were removed from the data set. The final set, included 77 RNA-ligand structures. Among them we selected 33 representative (non-redundant) pockets from 10 different RNA groups (highlighted in gray in Table S1) for further analyses.
Apo and holo structures of binding pockets on RNA
Ten pairs of apo and holo structures of RNA (one representative from each RNA group) were obtained from the PDB. The structures were solved either by X-ray crystallography (< 3.3Å) or NMR.
All RNA
All RNA structures (excluding ribosomal structures) were obtained from the PDB (January 2011 Release). Data included RNA structures that were solved either by X-ray crystallography (< 3Å) or NMR. A total set of 776 structures was obtained, including 225 protein-RNA complexes. Consistent with the RNA-ligand data set, structures with < 12 nucleotides were removed from the “all RNA” control set. In addition, water molecules and edge nucleotides (the last nucleotide in each RNA chain) were not included in the structural analysis. Interface residues were calculated using the Intervor web serverCitation36 excluding water molecules.
Binding site extraction
A binding site was defined to include all atoms within a radius of 6Å from any atom of the ligand. The water molecules were excluded, as well as all edge nucleotides.
Putative pockets extraction for the background model
Putative pockets were extracted from all RNA structures using two different methods:
1. Using the Solvent program: The Solvent program from the 3V packageCitation37 gets as an input a PDB file and the radius of a small and a large probe sphere, and outputs the void in the structures that can accommodate a small probe size (1.5Å) but not a large probe size (5.5Å). The void itself, which corresponds to a potential binding pocket, is represented by the oxygen atoms of water molecules that define the borders of the void. The procedure for extracting putative pockets was performed as in David-Eden et al.Citation10 The pocket was defined to include all atoms within a radius of 9Å from the computed oxygen atom. Only pockets that comprise 12 nucleotides and differ in at least one nucleotide were retained, resulting in 70,912 pockets.
2. Using the Fpocket program: The Fpocket program relies on the concept of α spheres.Citation38 The Fpocket core can be resumed in three major steps. During the first step, the whole ensemble of α spheres is determined from the structure, resulting in a pre-filtered collection of spheres. The second step involves identifying clusters of spheres close together to identify pockets and remove clusters of poor interest. The final scoring step in Fpocket calculates the atomic properties of the pocket and is not applied to RNA structures. The program takes a PDB file as input and outputs a PDB file containing only the atoms defining the pocket. Running Fpocket on all RNA structures resulted in 2,938 computed pockets.
Sensitivity and positive predictive value (PPV) of binding site identification
Sensitivity and PPV were calculated based on the degree of overlap between computed pockets and known binding sites (Tables S2 and S3). Sensitivity equals the fraction of common nucleotides divided by the number of nucleotides in the known binding site. PPV equals the fraction of common nucleotides divided by the number of nucleotides included in the putative (computed) pocket.
Physicochemical and structural properties calculations
The nitrogen and oxygen atoms in the base were classified as donors or acceptors. Backbone oxygen atoms were labeled as sugar or phosphate. The MC-Annotate programCitation15 was employed for classifying the RNA structural properties, including base-pairing and base-base stacking interactions.Citation39,Citation40 Base-pairing was defined according to three edges of the corresponding RNA bases available for H-bonding interactions: the Watson-Crick (W) edge, the Hoogsteen edge (H) and the sugar edge (S).Citation41 Base-base stacking interactions were categorized as stacking interactions between adjacent and non-adjacent nucleotides.
The features were extracted as described in reference Citation42 using an in-house Perl script converting the MC-Annotate output files into binary format, i.e., each nucleotide was given a score of “1” when a specific property was present and a score of “0” when it was absent. To calculate the relative abundance of a specific property, the fraction of nucleotides in the pocket possessing that property was calculated.
Analysis of RNA-ligand interactions
Intermolecular hydrogen bonds and VDW, hydrophobic and aromatic contacts were calculated for each RNA-ligand complex using the LIGPLOT program.Citation43 LIGPLOT uses the HBPLUSCitation44 program for calculating the hydrogen bonds. The program computes all possible positions of hydrogen atoms (H) attached to donor atoms (D) that satisfy specified geometrical criteria with acceptor atoms (A) in the vicinity. The criteria used to define a hydrogen bond were H-A distance < 2.7Å, D-A distance < 3.35Å and D-H-A angle > 90̊. VDW contacts were defined as all contacts between carbon atoms of the RNA and atoms of the ligand not involved in hydrogen bonds that were < 3.9Å apart. Hydrophobic contacts were defined as all contacts between carbon atoms of the RNA and carbon atoms of the ligand not involved in hydrogen bonds that were < 3.9Å apart. Aromatic contacts were defined as all contacts between aromatic carbon atoms of the RNA and aromatic carbon atoms of the ligand not involved in hydrogen bonds that were < 3.9Å apart. Aromatic carbon atoms of the ligand were determined according to the PDB chemical component dictionary.
Enrichment analysis
The relative abundance of each property in a known site was evaluated relative to the background of putative pockets (from the same technique X-ray or NMR) calculated by Solvent.Citation37 An individual score of each property was standardized by the Z score. Scores were scaled to range from -1 to 1. The statistical significance of the property enrichment was evaluated based on the hyper geometric distribution using the Fisher's exact test.
Clustering was performed using MeV software,Citation45 applying hierarchical clustering with average linkage.
Conclusions
In this study, we show that ligand-binding sites on RNA, mainly the adaptive pockets, such as artificial aptamers and natural riboswitches, are characterized by unique RNA structural properties. Specifically, we noticed a strong bias in favor of unusual syn conformation of RNA bases, an unusual sugar pucker, non-canonical base pairing, non-paired bases and an increased representation of atoms belonging to nitrogen bases. Interestingly, these preferred features were found to be unique to ligand-binding sites and are not common to RNA-protein interfaces in general. The only property found in excess in both ligand binding pockets on RNA and RNA-protein interfaces was the non-paired bases that had been previously shown to play an important role in RNA recognition by both small ligands and proteins. Further examination of RNA-ligand interactions confirmed that ligands interact preferentially with nucleotides possessing these unique properties. Specifically, the non-paired bases were found to be involved in direct contacts with the ligand via hydrogen bonds, while nucleotides possessing other unique properties did not show preference for a specific type of interaction and were generally found to be in contact with ligands either by electrostatic or hydrophobic contacts. These results as well as the noticeable enrichment of the rare conformations in the available apo structures support the hypothesis that ligand-binding sites on RNA are commonly recognized by their unusual conformations, which tend to be energetically higher and intrinsically more flexible. We term the recognition between the ligand and the nucleotides which adopt unusual conformation a “conformational readout.” We propose that conformational readout is a general way by which binding sites on RNA are recognized specifically by small ligands and are selected from the ensemble of different RNA states. This hypothesis is supported by many other RNA dynamics studies suggesting that the RNA is not frozen in a certain conformation but is rather found in multiple pre-existing conformations from which the ligand selects the competent conformation.
Additional material
Download Zip (277.3 KB)Acknowledgments
We would like to thank Hilda David-Eden and Inbal Paz for support with the programs and the other members of the lab for many helpful comments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Israeli Science Foundation, ISF (Grant number 1297/09 granted to Y.M.G.).
References
- Gallego J, Varani G. Targeting RNA with small-molecule drugs: therapeutic promise and chemical challenges. Acc Chem Res 2001; 34:836 - 43; http://dx.doi.org/10.1021/ar000118k; PMID: 11601968
- Sucheck SJ, Wong CH. RNA as a target for small molecules. Curr Opin Chem Biol 2000; 4:678 - 86; http://dx.doi.org/10.1016/S1367-5931(00)00142-3; PMID: 11102874
- Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem Rev 2008; 108:1171 - 224; http://dx.doi.org/10.1021/cr0681546; PMID: 18361529
- Schneider B, Morávek Z, Berman HM. RNA conformational classes. Nucleic Acids Res 2004; 32:1666 - 77; http://dx.doi.org/10.1093/nar/gkh333; PMID: 15016910
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res 2000; 28:235 - 42; http://dx.doi.org/10.1093/nar/28.1.235; PMID: 10592235
- Hermann T, Tor Y. RNA as a target for small-molecule therapeutics. Expert OpinTherPat 2005; 15:49 - 62
- Chow CS, Bogdan FM. A Structural Basis for RNAminus signLigand Interactions. Chem Rev 1997; 97:1489 - 514; http://dx.doi.org/10.1021/cr960415w; PMID: 11851457
- Aboul-ela F. Strategies for the design of RNA-binding small molecules. Future Med Chem 2010; 2:93 - 119; http://dx.doi.org/10.4155/fmc.09.149; PMID: 21426048
- Kondo J, Westhof E. Base pairs and pseudo pairs observed in RNA-ligand complexes. J Mol Recognit 2010; 23:241 - 52; PMID: 19701919
- David-Eden H, Mankin AS, Mandel-Gutfreund Y. Structural signatures of antibiotic binding sites on the ribosome. Nucleic Acids Res 2010; 38:5982 - 94; http://dx.doi.org/10.1093/nar/gkq411; PMID: 20494981
- Sokoloski JE, Godfrey SA, Dombrowski SE, Bevilacqua PC. Prevalence of syn nucleobases in the active sites of functional RNAs. RNA 2011; 17:1775 - 87; http://dx.doi.org/10.1261/rna.2759911; PMID: 21873463
- Patel DJ, Suri AK, Jiang F, Jiang L, Fan P, Kumar RA, et al. Structure, recognition and adaptive binding in RNA aptamer complexes. J Mol Biol 1997; 272:645 - 64; http://dx.doi.org/10.1006/jmbi.1997.1281; PMID: 9368648
- Mandal M, Breaker RR. Gene regulation by riboswitches. Nat Rev Mol Cell Biol 2004; 5:451 - 63; http://dx.doi.org/10.1038/nrm1403; PMID: 15173824
- Nudler E, Mironov AS. The riboswitch control of bacterial metabolism. Trends Biochem Sci 2004; 29:11 - 7; http://dx.doi.org/10.1016/j.tibs.2003.11.004; PMID: 14729327
- Gendron P, Lemieux S, Major F. Quantitative analysis of nucleic acid three-dimensional structures. J Mol Biol 2001; 308:919 - 36; http://dx.doi.org/10.1006/jmbi.2001.4626; PMID: 11352582
- Zhang Q, Kang M, Peterson RD, Feigon J. Comparison of solution and crystal structures of preQ1 riboswitch reveals calcium-induced changes in conformation and dynamics. J Am Chem Soc 2011; 133:5190 - 3; http://dx.doi.org/10.1021/ja111769g; PMID: 21410253
- Bloomfield VA, Crothers DM, Ignacio T, Hearst JE, Wemmer DE, Killman PA, et al. Nucleic acids: structures, properties, and functions. University Science Books. 2000.
- Kowalak JA, Bruenger E, McCloskey JA. Posttranscriptional modification of the central loop of domain V in Escherichia coli 23 S ribosomal RNA. J Biol Chem 1995; 270:17758 - 64; http://dx.doi.org/10.1074/jbc.270.30.17758; PMID: 7629075
- Dalluge JJ, Hashizume T, Sopchik AE, McCloskey JA, Davis DR. Conformational flexibility in RNA: the role of dihydrouridine. Nucleic Acids Res 1996; 24:1073 - 9; http://dx.doi.org/10.1093/nar/24.6.1073; PMID: 8604341
- Mortimer SA, Weeks KM. C2′-endo nucleotides as molecular timers suggested by the folding of an RNA domain. Proc Natl Acad Sci USA 2009; 106:15622 - 7; http://dx.doi.org/10.1073/pnas.0901319106; PMID: 19717440
- LaGrandeur TE, Hüttenhofer A, Noller HF, Pace NR. Phylogenetic comparative chemical footprint analysis of the interaction between ribonuclease P RNA and tRNA. EMBO J 1994; 13:3945 - 52; PMID: 7521296
- Leontis NB, Westhof E. A common motif organizes the structure of multi-helix loops in 16 S and 23 S ribosomal RNAs. J Mol Biol 1998; 283:571 - 83; http://dx.doi.org/10.1006/jmbi.1998.2106; PMID: 9784367
- Kolev NG, Steitz JA. In vivo assembly of functional U7 snRNP requires RNA backbone flexibility within the Sm-binding site. Nat Struct Mol Biol 2006; 13:347 - 53; http://dx.doi.org/10.1038/nsmb1075; PMID: 16547514
- Fulle S, Gohlke H. Statics of the ribosomal exit tunnel: implications for cotranslational peptide folding, elongation regulation, and antibiotics binding. J Mol Biol 2009; 387:502 - 17; http://dx.doi.org/10.1016/j.jmb.2009.01.037; PMID: 19356596
- Davidovich C, Bashan A, Auerbach-Nevo T, Yaggie RD, Gontarek RR, Yonath A. Induced-fit tightens pleuromutilins binding to ribosomes and remote interactions enable their selectivity. Proc Natl Acad Sci USA 2007; 104:4291 - 6; http://dx.doi.org/10.1073/pnas.0700041104; PMID: 17360517
- Heus HA, Hilbers CW. Structures of non-canonical tandem base pairs in RNA helices: review. [review] Nucleosides Nucleotides Nucleic Acids 2003; 22:559 - 71; http://dx.doi.org/10.1081/NCN-120021955; PMID: 14565230
- Garst AD, Héroux A, Rambo RP, Batey RT. Crystal structure of the lysine riboswitch regulatory mRNA element. J Biol Chem 2008; 283:22347 - 51; http://dx.doi.org/10.1074/jbc.C800120200; PMID: 18593706
- Rieder U, Kreutz C, Micura R. Folding of a transcriptionally acting preQ1 riboswitch. Proc Natl Acad Sci USA 2010; 107:10804 - 9; http://dx.doi.org/10.1073/pnas.0914925107; PMID: 20534493
- Liberman JA, Wedekind JE. Riboswitch structure in the ligand-free state. Wiley Interdiscip Rev RNA 2012; 3:369 - 84; http://dx.doi.org/10.1002/wrna.114; PMID: 21957061
- Serganov A, Patel DJ. Molecular recognition and function of riboswitches. Curr Opin Struct Biol 2012; 22:279 - 86; http://dx.doi.org/10.1016/j.sbi.2012.04.005; PMID: 22579413
- Gupta A, Gribskov M. The role of RNA sequence and structure in RNA--protein interactions. J Mol Biol 2011; 409:574 - 87; http://dx.doi.org/10.1016/j.jmb.2011.04.007; PMID: 21514302
- Treger M, Westhof E. Statistical analysis of atomic contacts at RNA-protein interfaces. J Mol Recognit 2001; 14:199 - 214; http://dx.doi.org/10.1002/jmr.534; PMID: 11500966
- Bahadur RP, Zacharias M, Janin J. Dissecting protein-RNA recognition sites. Nucleic Acids Res 2008; 36:2705 - 16; http://dx.doi.org/10.1093/nar/gkn102; PMID: 18353859
- Daubner GM, Cléry A, Jayne S, Stevenin J, Allain FH. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J 2012; 31:162 - 74; http://dx.doi.org/10.1038/emboj.2011.367; PMID: 22002536
- Hermann T, Patel DJ. Adaptive recognition by nucleic acid aptamers. Science 2000; 287:820 - 5; http://dx.doi.org/10.1126/science.287.5454.820; PMID: 10657289
- Cazals F, Proust F, Bahadur RP, Janin J. Revisiting the Voronoi description of protein-protein interfaces. Protein Sci 2006; 15:2082 - 92; http://dx.doi.org/10.1110/ps.062245906; PMID: 16943442
- Voss NR, Gerstein M, Steitz TA, Moore PB. The geometry of the ribosomal polypeptide exit tunnel. J Mol Biol 2006; 360:893 - 906; http://dx.doi.org/10.1016/j.jmb.2006.05.023; PMID: 16784753
- Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics 2009; 10:168; http://dx.doi.org/10.1186/1471-2105-10-168; PMID: 19486540
- Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA 2001; 7:499 - 512; http://dx.doi.org/10.1017/S1355838201002515; PMID: 11345429
- Gabb HA, Sanghani SR, Robert CH, Prévost C. Finding and visualizing nucleic acid base stacking. J Mol Graph 1996; 14:6 - 11, 23-4; http://dx.doi.org/10.1016/0263-7855(95)00086-0; PMID: 8744567
- Leontis NB, Stombaugh J, Westhof E. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res 2002; 30:3497 - 531; http://dx.doi.org/10.1093/nar/gkf481; PMID: 12177293
- Banatao DR, Altman RB, Klein TE. Microenvironment analysis and identification of magnesium binding sites in RNA. Nucleic Acids Res 2003; 31:4450 - 60; http://dx.doi.org/10.1093/nar/gkg471; PMID: 12888505
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 1995; 8:127 - 34; http://dx.doi.org/10.1093/protein/8.2.127; PMID: 7630882
- McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol 1994; 238:777 - 93; http://dx.doi.org/10.1006/jmbi.1994.1334; PMID: 8182748
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, et al. TM4 microarray software suite. Methods Enzymol 2006; 411:134 - 93; http://dx.doi.org/10.1016/S0076-6879(06)11009-5; PMID: 16939790