Abstract
Eukaryotic cells rapidly adjust the levels of mRNAs in response to environmental stress primarily by controlling transcription and mRNA turnover. How different stress conditions influence the fate of stress-responsive mRNAs, however, is relatively poorly understood. This is largely due to the fact that mRNA half-life assays are traditionally based on interventions (e.g., temperature-shifts using temperature-sensitive RNA polymerase II alleles or treatment with general transcription inhibitory drugs), which, rather than blocking, specifically induce transcription of stress-responsive genes. To study the half-lives of the latter suite of mRNAs, we developed and describe here a minimally perturbing alternative method, coined CEO, which is based on discontinuance of transcription following the conditional excision of open reading frames. Using CEO, we confirm that the target of rapamycin complex I (TORC1), a nutrient-activated, central stimulator of eukaryotic cell growth, favors the decay of mRNAs that depend on the stress- and/or nutrient-regulated transcription factors Msn2/4 and Gis1 for their transcription. We further demonstrate that TORC1 controls the stability of these mRNAs via the Rim15-Igo1/2-PP2ACdc55 effector branch, which reportedly also controls Gis1 promoter recruitment. These data pinpoint PP2ACdc55 as a central node in homo-directional coordination of transcription and post-transcriptional mRNA stabilization of a specific array of nutrient-regulated genes.
Introduction
mRNA turnover is a key factor in the regulation of eukaryotic gene expression, which, in parallel to transcriptional activation/repression, allows cells to rapidly adjust their levels of specific transcripts in response to environmental stress conditions. Information on how different stress conditions impact on the post-transcriptional fate of stress-responsive mRNAs, however is still quite limited. Studies in Saccharomyces cerevisiae for instance suggest that heat shock favors the stabilization of heat-shock inducible mRNAs, while pre-existing mRNAs may suffer from accelerated degradation via the 5′-3′ mRNA decay pathway or be shifted from polysomes toward a repressing mRNA state within P bodies.Citation1-Citation3 Similarly, nutrient limitation or inactivation of the nutrient-regulated target of rapamycin complex 1 (TORC1) broadly stimulates 5′-3′ mRNA decay,Citation4 while specifically endorsing the protection of newly expressed mRNAs of genes (e.g., HSP26) that are controlled by the stress-responsive and/or nutrient-regulated transcription factors Msn2, Msn4 and Gis1.Citation5
The limited amount of information on mRNA half-lives of stress-responsive genes is primarily due to the fact that most of the currently available methods to assess mRNA half-lives suffer from caveats that specifically affect stress-responsive mRNAs. For instance, assays that allow determination of mRNA half-lives are traditionally based on inhibition of general transcription using either a temperature-sensitive allele of the catalytic subunit of RNA polymerase II (RNA Pol II; i.e., rpb1-1),Citation6 or a variety of transcription inhibitors (e.g., thiolutin and 1,10-phenanthroline).Citation7 However, both interventions per se, namely temperature-shift (when using rpb1-1 strains) and thiolutin/1,10-phenanthroline treatment, can alter mRNA stability and, rather than blocking, in fact strongly induce transcription of heat-shock and/or stress-responsive genes.Citation8-Citation12 In addition, since stress-responsive genes depend on their native promoters for normal induction under stress conditions, alternative methods that make use of conditionally repressible promoters to downregulate transcription (such as the Tetoff system)Citation7 cannot be applied to assess the half-life of the respective mRNAs. Most recent mRNA half-life assays are based on metabolic labeling of newly transcribed mRNAs with 4-thiouracil (4-tU) or 4-thiouridine (4-sU), which requires the heterologous expression of a human nucleoside transporter (hENT1) to enable 4-sU uptake by yeast cells.Citation13,Citation14 Potential concerns regarding the applicability of 4-tU-pulse/uracil-chase experiments to assess the half-lives of newly expressed mRNAs under dynamic stress conditions, however, include the required long pulse/labeling period (i.e. > 4 h) and the possibility of uncontrolled 4-tU recycling within cells.Citation15 Finally, dynamic transcriptome analysis (DTA), which combines metabolic mRNA labeling with dynamic kinetic modeling, is applicable on a genome-wide level and may currently be best suited to estimate mRNA half-lives under stress conditions.Citation13,Citation16 A theoretical concern of this method, which is technically challenging,Citation15 may be the fact that it is built on the assumption that mRNA synthesis and decay rates remain constant during the labeling time.
To specifically study the mRNA half-life of individual stress-responsive mRNAs, we developed a minimally perturbing method, which is not susceptible to any of the caveats of the classical mRNA half-life studies outlined above. Our method is based on the excision from the genome of a given loxP-flanked open reading frame, and hence discontinuance of transcription, following conditional nuclear targeting of the Cre recombinase. Using this assay, which we coin CEO (for conditional excision of the open reading frame [ORF]), we confirm our previous data, which indicated that inactivation of TORC1 results in stabilization of newly expressed mRNAs of Msn2-, Msn4- and Gis1-controlled genes.Citation5,Citation17-Citation19 Using CEO, we extend our earlier studies and demonstrate that TORC1 controls mRNA stability via the Cdc55-protein phosphatase 2A (PP2ACdc55), which we recently found to be also implicated in direct regulation of the transcription factor Gis1.Citation18 Thus, our data indicate the existence of a mechanism that allows cells to tightly coordinate transcription with the post-transcriptional fate of specific mRNAs under nutrient stress conditions.
Results
The experimental system
To design our method, we used a Cre recombinase that is fused to the estradiol-binding domain (EBD) of the murine estrogen receptor (Cre-EBD78),Citation20,Citation21 which mediates the cytoplasmic to nuclear translocation of the fusion protein following its association with estradiol. This construct can therefore be used to conditionally grant the Cre recombinase access to loxP sites in the genome.Citation22 Stable and constitutive expression (from the GPD1 promoter) of the Cre-EBD78 fusion protein was achieved by integration of the corresponding fusion gene at the ADE3 locus in the genome (; of note, expression of this fusion protein as such did not measurably alter the expression profile of HSP26 [or of RTN2 or CIT2; see below] in exponentially growing and rapamycin-treated wild-type cells; data not shown). In parallel, loxP sites were introduced just downstream of and in-frame with the ATG as well as downstream of the 3′ untranslated region (UTR) of the HSP26 gene using the single-step PCR-based gene replacement technique (Material and Methods; ).Citation23 Addition of estradiol to yeast cells containing both reporter constructs should therefore trigger nuclear transfer of the Cre-EBD78 fusion protein and consequent excision of the HSP26 ORF (including the HSP26 - 3′ (a) region; ). To verify this assumption, exponentially growing wild-type cells harboring the two reporter constructs (referred to as “reporter cells/strain” hereafter) were treated with rapamycin for 60 min to induce the expression of HSP26 (which was normal when compared with wild-type cells bearing no reporter constructs; ). The culture was then equally split and the two subcultures were either treated with vehicle alone or with estradiol, which per se did not significantly alter the HSP26 expression levels in rapamycin-treated wild-type cells carrying no reporter genes (). As expected, estradiol treatment, but not treatment with the vehicle alone, triggered the excision of the ORF at the HSP26 locus in reporter cells (). Notably, most of the HSP26 loci (i.e. > 96%) were excised within 20 min following a 50 min lag period () and the remaining fraction of HSP26 loci contributed only marginally (i.e. < 3%) to the existing pool of HSP26 mRNAs (as assessed in separate experiments in which HSP26 mRNA expression was quantified during a 1 h rapamycin treatment in reporter cells that had been first exposed to estradiol for 70 min; data not shown). To determine the half-life of HSP26 mRNAs, we have therefore started to collect samples (in 10 min intervals) 70 min following estradiol treatment, at which point in time HSP26 mRNA levels were maximally high in this experiment. Using this method, we determined a half-life of 55 min for HSP26 mRNA in rapamycin treated (130 min) cells ().
Figure 1. Schematic representation of the relevant features of the various plasmids used in this study. The NdeI-SphI 10.4-kb fragment from plasmid pNT081 can be integrated at the ADE3 locus via homologous recombination and mediates constitutive expression of Cre-EBD78. Plasmids pNT082, pNT084 and pNT085 contain the HSP26, RTN2 and CIT2 reporter constructs that, following excision from the plasmids with XbaI/SpeI, KpnI or NaeI/KpnI as indicated, can be integrated at the respective endogenous loci via homologous recombination. The plasmid pNT083 serves as a backbone to facilitate construction of any reporter construct of choice. Please note that the loxP site codes for 12 additional amino acids and cloning of any ORF should be designed to be in-frame with the 5′ and 3′ ends of the indicated loxP-derived reading frame. For a more detailed description of plasmid constructions, please see the section Materials and Methods.
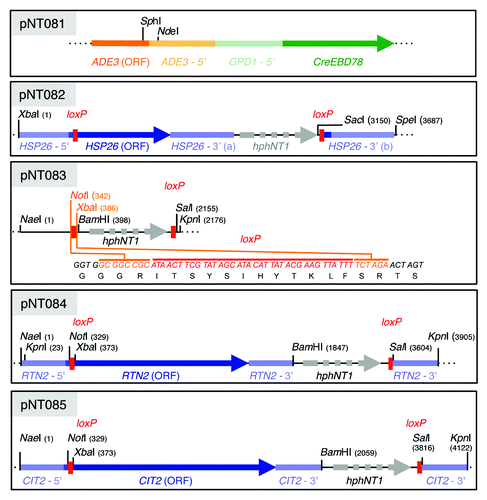
Figure 2. Description of the CEO method. (A) The Cre-EBD78 fusion protein (expressed under the control of the GPD1 promoter) is predominantly localized in the cytoplasm in the absence of estradiol, but shuttles into the nucleus following estradiol (1 µM) addition to reporter cells. Nuclear Cre-EBD78 mediates recombination between loxP sites that are introduced (in tandem orientation) in the genome just downstream of and in-frame with the ATG as well as downstream of the 3′UTR of target genes using the single-step PCR-based gene replacement technique (and the hygromycin-resistance gene (hphNT1) as selectable marker). As a proof of principle, exponentially growing wild-type cells harboring the Cre-EBD78 and loxP-HSP26-loxP reporter constructs (NT393-6D) were treated with rapamycin (200 ng ml−1) to induce the expression of HSP26, followed by addition of estradiol (at time 60 min) to trigger excision of the HSP26 ORF and the hphNT1 cassette from the genome (as illustrated). The efficacy of HSP26 excision was assessed by qPCR using the indicated primers (P1 and P2) and genomic DNA as template. Bar graphs show the mean levels of three independent experiments (± S.D.) of PCR-amplified HSP26 (normalized to 1.0 for exponentially growing, untreated cells) in cells treated with either vehicle alone (v; on the left) or with estradiol (on the right) for the times indicated. Samples to determine the half-life of HSP26 mRNA were collected (at 10 min intervals) 70 min following vehicle or estradiol treatment and the respective data are presented in . (B) qRT-PCR analysis of HSP26 mRNA induction in wild-type (WT) and loxP-HSP26-loxP reporter cells (same as in (A)) treated for the times indicated with 200 ng ml−1 rapamycin. Mean HSP26 mRNA levels of three independent experiments (± S.D.), normalized to the values at 60 min, are shown. Notably, in terms of absolute levels, the rapamycin-induced accumulation of HSP26 mRNA (and of Hsp26 protein) did not significantly differ in loxP-HSP26-loxP reporter cells when compared with wild-type cells (data not shown). (C) Estradiol per se (added at time 60 min), like vehicle alone, does not interfere with rapamycin-induced HSP26 expression (quantified by qRT-PCR as in (B)) in wild-type cells.
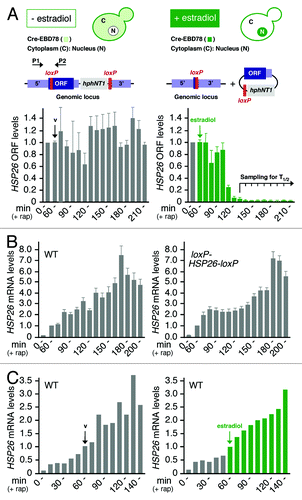
Figure 3. TORC1 controls mRNA stability via the PP2ACdc55-inhibitory endosulfines. (A–E) qRT-PCR analysis of HSP26 (A and D), RTN2 (B and E) and CIT2 (C) mRNA levels in rapamycin-treated cells. mRNA samples were harvested from cells, which harbored the Cre-EBD78 and an appropriate loxP-ORF-loxP reporter construct (see ) and that were treated with rapamycin and estradiol following the protocol outlined in . The values for the reference samples at time point 0 (corresponding to the time point 130 min of the rapamycin treatment) were normalized to 1.0 for each strain. Notably, the Rim15-Igo1/2-PP2ACdc55 signaling branch also regulates transcription (in part via Gis1) of HSP26 and RTN2, but not of CIT2 (see also the model in ).Citation5,Citation18,Citation31 Before their normalization to 1.0, the relative levels of HSP26 and RTN2 mRNAs, but not the ones of CIT2, therefore differed significantly between the various mutant strains. Accordingly, the HSP26 mRNA levels in rim15∆, igo1/2∆, cdc55∆, cdc55∆ rim15∆ and cdc55∆ igo1/2∆ cells were 23%, 13%, 225%, 170% and 196%, respectively, when compared with those in wild-type cells (in (A and D)). Similarly, RTN2 mRNA levels in rim15∆, igo1/2∆, cdc55∆, cdc55∆ rim15∆ and cdc55∆ igo1/2∆ cells were 17%, 25%, 155%, 150% and 132%, respectively, when compared with those in wild-type cells (in (B and E)). Estradiol-induced excision of the loxP-ORF-loxP loci was verified independently and found to be at least 85% complete for each strain at time point 0 (i.e. 70 min following estradiol addition). Data points represent means ± S.D. of three independent experiments.
To demonstrate that CEO is more broadly applicable, we also studied two additional genes (i.e., RTN2 and CIT2) and their respective mRNAs, which we previously found to be strongly induced following rapamycin treatment.Citation5 To this end, we constructed an additional hygromycin-selectable plasmid (pNT083) containing the two loxP sites and several restriction sites that facilitate construction of the DNA fragments that allow introduction of the loxP-reporter genes by single-step gene replacement in yeast (Materials and Methods; ). Like the HSP26 reporter gene described above, Cre-EBD78-excisable RTN2 and CIT2 reporter genes were efficiently (and with similar kinetics) removed from the genomes of the respective reporter strains following addition of estradiol, which on its own had no detectable effect on RTN2 or CIT2 expression (not shown). Using the same conditions as above for determination of the HSP26 mRNA half-life, we found that RTN2 and CIT2 mRNAs exhibited half-lives of 34 and 38 min, respectively, in rapamycin-treated cells ().
Validation of CEO
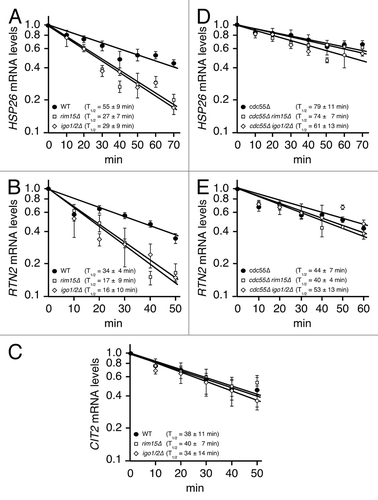
We have previously reported that inactivation of TORC1 results in stabilization of newly expressed mRNAs of Msn2-, Msn4- and Gis1-controlled genes via a process that depends on the greatwall protein kinase ortholog Rim15 and its targets Igo1/2.Citation5 In this study, the experimental caveats of mRNA half-life assays (outlined above) were circumvented by rendering HSP26 transcription doxycycline-repressible due to the insertion of a series of doxycycline-responsive tetO elements in the HSP26 promoter. Notably, identification of sites in the HSP26 promoter that tolerated the respective tetO insertions without interfering with normal transcriptional regulation of HSP26 was challenging and time-consuming. Using the much easier applicable CEO assay, we confirm here our earlier results that loss of Rim15 or of Igo1/2 reduced the HSP26 mRNA half-life in rapamycin-treated cells about 2-fold (). To further extend these data, we also studied the half-lives of two additional mRNAs, which were encoded by genes that either depended (i.e., RTN2), or did not depend (i.e., CIT2), on the presence of Rim15 and Igo1/2 for normal expression following TORC1 inactivation.Citation5 In line with our expectations, we found that the half-life of the RTN2 mRNA, but not the one of CIT2 mRNA, was reduced 2-fold in rapamycin treated rim15∆ and igo1∆ igo2∆ cells when compared with wild-type cells (). These data not only demonstrate the validity of CEO for mRNA half-life studies, but also corroborate our earlier conclusion that TORC1 controls the stability of a specific set of mRNAs via Rim15-Igo1/2.Citation5,Citation17,Citation24,Citation25
TORC1 controls mRNA stability via the PP2ACdc55-inhibitory endosulfines
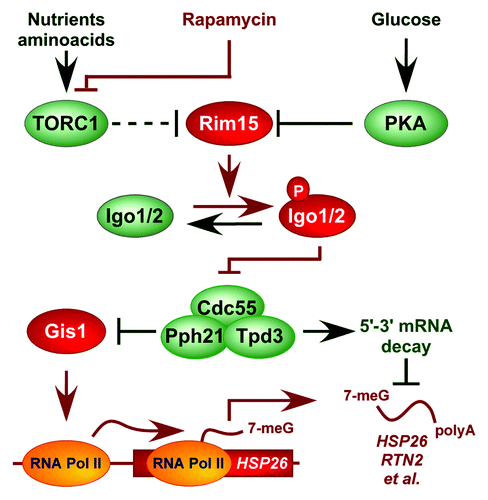
We recently discovered that Rim15, analogous to the greatwall kinase in Xenopus, phosphorylates endosulfines (i.e. Igo1/2) to directly inhibit the Cdc55-protein phosphatase 2A (PP2ACdc55).Citation18 Accordingly, when phosphorylated by Rim15 on Ser64, Igo1 directly binds to and prevents PP2ACdc55 from dephosphorylating various target proteins among which we identified the transcription factor Gis1. Inhibition of PP2ACdc55 preserves Gis1 in a phosphorylated state and consequently promotes its recruitment to and activation of transcription from promoters of specific nutrient-regulated genes such as RTN2. Together with our earlier studies, these recent data suggest that Igo1/2, following their activation by Rim15, stimulate both transcriptional activation of Gis1-dependent genes and post-transcriptional stability of the respective mRNAs by inhibiting PP2ACdc55. To address this assumption further, using the CEO method, we studied whether loss of Cdc55 may suppress the defect in HSP26 and RTN2 mRNA stability that we observed in rapamycin-treated rim15∆ and igo1∆ igo2∆ cells (). Mutant cdc55∆ cells exhibited slightly enhanced (i.e. 1.3–1.4-fold) HSP26 and RTN2 mRNA half-lives when compared with wild-type cells treated with rapamycin (). Interestingly, in contrast to the situation in wild-type cells, loss of Rim15 or of Igo1/2 had no significant impact on the observed HSP26 and RTN2 mRNA half-lives in cdc55∆ cells under the same conditions (). Thus, the enhanced turnover of HSP26 and RTN2 mRNAs in rapamycin-treated rim15∆ and igo1/2∆ cells can be suppressed by loss of Cdc55. These data extend our previous model and suggest that TORC1 controls in fact both transcriptional activation of Gis1-dependent genes and post-transcriptional stability of the respective mRNAs via the Rim15-Igo1/2- PP2ACdc55 effector branch ().
Figure 4. Model for the role of Igo1/2-PP2ACdc55 in controlling expression of nutrient-regulated genes. Arrows and bars denote positive and negative interactions, respectively. TORC1 regulates Rim15 indirectly via Sch9.Citation25 For details, see text.
Discussion
Here we describe an alternative method to study the mRNA half-life of stress-responsive genes in yeast that is based on the conditional excision of ORFs (CEO). CEO offers a distinct advantage over conventional mRNA half-life assays that build on the inactivation of transcription using either the temperature-sensitive rpb1-1 allele or a variety of specific transcription inhibitors (see Introduction). Accordingly, CEO relies on estradiol treatment (to induce cytoplasmic to nuclear transfer of the Cre-EBD78 fusion protein), which, unlike heat-shock (when using rpb1-1 mutants) or various treatments with transcription inhibitory drugs, per se does neither induce the expression nor interfere with the normal (rapamycin-mediated) regulation of stress-responsive genes such as HSP26, RTN2 and CIT2 ( and data not shown). Based on our observation that estradiol-induced excision of various target ORFs was exerted to a large extent within a time frame of 10–20 min (following a lag period of 50 min), we infer that CEO represents a valid method to determine the stability of those mRNA species that exhibit half-lives of at least 10–20 min. An additional advantage of CEO is that it preserves the endogenous structures of the 5′/3′ UTRs and the promoters of the genes under study, which makes CEO less likely to alter the fate of mRNAs than traditional methods that are based on the use heterologous repressible promoters.Citation26-Citation30 Although it remains theoretically possible that the insertion of a loxP site within the ORF of a given target gene may affect its expression, our control experiments, in which we compared the level of HSP26 mRNAs between rapamycin-treated wild-type and loxP-HSP26-loxP cells, indicated that the introduction of a loxP site within the HSP26 ORF per se is minimally invasive with no significant effect on the expression and/or stability of HSP26 mRNAs. Taken together, despite a few limitations (regarding the resolution of half-lives of very short-lived mRNAs and the unsuitability for genome-wide analyses), CEO offers a valid alternative to sample the mRNA half-life of stress-responsive genes, which, together with the toolbox offered here, only requires relatively easily applicable standard yeast genetic methods. Finally, although not specifically studied here, we would like to point out that CEO may in principle also be applied to the analysis of gene function as it allows conditional inactivation of specific genes. Provided that the respective gene products have a relatively limited half-life, CEO could be particularly useful to replace and/or complement analyses of essential genes that have hitherto relied on the use of temperature-sensitive alleles.
The use of CEO allowed us to confirm our previous finding that the protein kinase Rim15, which is indirectly and negatively controlled by TORC1,Citation24,Citation25,Citation31,Citation32 plays a key role in stabilizing mRNAs of Msn2/4- and Gis1-controlled genes via its direct targets Igo1/2. More recently, we discovered that, following their phosphorylation by Rim15, Igo1/2 serve to inhibit PP2ACdc55, thereby favoring promoter recruitment of and transcription by the transcription factor Gis1.Citation18 These new data raised the question whether Igo1/2-mediated inhibition of PP2ACdc55 may, in addition to its role in transcription, also be implicated in post-transcriptional control of mRNA stability. Our data presented here indicate that this is the case. Thus, Igo1/2-dependent regulation of PP2ACdc55 appears to represent a key node in nutrient-sensitive, homo-directional coordination of transcription and post-transcriptional mRNA stabilization of a specific array of genes (). Interestingly, loss of components of the 5′-3′ mRNA decay pathway, like loss of Cdc55, suppresses the defect in HSP26 expression in rapamycin-treated igo1/2∆ cells.Citation5,Citation17,Citation18 It is therefore possible that PP2ACdc55 regulates, in addition to Gis1, a specific protein(s) within the 5′-3′ mRNA decay pathway. In this context, our previously published phosphoproteome studies pinpointed, among others, Vts1 as a potential PP2ACdc55 target.Citation18 Vts1 is a member of the Smaug family of proteins, which directly binds a defined RNA motif in target mRNAs of nutrient-regulated genes and controls their stability by interfering with the 5′-3′ mRNA decay pathway.Citation33,Citation34 It will therefore be interesting to explore in future studies whether parallel, temporally coordinated regulation by the Igo1/2-PP2ACdc55 module of both Gis1 and Vts1 (or of other candidate proteins) may allow cells to coordinate transcription and posttranscriptional mRNA stability.
Materials and Methods
Strains and growth conditions
S. cerevisiae strains () were grown at 30°C in standard rich medium (YPD) with 2% glucose or in synthetic defined (SD) medium (0.17% yeast nitrogen base, 0.5% ammonium sulfate, and 2% glucose) complemented with the appropriate nutrients for plasmid maintenance. Rapamycin and estradiol were used at a concentration of 200 ng ml−1 and 1 µM, respectively.
Table 1. Strains used in this study
Plasmid constructions
The plasmids used in this study are depicted in . pNT081, containing part of the ADE3 gene (i.e. 600 bp upstream and 400 bp downstream of the ATG), is derived from pGPD1:Cre-EBD78.Citation20,Citation21 Digestion of pNT081 with NdeI and SphI generates a 10.4 kb fragment that drives integration of the GPD1:Cre-EBD78 cassette into the genomic ADE3 locus in yeast. Positive clones can be selected using the LEU2 marker that is also present on this fragment. pNT082 was constructed by inserting, via homologous recombination, elements of the HSP26 gene into the pFA6a-CEN4-ASK1-loxP-HphNT1 plasmid (kind gift of Yves Barral). Digestion of pNT082 with XbaI and SpeI generates two fragments (3,686 and 3,240 bp) of which the 3,686 bp fragment can be inserted at the HSP26 locus by homologous recombination. As illustrated in , this fragment contains sequentially the 347 bp upstream of and including the ATG plus the first 18 bp of the HSP26 ORF (HSP26-5′), a first loxP site that preserves the reading frame with respect to the preceding ATG and the following HSP26 ORF (encoding amino acids 2–214), the 428 bp downstream of and including the stop codon of HSP26 (HSP26 - 3′ (a)), the hphNT1 cassette, a second loxP site (in tandem orientation with the first loxP site) and the last 42 bp of the HSP26 ORF, including the stop codon followed by 343 bp of the 3′ region (HSP26 - 3′ (b)). The hphNT1 cassette allows expression of the hygromycin B resistance gene under the control of the TEF1 promoter. pNT083 is based on pRS316 and was designed to facilitate construction of reporter cassettes for use in CEO assays. To this end, a fragment containing the 300 bp upstream of and including the ATG plus the first 15 bp of the ORF of a given gene can be cloned at the NaeI and NotI sites (or only at NotI) in pNT083 (in-frame with the subsequent loxP-encoded sequence indicated in ). A second fragment coding for the entire ORF (except the ATG) plus at least 300 bp downstream of and including the stop codon can be cloned at the XbaI and BamHI sites (or only at XbaI; in frame with the preceding loxP-encoded sequence). Finally, a third fragment including at least the first 300 bp of the 3′ region following the stop codon can be cloned at the SalI and/or KpnI sites. For homologous recombination in yeast, the entire cassette can then be removed from the plasmid by digestion with NaeI and KpnI. Both plasmids pNT084 and pNT085 were constructed using this approach and pNT083 as template. Accordingly, pNT084 contains sequentially the 300 bp upstream of and including the ATG plus the first 18 bp of RTN2 (RTN2-5′), a first loxP site that preserves the reading frame with respect to the preceding ATG and the following RTN2 ORF (encoding amino acids 4–393), the 295 bp downstream of and including the RTN2 stop codon (RTN2-3′), the hphNT1 cassette, a second loxP site (in tandem orientation with the first loxP site) and the 295 bp downstream of the RTN2 stop codon (RTN2-3′). Digestion of pNT084 with KpnI generates two fragments (3,882 and 4,482 bp) of which the 3,882 bp fragment can be inserted at the RTN2 locus by homologous recombination. Similarly, the pNT085 contains sequentially the 295 bp upstream of and including the ATG plus the first 18 bp of CIT2 (CIT2-5′), a first loxP site that preserves the reading frame with respect to the preceding ATG and the following CIT2 ORF (encoding amino acids 2–460), the 300 bp downstream of and including the CIT2 stop codon (CIT2-3′), the hphNT1 cassette, a second loxP site (in tandem orientation with the first loxP site) and the 300 bp downstream of the CIT2 stop codon (CIT2-3′). Digestion of pNT085 with NaeI and KpnI generates two fragments (4,121 and 4,460 bp) of which the 4,121 bp fragment can be inserted at the CIT2 locus by homologous recombination.
Real-time qPCR and qRT-PCR
High-quality yeast genomic DNA preparation was mainly performed as described previously with minor modifications.Citation35 At each step, one-tenth of the indicated values was used. Total RNA was extracted using the hot acidic phenol method. DNA was removed with the DNA-free Kit (Ambion) and first-strand cDNA was synthesized with the PrimeScript RT Reagent Kit (TaKaRa). Quantitative RT-PCR (qRT-PCR) was performed on a Rotor-Gene 6000 machine (Corbett Life Science), with the 5 x EvaGreen QPCR Mix II no Rox (Bio&SELL) or with the QuantiTect SYBR Green PCR Kit (Qiagen). HSP26, RTN2 and CIT2 DNA and mRNA levels were normalized with respect to NSR1 DNA or mRNA. Oligonucleotides used for qPCR and qRT-PCR were as follows: AAGGCCGTCTCTTCCTCTTC (NSR1qF2); GGTTTCAGCTTCACTTTCGC (NSR1qR2); TGTTTTTAATCCATTCACCTTTCAT (HSP26 gF2; P1); CCTTCACCCAGCAATCTGTT (HSP26 gR2; P2); AGTTAGCAAACACACCCGCAAAGG (HSP26qF1); TGCTCTCCTTGACCTTGACC (HSP26qR1); TCTGGTGAGTTTGCTGATCC (RTN2qF1); GCGGTCGATATGAGGCTTTAAG (RTN2qR1); ATCACCTTATCTGTCCCTTGC (CIT2qF1); ACAGCATGACCATAACCGG (CIT2qR1).
| Abbreviations: | ||
| CEO | = | conditional excision of open reading frames |
| TORC1 | = | target of rapamycin complex I |
| RNA | = | ribonucleic acid |
| mRNA | = | messenger RNA |
| Tet | = | tetracycline |
| 4-tU | = | 4-thiouracil |
| 4-sU | = | 4-thiouridine |
| DTA | = | dynamic transcriptome analysis |
| loxP | = | locus of X-over P1 |
| Cre | = | causes recombination |
| PP2A | = | protein phosphatase type 2A |
| YPD | = | yeast peptone dextrose |
| SD | = | synthetic defined |
| bp | = | base pair |
| ORF | = | open reading frame |
| PCR | = | polymerase chain reaction |
| qPCR | = | quantitative PCR |
| qRT-PCR | = | quantitative reverse transcription PCR |
| DNA | = | desoxyribonucleic acid |
| cDNA | = | complementary DNA |
| EBD | = | estradiol-binding |
| UTR | = | untranslated region |
Acknowledgments
We thank Yves Barral for providing plasmids and Marie-Pierre Péli-Gulli for critical comments on the manuscript. This research was supported by the Canton of Fribourg and the Swiss National Science Foundation (C.D.V.).
Submitted
04/30/13
Revised
06/06/13
Accepted
06/10/13
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author’s Contribution
N.T. designed and performed the experiments. All authors contributed to the data analysis and creation of figures and commented on the manuscript, which was written by C.D.V.
References
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA 2002; 99:5860 - 5; http://dx.doi.org/10.1073/pnas.092538799; PMID: 11972065
- Bond U. Stressed out! Effects of environmental stress on mRNA metabolism. FEMS Yeast Res 2006; 6:160 - 70; http://dx.doi.org/10.1111/j.1567-1364.2006.00032.x; PMID: 16487339
- Gasch A. The environmental stress response: a common yeast response to environmental stresses. In: Hohmann S, Mager P (eds). Yeast stress responses. Springer, Berlin Heidelberg, New York. 2002
- Albig AR, Decker CJ. The target of rapamycin signaling pathway regulates mRNA turnover in the yeast Saccharomyces cerevisiae.. Mol Biol Cell 2001; 12:3428 - 38; PMID: 11694578
- Talarek N, Cameroni E, Jaquenoud M, Luo X, Bontron S, Lippman S, et al. Initiation of the TORC1-regulated G0 program requires Igo1/2, which license specific mRNAs to evade degradation via the 5′-3′ mRNA decay pathway. Mol Cell 2010; 38:345 - 55; http://dx.doi.org/10.1016/j.molcel.2010.02.039; PMID: 20471941
- Nonet M, Scafe C, Sexton J, Young R. Eucaryotic RNA polymerase conditional mutant that rapidly ceases mRNA synthesis. Mol Cell Biol 1987; 7:1602 - 11; http://dx.doi.org/10.1128/MCB.7.5.1602; PMID: 3299050
- Coller J. Methods to determine mRNA half-life in Saccharomyces cerevisiae.. Methods Enzymol 2008; 448:267 - 84; http://dx.doi.org/10.1016/S0076-6879(08)02614-1; PMID: 19111181
- Adams CC, Gross DS. The yeast heat shock response is induced by conversion of cells to spheroplasts and by potent transcriptional inhibitors. J Bacteriol 1991; 173:7429 - 35; PMID: 1938939
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, et al. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 1998; 95:717 - 28; http://dx.doi.org/10.1016/S0092-8674(00)81641-4; PMID: 9845373
- Li B, Nierras CR, Warner JR. Transcriptional elements involved in the repression of ribosomal protein synthesis. Mol Cell Biol 1999; 19:5393 - 404; http://dx.doi.org/10.1152/ajpgi.00315.2009; PMID: 10409730
- Herruer MH, Mager WH, Raué HA, Vreken P, Wilms E, Planta RJ. Mild temperature shock affects transcription of yeast ribosomal protein genes as well as the stability of their mRNAs. Nucleic Acids Res 1988; 16:7917 - 29; http://dx.doi.org/10.1093/nar/16.16.7917; PMID: 3047675
- Pelechano V, Pérez-Ortín JE. The transcriptional inhibitor thiolutin blocks mRNA degradation in yeast. Yeast 2008; 25:85 - 92; http://dx.doi.org/10.1002/yea.1548; PMID: 17914747
- Miller C, Schwalb B, Maier K, Schulz D, Dümcke S, Zacher B, et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol Syst Biol 2011; 7:458; http://dx.doi.org/10.1038/msb.2010.112; PMID: 21206491
- Munchel SE, Shultzaberger RK, Takizawa N, Weis K. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell 2011; 22:2787 - 95; http://dx.doi.org/10.1091/mbc.E11-01-0028; PMID: 21680716
- Pérez-Ortín JE, Alepuz P, Chávez S, Choder M. Eukaryotic mRNA Decay: Methodologies, Pathways, and Links to Other Stages of Gene Expression. J Mol Biol 2013; In press http://dx.doi.org/10.1016/j.jmb.2013.02.029; PMID: 23467123
- Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Larivière L, et al. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 2012; 22:1350 - 9; http://dx.doi.org/10.1101/gr.130161.111; PMID: 22466169
- Luo X, Talarek N, De Virgilio C. Initiation of the yeast G0 program requires Igo1 and Igo2, which antagonize activation of decapping of specific nutrient-regulated mRNAs. RNA Biol 2011; 8:14 - 7; http://dx.doi.org/10.4161/rna.8.1.13483; PMID: 21289492
- Bontron S, Jaquenoud M, Vaga S, Talarek N, Bodenmiller B, Aebersold R, et al. Yeast endosulfines control entry into quiescence and chronological life span by inhibiting protein phosphatase 2A. Cell Rep 2013; 3:16 - 22; http://dx.doi.org/10.1016/j.celrep.2012.11.025; PMID: 23273919
- Cameroni E, Hulo N, Roosen J, Winderickx J, De Virgilio C. The novel yeast PAS kinase Rim 15 orchestrates G0-associated antioxidant defense mechanisms. Cell Cycle 2004; 3:462 - 8; http://dx.doi.org/10.4161/cc.3.4.791; PMID: 15300954
- Lindstrom DL, Gottschling DE. The mother enrichment program: a genetic system for facile replicative life span analysis in Saccharomyces cerevisiae.. Genetics 2009; 183:413 - 22; http://dx.doi.org/10.1534/genetics.109.106229; PMID: 19652178
- Hotz M, Leisner C, Chen D, Manatschal C, Wegleiter T, Ouellet J, et al. Spindle pole bodies exploit the mitotic exit network in metaphase to drive their age-dependent segregation. Cell 2012; 148:958 - 72; http://dx.doi.org/10.1016/j.cell.2012.01.041; PMID: 22385961
- Cheng TH, Chang CR, Joy P, Yablok S, Gartenberg MR. Controlling gene expression in yeast by inducible site-specific recombination. Nucleic Acids Res 2000; 28:E108; http://dx.doi.org/10.1093/nar/28.24.e108; PMID: 11121495
- Wach A, Brachat A, Pöhlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae.. Yeast 1994; 10:1793 - 808; http://dx.doi.org/10.1002/yea.320101310; PMID: 7747518
- Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae.. Mol Cell 2007; 26:663 - 74; http://dx.doi.org/10.1016/j.molcel.2007.04.020; PMID: 17560372
- Wanke V, Cameroni E, Uotila A, Piccolis M, Urban J, Loewith R, et al. Caffeine extends yeast lifespan by targeting TORC1. Mol Microbiol 2008; 69:277 - 85; http://dx.doi.org/10.1111/j.1365-2958.2008.06292.x; PMID: 18513215
- Trcek T, Larson DR, Moldón A, Query CC, Singer RH. Single-molecule mRNA decay measurements reveal promoter- regulated mRNA stability in yeast. Cell 2011; 147:1484 - 97; http://dx.doi.org/10.1016/j.cell.2011.11.051; PMID: 22196726
- Bregman A, Avraham-Kelbert M, Barkai O, Duek L, Guterman A, Choder M. Promoter elements regulate cytoplasmic mRNA decay. Cell 2011; 147:1473 - 83; http://dx.doi.org/10.1016/j.cell.2011.12.005; PMID: 22196725
- Shalgi R, Lapidot M, Shamir R, Pilpel Y. A catalog of stability-associated sequence elements in 3′ UTRs of yeast mRNAs. Genome Biol 2005; 6:R86; http://dx.doi.org/10.1186/gb-2005-6-10-r86; PMID: 16207357
- Tuller T, Ruppin E, Kupiec M. Properties of untranslated regions of the S. cerevisiae genome. BMC Genomics 2009; 10:391; http://dx.doi.org/10.1186/1471-2164-10-391; PMID: 19698117
- Beilharz TH, Preiss T. Widespread use of poly(A) tail length control to accentuate expression of the yeast transcriptome. RNA 2007; 13:982 - 97; http://dx.doi.org/10.1261/rna.569407; PMID: 17586758
- Pedruzzi I, Dubouloz F, Cameroni E, Wanke V, Roosen J, Winderickx J, et al. TOR and PKA signaling pathways converge on the protein kinase Rim15 to control entry into G0. Mol Cell 2003; 12:1607 - 13; http://dx.doi.org/10.1016/S1097-2765(03)00485-4; PMID: 14690612
- Wanke V, Pedruzzi I, Cameroni E, Dubouloz F, De Virgilio C. Regulation of G0 entry by the Pho80-Pho85 cyclin-CDK complex. EMBO J 2005; 24:4271 - 8; http://dx.doi.org/10.1038/sj.emboj.7600889; PMID: 16308562
- Riordan DP, Herschlag D, Brown PO. Identification of RNA recognition elements in the Saccharomyces cerevisiae transcriptome. Nucleic Acids Res 2011; 39:1501 - 9; http://dx.doi.org/10.1093/nar/gkq920; PMID: 20959291
- Rendl LM, Bieman MA, Smibert CA. S. cerevisiae Vts1p induces deadenylation-dependent transcript degradation and interacts with the Ccr4p-Pop2p-Not deadenylase complex. RNA 2008; 14:1328 - 36; http://dx.doi.org/10.1261/rna.955508; PMID: 18469165
- Philippsen P, Stotz A, Scherf C. DNA of Saccharomyces cerevisiae.. Methods Enzymol 1991; 194:169 - 82; http://dx.doi.org/10.1016/0076-6879(91)94014-4; PMID: 2005785