Abstract
The 3′ UTR of insulin has been identified as a critical region that confers mRNA stability, which is crucial for promoting transcription in response to glucose challenge. miRNAs are endogenously encoded non-coding RNAs that function as regulators of gene expression. This regulatory function is generally mediated by complementary binding to the 3′UTR of its mRNA targets that affects subsequent translational process. Genes involved in the regulation of glucose homeostasis, particularly in insulin production, have been found as targets of several miRNAs. Yet, no direct miRNA-based regulators of insulin biosynthesis have been identified. In this study, identification of possible miRNA-based regulators of insulin production is explored. Members of a miRNA family, miR-25 and miR-92a, are found as direct modulators of insulin expression. Overexpression of miR-25 or miR-92a reduced insulin expression while inhibition of miR-25 and miR-92a expression using corresponding antagomiRs promoted insulin expression and ultimately enhanced glucose-induced insulin secretion. Furthermore, suppression of insulin secretion by pre miR-9 could be attenuated by treatment with anti-miR-25 or miR-92a. Interestingly, we found the binding site of miR-25 and miR-92a to overlap with that of PTBP1, an important RNA binding molecule that stabilizes insulin mRNA for translation. Despite the increase in PTBP1 protein in the pancreas of diabetic rats, we observed insulin expression to be reduced alongside upregulation of miR-25 and miR-92a, suggesting an intricate regulation of insulin (bio)synthesis at its mRNA level.
Introduction
Glucose level in the system serves as the main regulator of pancreatic β cells’ function, which also coordinately governs insulin production.Citation1,Citation2 Besides controlling insulin production, glucose also induces insulin biosynthesis by promoting insulin transcription as well as insulin transcript stability.Citation3-Citation5 Nevertheless, prolonged exposure of islets to high glucose led to islet dysfunction and reduced insulin mRNA levels.Citation6-Citation8 The molecular mechanism of insulin biosynthesis is highly complex and tightly regulated by a network of transcriptional activators and repressors.Citation2 Since the characterization of the insulin gene in 1980s, intensive studies have been performed to identify the regulatory elements involved in its biosynthesis, including transcript stability.Citation9-Citation11 Synergistic efforts are required for regulation of the five main stages namely, insulin gene transcription, transcript stability, translation, post-translational processing, and insulin secretion. Although it was originally debated that the insulin mRNA contents are not affected by short-term glucose stimulation due to its high copy number, subsequent studies have challenged this view by reporting increased insulin mRNA levels in short-term glucose challenge.Citation11-Citation14 In fact, in vitro studies showed that insulin gene transcription is indeed enhanced by glucose within minutes of exposure but becomes suppressed by 48 h of high glucose treatment.Citation11,Citation13 In addition to glucose-induced regulation of insulin transcription, it has also been reported that glucose level affects the insulin transcript stability.Citation13 On the same note, the 3′ untranslated (UTR) region of the insulin mRNA has been identified as the critical region for glucose-mediated regulation of insulin mRNA stability.Citation15 It was later discovered that the polypyrimidine tract binding protein 1 (PTBP1) binds to the pyrimidine rich segments of the insulin 3′UTR to stabilize the transcript thereby accounting for the glucose-induced increase in insulin mRNA.Citation16,Citation17
It has long been known that microRNAs (miRNAs) participate in the regulation of gene expression by controlling mRNA stability as well as translation.Citation18,Citation19 This regulatory function of miRNAs is generally executed via complementary binding to the 3′UTR of its corresponding mRNA targets. miRNA action usually results in suppression of the target expression either through degradation (perfect complementarity) or translational repression (imperfect complementarity). A handful of miRNAs has been identified as important indirect regulators of insulin production machinery.Citation20-Citation22 Among them, miR-30d has previously been reported to regulate insulin gene transcription in MIN6 cells but its underlying mechanism remained unexplored.Citation20 It was only recently that Zhao et al.Citation22 reported MAP4K4 as a direct target of miR-30d. Overexpression of miR-30d was found to suppress MAP4K4 expression which, in turn, induces activation of MafA, a β cell-specific transcription factor. This ultimately promotes insulin gene transcription. Another recent study has also demonstrated that inhibiting miR-24, -26, -148, and -182 reduces insulin expression in primary cultured mouse islets and that inactivation of these miRNAs in β cells of adult mice results in diabetic phenotype.Citation21 Nevertheless, these studies mainly discussed about the indirect implication of miRNA in regulating insulin expression. To date, there are no reports available on the direct regulatory effects of miRNAs on the insulin (bio)synthesis. In this study, we aim to identify potential miRNA(s) that bind to the 3′UTR of insulin mRNA and understand how insulin biosynthesis can be regulated by these miRNAs.
Results
Bioinformatics predictions of miRNAs targeting rat insulin I
Initial screening for putative miRNAs targeting the 3′UTR of rat insulin I was performed using the web-based miRNA prediction database miRWalk (http://www.ma.uni-heidelberg.de/apps/zmf/mirwalk/), which encompasses other programs including miRanda, miRDB, RNA22, and Targetscan. Of the five programs, only miRWalk and miRanda have predicted miRNAs targeting rat insulin I. Either miRanda or miRWalk or both predicted 12 miRNAs to target the 3′UTR of rat insulin I (). Interestingly among these, miR-25/32/363/92a/92b were identified as a miRNA family that shares the same seed sequence (highlighted in gray in ) and target a common region on the 3′UTR of insulin I. As miRNAs belonging to the same family are postulated to share similar biological functions, the relevance of this miRNA family in insulin I regulation was further explored. miRNA profiling of rat pancreatic tissue and isletsCitation23 showed highest expression for miR-25 and miR-92a in both cases (). Since miR-92a and miR-92b differed only by three nucleotides at the 3′ end of the sequence, miR-92a, which was detected at much higher intensity in the islets, was selected along with miR-25 for subsequent interaction studies. The binding of miR-25 and miR-92a was also predicted to be conserved in rat insulin II, mouse insulin I, and II ().
Table 1. miRNAs predicted to target the 3′UTR rat insulin I
Figure 1. Sequence of rat insulin I mRNA. (A) The complete sequence of rat insulin I mRNA (463 nucleotides long) was obtained from NCBI (NM_019129.3). The 3′UTR region is located between 391–463 nucleotides (underlined). The seed region of the miR-25/32/363/92a/92b family is highlighted in gray. Sequence of specific primers used to amplify the 3′UTR region is shown in bold italics. Core binding region of PTBP1 is shown in a box. (B) miR-25 and miR-92a consensus binding sequence in rat and mouse. Binding of miR-25 and miR-92a at the 3′UTR of rat and mouse insulin are predicted by either miRWalk or miRanda or both. Although the programs did not include prediction for rat insulin II, the seed region of miR-25 and miR-92a is conserved in rat and mouse for both insulin I and II. The PTBP1 binding site is underlined. The sequences corresponding to human insulin and the partially overlapping seed region and PTBP1 binding site are also shown.

Table 2. Expression of selected miRNAs in the rat pancreatic tissue and islets
miR-25 and miR-92a directly targets rat insulin I
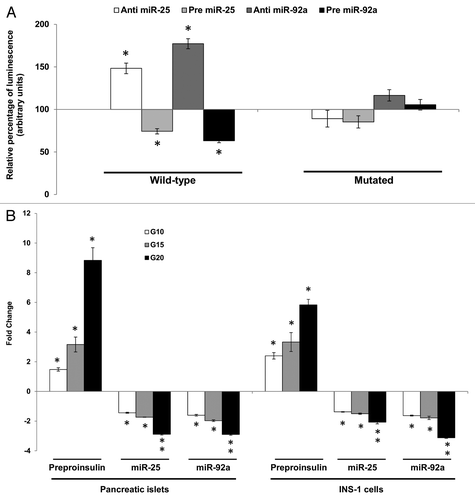
Prior to establishing interaction between the selected miRNAs and the 3′UTR of insulin I in an in-vitro setting, co-expression studies were performed using INS-1, an insulin-secreting pancreatic cell line. Expression values of insulin I (preproinsulin) and miR-25 and miR-92a in this cell line were determined via qPCR (Table S1). The 3′UTR of the rat insulin I gene () was amplified using specific primers and subcloned into luciferase reporter vectors. The reporter construct containing the wild-type 3′UTR of insulin I was then co-transfected together with their respective anti- or pre-miR-25 or miR-92a into INS-1 cells independently. Changes in the luciferase reporter activity were observed at significant levels (). When the wild-type insulin I 3′UTR construct was co-transfected with anti-miR-25, an approximate 48 ± 6.2% increase in relative luminescence was noticed. In contrast, the introduction of pre miR-25 resulted in a significant suppression of the reporter gene expression. A stronger regulatory effect was seen when miR-92a expression was modulated. In the presence of anti-miR-92a, the relative luminescence was greatly enhanced by about 77 ± 5.9%, while transfection of pre miR-92a suppressed the reporter gene activity by approximately 40% ± 2.4 (). Site-directed mutagenesis of the miR-25 and miR-92a recognition site abolished interactions between the miRNAs and their target. These results indicate that miR-25 and miR-92a are direct regulators of insulin I expression and also suggested miR-92a as a stronger modulator in comparison to miR-25. While direct negative interaction has been shown, further investigations are needed to determine the regulatory potential of these miRNAs in a biological setting.
Figure 2A–B. Direct inhibitory effect of miR-25 and miR-92a on Ins1. (A) Dual-Luciferase ® reporter assay quantitation of the effects of anti- or pre-miR-25 or miR-92a interaction with the wild-type and mutated 3′ UTR of Ins1. For the mutated construct, the target sequence 5′-TGCAAT-3′ for the miRNA seed region was mutated to 5′-GCGCCG-3′. The plasmid constructs together with anti or pre miR-25 or miR-92a were co-transfected independently into INS-1 cells. Relative luminescence for luciferase gene activity in treated samples were obtained 48 h post-transfection by normalizing the values against control plasmids transfected with corresponding anti- or pre-scrambled miRNAs. The luminescence for luciferase activity of the controls is considered to be 100%. All transfections of miRNAs were done at 30 nM concentration. Data are presented as mean ± SEM (n = 3) against control. Statistically significant differences are tested using t-test at *p < 0.05 significance. (B) Fold change in the expression of preproinsulin, miR-25 and miR-92a in pancreatic islets and INS-1 cells subjected to increasing glucose concentration (G10, 15, and 20 mM). Data presented as mean ± SEM (n = 3) against control at 5 mM glucose concentration. Statistically significant differences are tested using t-test at p < 0.05 significance. *p < 0.05, **p < 0.01.
Co-regulation of insulin I mRNA synthesis using glucose as well as miR-25 and miR-92
Thus far, the study has demonstrated the potential of miR-25, miR-92a to negatively regulate insulin I expression via 3′UTR binding. Nevertheless, glucose itself is a known regulator of insulin biosynthesis, which can stimulate insulin gene transcription within minutes of high glucose exposure.Citation11,Citation13 In fact, exposure of INS-1 cells to high glucose for minutes to hours resulted in increased rate of insulin gene transcription, which was then inhibited by 48 h.Citation11 Thus, an attempt was made to understand the co-regulatory potential of miR-25, miR-92a, and glucose in a biological setting. Two different set ups were done for this study, rat primary pancreatic islets as well as INS-1 cells. Glucose-stimulated insulin secretion (GSIS) was performed at four different glucose concentrations 5, 10, 15, and 20 mM for a period of 2 h. Increasing concentration of glucose stimulated insulin I synthesis in a dose-dependent manner in both cell lines (). Correspondingly, expression levels of miR-25 and miR-92a displayed a decreasing pattern with increasing glucose concentration (). In fact, both miR-25 and miR-92a levels were decreased by more than 2-fold as insulin mRNA increased up to 8-fold and 5-fold in islets and INS-1 cells, respectively, at 20 mM glucose. This in vitro study highlights that increasing insulin I expression in response to glucose stimulation occurs alongside downregulation of miR-25 and miR-92a. These results were consistent with our earlier findings where miR-25 and miR-92a were identified as negative regulators of insulin I expression.
To understand whether miR-25 and miR-92a regulation can override GSIS, the INS-1 cells were transfected with anti- or pre-miR-25 and miR-92a at both basal (5 mM) and high glucose environment (20 mM) independently. To ensure successful modulation of miRNAs, the expression levels of both miR-25 and miR-92a were measured following transfection of their corresponding anti- and pre-miRNAs. Our results showed that miR-25 and miR-92a levels were reduced and enhanced by their corresponding anti- and pre-miRNA treatment, respectively (). At basal condition (5 mM glucose), modulation of miR-25 or miR-92a expression was found to modestly affect the preproinsulin level (anti-miR-25: 1.27 ± 0.13, pre-miR-25: 0.86 ± 0.23, anti-miR-92a: 1.53 ± 0.17, pre-miR-92a: 0.77 ± 0.14). Our results are somewhat comparable to that observed by Tang et al.,Citation20 where modulation of miR-30d at 1 mM glucose did not affect insulin mRNA level. This is expected since at basal glucose level there will be minimal requirement for insulin production. As for the high glucose condition (20 mM), cells transfected with scrambled miRNAs (anti or pre) followed by the same high glucose (20 mM) treatment were used as corresponding controls. By using these cells as controls, we can now study the effects of the miRNAs independent of glucose. As indicated in , inhibition of either miR-25 or miR-92a resulted in increased expression of the preproinsulin mRNA by high glucose (anti-miR-25: 1.63 ± 0.14, anti-miR-92a: 2.71 ± 0.14 as compared with cells subjected to 20 mM glucose alone). Consistent with this finding, introduction of pre miR-25 or pre miR-92a abolished glucose-induced expression of preproinsulin mRNA levels at 0.75 ± 0.15 and 0.56 ± 0.13, respectively (). Collectively, these data demonstrated the regulatory roles of miR-25 and miR-92a in insulin response upon glucose stimulation.
Table 3. Changes in expression of miR-25, miR-92a and Ins1 mRNA
miR-25 and miR-92 modulate insulin I protein
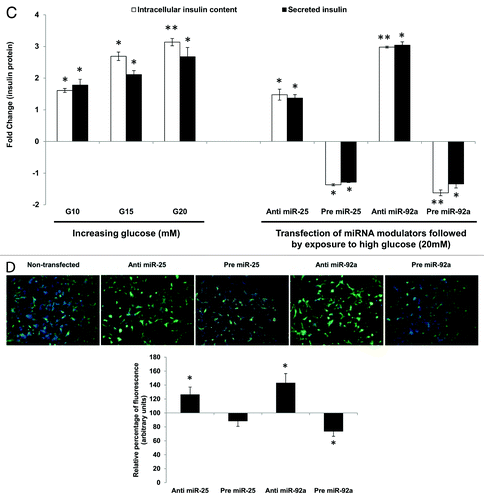
Having established that miR-25 and miR-92a could directly regulate insulin biosynthesis as well as insulin response by controlling insulin I mRNA (preproinsulin) expression (), the next attempt was to investigate whether such regulation was reflected in the downstream protein levels. Both intracellular and extracellular insulin protein content in INS-1 cells subjected to GSIS were monitored using ELISA to give a more accurate indicator of insulin synthesis. Intracellular insulin protein content increased in a dose-dependent manner with increasing glucose concentration (, left panel). The increase in the intracellular insulin protein also resulted in increased secretion into the extracellular environment. These results are in agreement with the observations made earlier at mRNA level (). Next, we were interested to know whether miRNA-based modulation of insulin I expression will ultimately affect the amount of translated insulin protein. To address this, we modulated the expression of miR-25 and miR-92a independently by using their corresponding precursors and anti-miRs. Following that, cells were subjected to high glucose (20 mM) and the resultant effects at protein level were determined by ELISA (, right panel). Cells transfected with scrambled miRNAs (anti or pre) followed by the same high glucose (20 mM) treatment were used as corresponding controls. INS-1 transfected with either anti-miR-25 or miR-92a exhibited significant increase in both intracellular insulin content and secreted insulin (, right panel). On the contrary, the presence of either pre miR-25 or miR-92a resulted in a marked reduction of insulin within and outside the cells (, right panel). The observed reduction in secreted insulin level despite glucose stimulation in INS-1 cells transfected with precursors of miR-25, miR-92a, could be attributed to the reduced insulin content in the cells. Regulation of insulin I by miR-25 and miR-92a was further verified by immunocytochemistry. The insulin I protein expression correlated with that of ELISA assays. Anti-miR-25 or miR-92a transfected INS-1 cells displayed enhanced fluorescence intensity for insulin I protein (), while the opposite was observed in pre miR-25 or miR-92a transfected cells ().
Figure 2C–D. (C) Changes in insulin protein in INS-1 cells. INS-1 cells were subjected to increasing glucose concentration (G10, 15, and 20 mM) to study relative changes in insulin protein (left panel). Data presented as mean ± SEM (n = 3) against control at 5 mM glucose concentration. Intracellular insulin content (in white bar) and secreted insulin (in black bar) are normalized to total protein content. Further, the effects of miR-25 and miR-92a on insulin protein were determined by modulating the expression of miRNAs independently (right panel). Following transfections, cells were subjected to high glucose (20 mM). Cells transfected with scrambled miRNAs (anti or pre) followed by the same high glucose (20 mM) treatment were used as corresponding controls. All transfections of miRNAs were done at 30 nM concentration. Data presented as mean ± SEM (n = 3) against control at 20 mM glucose concentration. Intracellular insulin content (in white bar) and secreted insulin (in black bar) are normalized to total protein content. Statistically significant differences are tested using t-test at p < 0.05 significance. *p < 0.05, **p < 0.01. (D) Insulin I immunoreactives in INS-1 cells transfected with either anti or pre miR-25 or miR-92a. The cells were fixed and immunolabeled with insulin I antibody (green) and nuclear stain Hoechst 33342 (blue). The data presented here is a representative of three independent experiments. Fluorescence signal quantitation was performed according to Su et al.Citation60 using Varioskan® Flash (Thermo Scientific), whereby FITC fluorescence is measured at excitation 490 nm and emission 525 nm. Each signal is normalized to the number of cells by Hoechst 33342 fluorescence measured at excitation 346 nm and emission 460 nm. Relative fluorescence were obtained 48 h post-transfection by normalizing the values against cells transfected with corresponding anti- or pre-scrambled miRNAs. The fluorescence of the controls is considered to be 100%. All transfections of miRNAs were done at 30nM concentration. Data are presented as mean ± SEM (n = 3) against control. Statistically significant differences are tested using t-test at *p < 0.05 significance.
Effect of miR-25 and miR-92 regulation on insulin secretion
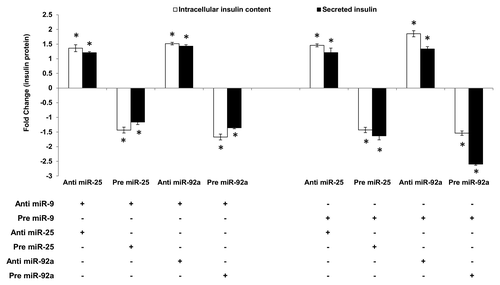
The ability of miR-25 and miR-92a to regulate insulin I even in the presence of glucose stimulation highlights its potential as a superior regulator. Thus, the study was extended to explore the capability of miR-25 and miR-92a-induced insulin I expression to override the insulin secretion process. miR-9 has been identified to inhibit insulin secretion by targeting Onecut-2.Citation24 Hence, miR-9 was used to test whether the regulatory function of miR-25 and miR-92a on insulin biosynthesis can supersede insulin secretion. In agreement with the published report, introduction of anti-miR-9 in INS-1 cells showed enhanced insulin secretion, while treatment with pre miR-9 suppressed insulin secretion significantly (Table S2). Following that, co-transfections using anti or pre of miR-25 or miR-92a were performed on these INS-1 cells. Subsequently, cells were subjected to high glucose and both intracellular insulin content as well as secreted insulin were determined using ELISA. Corresponding anti- and pre-miR-9-transfected cells were used as controls. In INS-1 cells co-transfected with anti-miR-9 and either anti-miR-25 or anti-miR-92a, high glucose stimulation significantly increased both the intracellular as well as secreted insulin content (, left panel). Conversely, cells that received subsequent treatment with precursors (pre) of miR-25 or miR-92a showed reduced insulin production. These findings implied that despite the presence of anti-miR-9, which supposedly increases insulin secretion, insulin production (intracellular and secreted) could be reduced by pre miR-25 or miR-92a. These observations were supported further in the next set of experiments in which INS-1 cells co-transfected with pre miR-9 (supposedly reducing insulin secretion) and either with anti-miR-25 or miR-92a showed a significant increase in intracellular insulin content (, right panel). Although collected media from the anti-miR-25-treated cells showed only minimal increase in secreted insulin, cells co-transfected with anti-miR-92a exhibited a marked increase in insulin secretion. Altogether, our results suggest that enhanced insulin synthesis in the presence of anti-miR-25 or miR-92a could attenuate suppression on insulin secretion by miR-9. On the other hand, cells transfected with pre-miR-25 or miR-92a exhibited reduced insulin content as well as secreted insulin (, right panel). In fact, a striking impairment in insulin secretion was observed in cells co-transfected with pre miR-9 and miR-92a. This could reflect the synergistic inhibition on both insulin biosynthesis and insulin secretion by miR-92a.
Figure 3. Regulatory effects of miR-25 and miR-92a on insulin synthesis overrides that of miR-9 in the downstream insulin secretion. Changes in insulin protein in INS-1 cells with the corresponding transfection as shown above followed by high glucose treatment (20 mM). In the left panel, cells were first transfected with anti-miR-9 followed by independent transfection of anti or pre miR-25 or miR-92a . Cells transfected with anti-miR-9 and corresponding scrambled miRNAs (anti or pre) followed by the same high glucose (20 mM) treatment were used as controls). In the right panel, cells were first transfected with pre miR-9 followed by independent transfection of anti or pre miR-25 or miR-92a. Cells transfected with pre miR-9 and corresponding scrambled miRNAs (anti or pre) followed by the same high glucose (20 mM) treatment were used as controls. All transfections of miRNAs were done at 30 nM concentration. Positive sign (+) indicates presence of corresponding anti- or pre-miRNAs while negative sign (–) indicates absence. Data presented as mean ± SEM (n = 3) against corresponding controls. Intracellular insulin content (in white bar) and secreted insulin (in black bar) are normalized to total protein content. Statistically significant differences are tested using t-test at *p < 0.05 significance.
Altered expression of miR-25, miR-92a and insulin I in the pancreas of diabetic rats
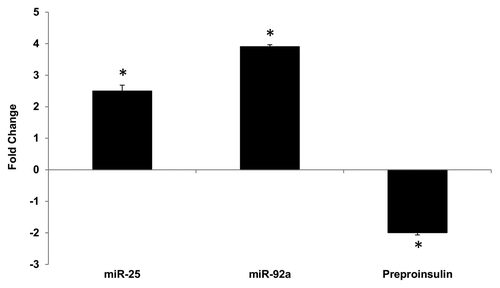
Reduced insulin expression has been shown to contribute to impaired insulin secretion in rat diabetic models.Citation25,Citation26 In order to verify the implication of miR-25 and miR-92a in diabetic condition, a diabetic rat model using a low dose streptozotocin (STZ) and high fat diet was created, according to Reed et al.,Citation27 with some modifications as described in Karolina et al.Citation26 Effects of the treatment on the body weight, glucose, insulin, cholesterol, and triglycerides levels as well as confirmation of diabetic condition by oral glucose tolerance test (OGTT) are summarized in Table S3. Total RNA from the pancreas of a diabetic rat model was isolated to examine the correlation between insulin I (preproinsulin) and miR-25, miR-92a expression. In comparison with the normal rat, significant upregulation of miR-25 and miR-92a was observed in the pancreas of diabetic rats (). These changes correlated with a corresponding decrease in the insulin I (preproinsulin) mRNA level. Here, we observed that overexpression of miR-25 and miR-92a in diabetic rat model is likely to play a role in the insulin deficiency, which may eventually contribute to impaired insulin secretion.
Figure 4. Altered expression of miR-25, miR-92a and preproinsulin in diabetic rat pancreas. Total RNA was isolated from pancreatic tissue of diabetic rat model. Expression of miR-25, miR-92a, and insulin I was measured by qPCR relative to healthy control rats. Data are presented as mean ± SEM with n = 6. Statistically significant differences are tested using t-test at *p < 0.05 significance.
Increased binding of miR-25 or miR-92a may be implicated in polypyrimidine tract-binding protein 1 (PTPB1) binding
Tillmar et al.Citation16 has highlighted the importance of PTBP1 protein binding in regulating insulin mRNA levels. In fact, the group has discovered that inhibition of PTBP1 protein binding to the pyrimidine rich sequence by mutation of the core binding site resulted in destabilization of insulin I mRNA. Of interest, we noticed that the binding site of miR-25 and miR-92a overlaps with that of PTBP1 (). This suggests the possibility that the binding of miR-25 or miR-92a not only suppresses insulin I mRNA expression but may also affect PTBP1 interaction, hence exacerbating the reduced stability of the insulin I mRNA.
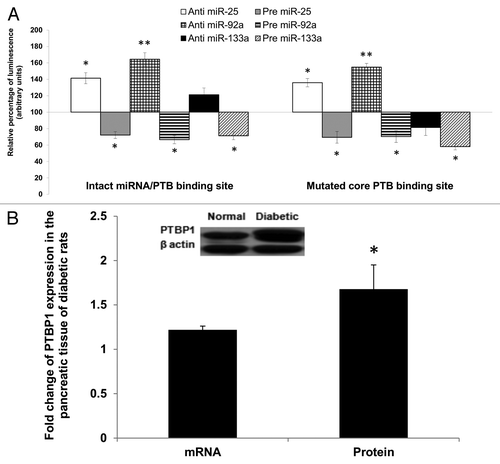
Hence, we compared the effects of anti- or pre-miR-25 or miR-92a in two different constructs of reporter gene vectors (Table S4A). The first construct consisted of the wild-type 3′UTR of insulin I, while in the second construct we introduced single base mutation of one of the pyrimidines (C) to a purine (A) in the PTBP1-core binding site. This mutation has been shown to reduce insulin mRNA stability by Tillmar et al.Citation16 Since the mutation is not within the recognition site of miR-25 and miR-92a seed sequence, this will only affect PTBP1 binding while conserving miR-25 and miR-92a interaction. In addition to miR-25 and miR-92a, we also explored the effects of anti- or pre-miR-133a, a regulator of Ptbp1 expression reported by Fred et al.,Citation28 which was also confirmed independently in this study (Table S4B). All experiments were performed using INS-1cells, which express both insulin I and PTBP1.
As observed previously, introduction of anti-miR-25 or anti-miR-92a resulted in a marked increase in the reporter gene activity containing the wild construct. It is noteworthy that inhibition of either miR-25 or miR-92a (by their respective antimiRs) was found to induce higher signal intensity as compared with treatment with anti-miR-133a. In fact, introduction of anti-miR-133a (to increase Ptbp1) showed only marginal increase in the reporter activity. In contrast, the presence of precursor forms of any of the three miRNAs (miR-25 or miR-92a or miR-133a) led to a decreased reporter gene activity (, left panel). Interestingly, we observed treatment with anti- or pre-miR-25 or miR-92a to be equally effective even in the construct with one base mutation in the core PTBP1 binding site. This could be due to the intact seed site for miRNA binding and therefore, the miRNA action is not affected. Conversely, introduction of either anti- or pre-miR-133a showed reduced activity implying inhibition of PTBP1 binding due to the mutation. These observations imply that effective regulatory action of miR-25 and miR-92a may occur independent of PTBP1 binding. We have also observed an increase in PTBP1 expression in the pancreas of diabetic rats at both mRNA and protein level (). Despite the significant increase in PTBP1 protein (1.68 ± 0.27), insulin expression was reduced in the diabetic rats (). Although these results may seem to suggest potential competition between the miRNAs (miR25 and miR-92a) and PTBP1, our data thus far is insufficient to make this claim and, hence, a more precise experiment is necessary to fully support this speculation. Nevertheless, based on the observations made thus far, we have provided evidence for miR-25 and miR-92a as direct negative regulators of insulin expression and that their overexpression upon diabetic condition is implicated in the impairment of insulin synthesis.
Figure 5. Regulatory effects of miR-25 and miR-92a occur independent of PTBP1 binding. (A) Wild-type and mutated PTBP1 core binding site in 3′UTR of rat insulin I were constructed into luciferase vector (Table S4). Dual luciferase assay (48 h post- transfection) was performed to study the effects of anti or pre miR-25 or miR-92a or miR-133a interaction with the 3′ UTR of Ins1. The plasmid constructs together with anti or pre miR-25 or miR-92a or miR-133a were co-transfected independently into INS-1 cells. Relative luminescence was obtained by normalizing the values against control plasmids transfected with corresponding anti or pre scrambled miRNAs. pMIR-REPORTTM without any 3′UTR insert. The luminescence for luciferase activity of the controls is considered to be 100%. All transfections of miRNAs were done at 30 nM concentration Data presented as mean ± SEM with n = 3. Statistically significant differences are tested using t-test at *p < 0.05 significance. (B) Relative expression of PTBP1 in the pancreas of diabetic rats. Expression of Ptbp1 mRNA was measured in the pancreas of diabetic rats relative to normal rats. Western blot analysis of PTBP1 protein in the pancreatic tissue of normal and diabetic rats is also shown (see insert). The data presented here is a representative of six independent experiments (n = 12, six normal rats and six diabetic rats). Statistically significant differences are tested using t-test at *p < 0.05 significance.
Discussion
A dynamic insulin biosynthesis is necessary for proper regulation of glucose homeostasis. In order to meet the demands for increased insulin production upon glucose challenge, insulin mRNA stores need to fluctuate accordingly. As sustained glucose stimulation progressively depletes the insulin stores, insulin biosynthesis must be activated almost immediately to replenish them. Since insulin biosynthesis boost is a prerequisite for prolonged insulin secretion, its deficit also contributes to insulin insufficiency. Reduced insulin (bio)synthesis in the pancreatic β-cells is closely associated with the onset and progression of diabetes. Moreover, insulin mRNA levels were found to be reduced in diabetic rat models which, in turn, result in inadequate insulin secretion.Citation25,Citation26,Citation29 Hence, it is important to look for ways to induce insulin production effectively.
Despite the intense efforts over the past few decades, the establishment of commercially available human pancreatic β cell lines has not been entirely successful. Several insulin-secreting cell-based studies for future therapeutic intervention in diabetes has mainly focused on rodent insulinoma cell lines.Citation30-Citation32 In particular, miRNA studies related to insulin production have been performed mostly using rodent insulinoma cell lines as well.Citation21,Citation22,Citation33 Due to limited access to functional human islets or derived β-cell lines,Citation34-Citation38 our study has been restricted to the more widely accepted rat β cell line, INS-1, to search for potential direct endogenous regulators of insulin biosynthesis.
In this study, we identified two miRNAs belonging to the same family, miR-25 and miR-92a, as direct negative regulators of insulin I expression. The seed site of these two miRNAs at the 3′UTR of the insulin gene was found to be conserved in rats and mice for both insulin I and II () and somewhat partially (4 out of 6 nucleotides) conserved between rodents and human insulin mRNA. The resultant effects of negative (inverse) miRNA:mRNA interaction were observed at protein level as well, where introduction of pre miR-25 or miR-92a not only reduced insulin I mRNA but also the total insulin protein (intracellular and extracellular) in INS-1 cells. Conversely, we were able to increase insulin synthesis (mRNA and protein) by inhibiting miR-25 and miR-92a expression. The important implication of miR-25 and miR-92a on insulin biosynthesis was further highlighted in our rat diabetic model, which showed a marked upregulation of miR-25 and miR-92a in the diabetic pancreatic tissue alongside reduced insulin expression. In support of our findings, a miRNA profiling study by Rosero et al.Citation39 has identified a miRNA signature, which also included both miR-25 and miR-92 to be implicated in pancreatic development. The degree of expression of miR-25 and miR-92a was found to change throughout the development. miR-92a and miR-25 were identified under the same group of miRNAs, which showed a decreasing expression profile in correlation to the increasing expression of their potential targets. Among them, NEUROD1, a key insulin transactivator, was highlighted as a target of miR-92a. Increased expression of NEUROD1, along with a corresponding downregulation of miR-92a is critical for the development of the endocrine pancreas. Moreover, islets lacking NEUROD1 have been found to show poor response to a high glucose environment and exhibit a glucose metabolic profile similar to that of immature β cells.Citation40,Citation41 Besides being a key regulator of pancreatic islet morphogenesis, NEUROD1 expression is also necessary for the insulin gene transcription and synthesis.Citation42,Citation43 In our study, we observed miR-92a to be a stronger suppressor of insulin expression as compared with miR-25. This could be attributed to its synergistic-suppressive effects on the transcription factor, NEUROD1 as well as insulin I. On another note, miR-92a has emerged as a principal regulator of angiogenesis,Citation44 an event that is impaired during diabetes and consequently contributes to the clinical manifestation of diabetes.Citation45,Citation46 This is in conjunction with our diabetic rat model, which showed an upregulation of miR-92a. On the other hand, miR-25 has been implicated to be involved in the regulation of apoptosis, which suggests its possible implication in diabetes pathogenesis.Citation47,Citation48 A key finding in our work is that independent overexpression of miR-25 or miR-92a was able to abolish glucose-induced insulin I production. This is evident in the experiment whereby the presence of anti-miR-25 or miR-92a increased total insulin protein (intracellular and secreted insulin) that is even higher than that induced by high glucose alone. Previous studies have indicated a few miRNAs, including miR-9, as regulators of insulin secretion.Citation24,Citation49 miR-9 reduced insulin secretion by targeting Onecut-2.Citation24 Onecut-2 is a transcription factor which controls the level of Granuphillin/Slp4, a Rab GTPase effector associated with β cell secretory granules. Interestingly, we observed that the positive effect of anti-miR-25 or anti-miR-92a on insulin synthesis could supersede the suppression on insulin secretion by miR-9. This was indicated by the increased insulin secretion in INS-1 cells that were co-transfected with pre-miR-9+anti-miR-25 or pre-miR-9+anti-miR-92a. Although significant increase in insulin secretion was only observed in cells transfected with anti-miR-92a, the presence of anti-miR-25 could perhaps partially restore insulin secretion to an observable level. In contrast, INS-1 cells co-transfected with anti-miR-9 (thus promoting insulin secretion) and either pre-miR-25 or miR-92a showed significant reduction in both intracellular insulin content as well as insulin secretion. This is rather expected since less insulin storage in cells (due to inhibitory effects of pre-miR-25 or miR-92a) would consequently reduce the amount of insulin available for secretion.
Another interesting finding in our study is the identification of overlapping binding site between miR-25 and miR-92a and PTBP1 protein on the 3′UTR of insulin I. We have also demonstrated that regulatory function of miR-25 and miR-92a is equally effective whether or not PTBP1 binding site is mutated. Moreover, the increase in PTBP1 protein in the pancreatic tissue of our diabetic model did not induce a corresponding increase in insulin, which could be attributable to the overexpression of miR-25 and miR-92a. Reduced PTBP1 binding in spite of the increase in its protein level has also been discussed by Ehehalt et al.Citation50 The study compared the amount of PTBP1 in the nuclear extracts from the islets of healthy and diabetic individuals.Citation50 It was demonstrated that diabetic patients displayed an increased nuclear retention of PTBP1 protein relative to non-diabetic islets. It was then postulated that the impaired redistribution of PTBP1 causes deficiency in total insulin levels upon glucose stimulation. Although there may be a potential competition between the two entities (miR-25, miR-92a, and PTBP1 protein on the 3′UTR of insulin), our present experimental data does not give a direct evidence for this claim. Hence, a more in-depth study is necessary to elucidate the postulated competition between miRNAs and protein for binding site at the targeted mRNA.
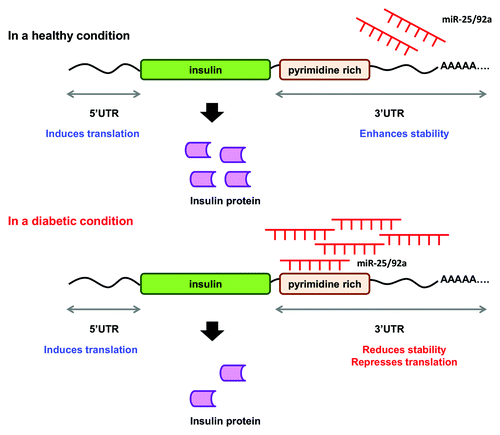
Thus far, the data collated in our study suggest a miRNA-based regulatory mechanism for insulin production (). Under normal conditions, increase in glucose level is accompanied by downregulation of miR-25 and miR-92a. At the same time, glucose-induced PTBP1 protein level will also remain high to promote insulin production.Citation16 On the other hand, in a diabetic condition, overexpression of miR-25 and miR-92a increases miRNA:mRNA interaction at the 3′UTR of insulin mRNA. This concerted regulation at the 3′UTR consequently impairs insulin synthesis resulting in deficiency in insulin production as observed in diabetic condition.
Figure 6. A proposed mechanism for the concerted regulatory function of miR-25 and miR-92a in insulin biosynthesis. miR-25 and miR-92a are direct negative regulators of insulin I expression. In a healthy condition, glucose stimulation promotes downregulation of miR-25 and miR-92a. In a diabetic condition, upregulation of miR-25 and miR-92a suppresses insulin expression by binding to the 3′UTR. Consequently, this results in reduced insulin synthesis as well as insulin secretion.
Interestingly, studies have also reported the implications of INS gene mutations in the rare diabetic conditions, particularly permanent neonatal diabetes and mutant INS-gene induced diabetes of the youth.Citation51-Citation53 Mutations are commonly found in the critical regions of the preproinsulin molecule, which will have implications for the maintenance of normal folding, processing, and bioactivity of mature insulin. In such cases, although insulin mRNA transcription may be increased in response to glucose challenge, it does not necessarily translate to increase in insulin production. It would be of interest to investigate the possibility of miRNA binding to these critical sites, which may contribute to the pathogenesis of these forms of diabetes. Nevertheless, we postulate that miRNA:mRNA binding at any part of the CDS region (i.e., signal peptide, α chain, β chain, or C-peptide) may effectively inhibit translation.Citation54
It is known that glucose level serves as a regulator of insulin transcription and secretion in the body. In this study, we discussed the significance of miRNAs as an additional important regulator of the insulin (bio)synthesis process, which act at the post-transcriptional level and independently of the upstream transcriptional process. We have identified miR-25 and miR-92a as direct regulators of insulin I expression. Hence, increased insulin transcription in the presence of glucose stimulation may not necessarily translate to proportional increase in the insulin I mRNA. This is because the availability of the net insulin I mRNA for translation also depends on the presence of the miR-25 and miR-92a, which target the insulin I mRNA. In other words, the regulatory action of miR-25 and miR-92a serves as an additional fine tuning step that will determine the final insulin I mRNA level proceeding to translation.
Although the seed sequence of miR-25 and miR-92a is not conserved in the human insulin gene, both miRNAs are conserved in human and rats. Hence, the two miRNAs are potentially implicated in diabetes via interaction with other targets. In fact, van de Bunt et al.Citation55 has identified a variant (rs3802177) within the 3′UTR of the SLC30A8 gene, the variant with the strongest association with Type 2 diabetes,Citation56 to map to predicted target site for islet-expressed miR-92a. Furthermore, miR-92a was found to be highly upregulated in stable angina pectoris patients complicated with diabetes as compared with those without diabetes.Citation57 In this study, we propose that the miRNA-based regulatory mechanism of insulin biosynthesis in rats could as well be adopted in humans via other miRNAs (Table S5). Using the same prediction software, a total of 11 miRNAs was predicted to target the 3′UTR of the human insulin gene. Of which, four miRNAs, namely miR-1286, miR-1908, miR-650, and miR-886-3p have their binding sites overlap with that of PTBP1. These miRNAs could be of importance in the regulation of insulin (bio)synthesis in human. We could not demonstrate this due to lack of suitable human cell lines for the study. Hence, the establishment of functional human-derived β-cell lines will be of utmost importance for future translational study of our findings. For now, to our knowledge, this is the first study to identify two miRNAs (miR-25 and miR-92a) that could control insulin synthesis directly by targeting the 3′UTR of insulin gene. Furthermore, we have shown that increased insulin synthesis mediated by introduction of anti-miR-25 or miR-92a was able to restore insulin secretion, which was suppressed by miR-9. These observations were comparable Zhao et al.,Citation22 where overexpression of miR-30d could attenuate suppression of insulin secretion. Hence, suppressing the expression of miR-25 and miR-92a serves as a possible approach that warrants further investigation and may turn out to have potential application in the treatment of diabetes.
Materials and Methods
miRNA array profiling from rat islets and pancreatic tissue
Expression values for the miRNAs of interest in the rat islets (n = 4) by miRNA profiling was obtained from Jacovetti et al.Citation23 Total RNA was isolated from the pancreatic tissue of normal rats (n = 6). After which, LNA-modified oligonucleotide (Exiqon) probes for human, mouse, and rat miRNAs annotated in miRBase version 11.0 were used in the microarray. Total RNA (1 µg) was 3′-end-labeled with Hy3 dye using the miRCURY LNATM Power Labeling Kit (Exiqon) and hybridized on miRCURY LNATM Arrays, using MAUI hybridization system. Data shown were an average of log2 normalized signal intensities ± SEM.
Transfection of miRNAs in INS-1 cells
INS-1 cells (kind gift from Professor Li Guo Dong from the Department of Clinical Research at Singapore General Hospital) were grown in 5% CO2 at 37 °C in DMEM containing 11.1 mM glucose supplemented with 10% FBS, 100U penicillin/ml, 100 g streptomycin/ml, 1 mM sodium pyruvate, 10 mM HEPES, and 0.05 mM 2-mercaptoethanol. Transfection of miRNAs was performed according to Sepramaniam et al.Citation58 Anti- or pre-miR-25, miR-92a, miR-9, miR-133a and scrambled miRNAs were purchased from Ambion. Briefly, respective miRNAs were diluted in 50 µl of Opti-MEM® (Ambion) to a final concentration of 30 nM. The mix was then complexed with 1 µl of siPORTTM NeoFXTM in 50 µl of Opti-MEM®. The cells were transfected with these complexes and maintained for 48 h prior to subsequent RNA/protein extraction.
Primary islets isolation
Primary islets were isolated from adult Wistar rats according to Wu et al.Citation59 Briefly, the pancreas was perfused with a solution of 0.5 mg/ml Collagenase Type V (Sigma) dissolved in Hanks Balanced Salt Solution (HBSS). The digestion was performed at 37 °C for 15–20 min after which the collagenase was neutralized with HBSS supplemented with 1% FBS. The collagenase treated pancreas was then sequentially filtered through 1.5 mm and 0.8 mm metal mesh filters. Islets were subsequently enriched by centrifugation in Histopaque 1077 (Sigma) and picked under direct light microscopic visualization. The islets were then cultured at 100 islets/well in 24-well plates supplemented with 500 μl of complete DMEM.
Extraction of total RNA and protein
Total RNA (+miRNAs) was extracted from cells by a single-step method using TRIzol® (Invitrogen Life Technologies) according to the manufacturer’s protocol. The concentration and integrity of RNA were determined using Nanodrop ND-1000 spectrophotometry (Nanodrop Tech) and denaturing agarose gel electrophoresis. For protein extraction, cells were lysed using non-denaturing lysis buffer that is compatible for subsequent ELISA assay.
Reverse transcription and real-time quantitative PCR
Reverse transcription followed by real-time quantitative PCR (qPCR) were performed according Karolina et al.Citation26 Quantitation of rat preproinsulin and Ptbp1 mRNA was performed using SYBR green assay. Specific primer sequences for rat preproinsulin (5′-AGTGACCAGC TACAATCATA G-3′ and 5′-GGTGCTCATT CAAAGGCTTT A-3′) and rat Ptbp1 (5′-TTCGGCGTCT ACGGTGATG-3′ and 5′-AGTGCGTTCT CCTTCTTGTT GAA-3′) were generated using Primer-Express (Applied Biosystems). For miRNA detection, reverse transcription followed by stem-loop real-time quantitative PCRs were performed according to the manufacturer’s protocols using miR-25 and miR-92a-specific primer probes (Applied Biosystems). qPCR was performed on an Applied Biosystems 7900 sequence detection system.
Luciferase assays
Dual-Luciferase® reporter assay allows the simultaneous expression and measurement of two individual reporter enzymes within a single system. The Firefly luciferase expression is used to correlate the effect of miRNA and its target interaction while the activity of the co-transfected Renilla luciferase provides an internal control that serves as the baseline response. Gene-specific primers were used to amplify the 3′UTR of rat insulin I mRNA (). For the mutated construct, the target sequence 5′-TGCAAT-3′ for the miRNA seed region was mutated to 5′-GCGCCG-3′ to abolish any interaction. The PCR products were cloned into the Firefly luciferase expressing vector (pMIR-REPORTTM; Ambion, USA) at the SpeI and HindIII sites. Plasmid transfection procedure was adapted from Sepramaniam et al.Citation58 INS-1 cells were transfected with 30 nM anti- or pre-miR-25 or miR-92a for 3 h followed by 100 ng/well pMIR-REPORTTM (+ wild-type 3′UTR of Ins1 or mutated seed sequence) for another 3 h. The cells were lysed 48 h later for measurement of luciferase activity. Dual-luciferase® assay (Promega) was performed to quantitate the effects of anti- or pre-miR-25 or miR-92a interaction with the 3′UTR of rat insulin I. The assay was performed according to the manufacturer’s protocol. In all experiments, transfection efficiencies were normalized to those of cells co-transfected with the Renilla luciferase expressing vector (pRLCMV; Promega,USA) at 10 ng/well.
Immunocytochemistry
Immunocytochemistry was performed on INS-1 cells transfected with either anti- or pre-miR-25 or miR-92a. The cells were fixed with 4% paraformaldehyde in PBS and blocked with 5% FBS before incubation with mouse anti-rat antibody Ins1 for 1 h at room temperature. Rabbit anti-mouse FITC labeled antibody was used as secondary antibody. Cells were mounted and nuclei were counterstained with Hoechst 33342 and viewed under a fluorescence microscope (Carl Zeiss LSM 510 META). Fluorescence signal quantitation was performed according to Su et al.Citation60 using Varioskan® Flash (Thermo Scientific) whereby FITC fluorescence is measured at excitation 490 nm and emission 525 nm. Each signal is normalized to the number of cells by Hoechst 33342 fluorescence measured at excitation 346 nm and emission 460 nm.
Measurement of preproinsulin mRNA, intracellular insulin protein content, and insulin secretion
INS-1 cells were seeded at a density of 7 × 104 per well in 24-well plates and incubated at basal glucose concentration (5 mM) for 24 h prior to glucose treatment. Cells were washed twice with sterile Krebs Ringer buffer (KRB) followed by pre-incubation with 500 μl of the same buffer supplemented with 2.8 mM glucose at 37 °C for 30 min. Subsequently, the medium was replaced by 500μl of sterile KRB containing varying concentrations of glucose (5–20 mM) for 2 h. At the end of incubation, both cell lysate and supernatants were collected for measurements of insulin content in the cell lysate and secreted insulin in the media, respectively. Insulin was assayed by ELISA (Crystal Chem, Inc.) method according to the manufacturer’s protocol and normalized to the total protein. Following that, changes in the amount of insulin protein is calculated relative (as a ratio) to the corresponding control used in the experiment.
Rat model of diabetes mellitus
Male Wistar rats, 6 wk of age and weighing approximately 150 g were used for all studies. The animals were handled according to the guidelines of the Council for International Organization of Medical Sciences on Animal Experimentation (World Health Organization) and the National University of Singapore. The animal protocols were approved (Protocol number 062/09) by the National University of Singapore Institutional Animal Care and Use committee (NUS IACUC). Induction of Type 2 diabetes (T2D) was done according to Reed et al.Citation27with some modifications as described by Karolina et al.Citation26 Briefly, 12 rats were randomly divided into two groups (n = 6). One group was fed with normal fat diet (Harlan Teklad, 2018) and the other with high fat diet (Specialty Feeds, SF10-019). After 2 wk, the animals on high fat diet were injected intra-peritoneally with low dose of streptozotocin STZ (40 mg/kg) after an overnight fast. Both groups of animals were continued on their respective diets for another week. This combination method is preferred to administration of high dose of STZ, which will completely damage the β cells. A low dose of STZ will damage the islets to a lesser extent in which insulin production still occurs at moderate level which will give a better representation of T2D. At the end of the experimental period (on the 8th day after STZ administration), animals were euthanized using CO2, and pancreatic tissue was harvested. Effects of treatment on body weight, glucose, insulin, cholesterol, and triglycerides concentrations as well as oral glucose tolerance test to ensure a successful T2D model are shown in Table S3.Citation26
SDS-PAGE and western blot analysis
Protein was extracted from pancreatic tissue of both normal and diabetic rats. Protein samples (40 ug) were resolved using 10% SDS-PAGE. Western blot was performed as described in Karolina et al.Citation26 The primary rabbit monoclonal anti-rat antibodies against PTBP1 were added at a 1:5,000 dilution in PBST containing 2.5% non-fat dry milk for 60 min to identify the specific protein. The membranes were washed in PBST followed by incubation in secondary goat anti-rabbit antibody (1:5,000 dilution) in PBST containing 2.5% non-fat dry milk for 60 min. β-actin was used as loading control. The membranes were washed again with PBST and visualized with Super-Signal® West-Dura Extended Duration Substrate (Pierce, Biotechnology) and developed in Kodak Biomax film. Films of western blots were scanned (Acer SWZ3300U), and the labeling intensities of the bands were quantified using ImageJ software (National Institutes of Health). Relative expression of protein is the quantity of band intensity expressed as a ratio to the control samples normalized to β-actin. All band images were representatives of at least six independent experiments (n = 6).
| Abbreviations: | ||
| 3′UTR | = | 3′ untranslated region |
| miRNA(s) | = | microRNA(s) |
| PTBP1 | = | polypyrimidine tract-binding protein 1 |
| MafA | = | v-maf musculoaponeurotic fibrosarcoma oncogene homolog A |
| GSIS | = | glucose-stimulated insulin secretion |
| MAP4K4 | = | mitogen-activated protein kinase kinase kinase kinase 4 |
| NEUROD1 | = | neuronal differentiation 1 |
| STZ | = | streptozotocin |
| OGTT | = | oral glucose tolerance test |
| CDS | = | coding region |
| SLC30A8 | = | Solute carrier family 30 (zinc transporter), member 8 |
Additional material
Download Zip (410.7 KB)Acknowledgments
This work was supported by research grants, R-184-002-165-281 and R-183-000-290-213 from the National Research Foundation (CRP Program) and the National Medical Research Council, respectively.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Ashcroft SJ, Bunce J, Lowry M, Hansen SE, Hedeskov CJ. The effect of sugars on (pro)insulin biosynthesis. Biochem J 1978; 174:517 - 26; PMID: 361036
- Andrali SS, Sampley ML, Vanderford NL, Ozcan S. Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochem J 2008; 415:1 - 10; http://dx.doi.org/10.1042/BJ20081029; PMID: 18778246
- Welsh M, Nielsen DA, MacKrell AJ, Steiner DF. Control of insulin gene expression in pancreatic beta-cells and in an insulin-producing cell line, RIN-5F cells. II. Regulation of insulin mRNA stability. J Biol Chem 1985; 260:13590 - 4; PMID: 3902821
- Poitout V, Hagman D, Stein R, Artner I, Robertson RP, Harmon JS. Regulation of the insulin gene by glucose and fatty acids. J Nutr 2006; 136:873 - 6; PMID: 16549443
- LeRoith D, Taylor SI, Olefsky JM. Diabetes Mellitus: A Fundamental and Clinical Text. Philadelphia, Baltimore, New York, London, Buenos Aires, Hong Kong, Sydney, Tokyo: Lippincott Williams and Wilkins, 2000.
- Eizirik DL, Korbutt GS, Hellerström C. Prolonged exposure of human pancreatic islets to high glucose concentrations in vitro impairs the beta-cell function. J Clin Invest 1992; 90:1263 - 8; http://dx.doi.org/10.1172/JCI115989; PMID: 1401063
- Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest 1992; 90:320 - 5; http://dx.doi.org/10.1172/JCI115865; PMID: 1644911
- Marshak S, Leibowitz G, Bertuzzi F, Socci C, Kaiser N, Gross DJ, et al. Impaired beta-cell functions induced by chronic exposure of cultured human pancreatic islets to high glucose. Diabetes 1999; 48:1230 - 6; http://dx.doi.org/10.2337/diabetes.48.6.1230; PMID: 10342809
- Khoo S, Griffen SC, Xia Y, Baer RJ, German MS, Cobb MH. Regulation of insulin gene transcription by ERK1 and ERK2 in pancreatic beta cells. J Biol Chem 2003; 278:32969 - 77; http://dx.doi.org/10.1074/jbc.M301198200; PMID: 12810726
- Docherty HM, Hay CW, Ferguson LA, Barrow J, Durward E, Docherty K. Relative contribution of PDX-1, MafA and E47/beta2 to the regulation of the human insulin promoter. Biochem J 2005; 389:813 - 20; http://dx.doi.org/10.1042/BJ20041891; PMID: 15862113
- Lawrence MC, McGlynn K, Park BH, Cobb MH. ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem 2005; 280:26751 - 9; http://dx.doi.org/10.1074/jbc.M503158200; PMID: 15899886
- Giddings SJ, Chirgwin J, Permutt MA. Evaluation of rat insulin messenger RNA in pancreatic and extrapancreatic tissues. Diabetologia 1985; 28:343 - 7; http://dx.doi.org/10.1007/BF00283141; PMID: 2412922
- Leibiger B, Moede T, Schwarz T, Brown GR, Köhler M, Leibiger IB, et al. Short-term regulation of insulin gene transcription by glucose. Proc Natl Acad Sci USA 1998; 95:9307 - 12; http://dx.doi.org/10.1073/pnas.95.16.9307; PMID: 9689076
- Leibiger B, Wahlander K, Berggren PO, Leibiger IB. Glucose-stimulated insulin biosynthesis depends on insulin-stimulated insulin gene transcription. J Biol Chem 2000; 275:30153 - 6; http://dx.doi.org/10.1074/jbc.M005216200; PMID: 10913151
- Wicksteed B, Herbert TP, Alarcon C, Lingohr MK, Moss LG, Rhodes CJ. Cooperativity between the preproinsulin mRNA untranslated regions is necessary for glucose-stimulated translation. J Biol Chem 2001; 276:22553 - 8; http://dx.doi.org/10.1074/jbc.M011214200; PMID: 11297542
- Tillmar L, Carlsson C, Welsh N. Control of insulin mRNA stability in rat pancreatic islets. Regulatory role of a 3′-untranslated region pyrimidine-rich sequence. J Biol Chem 2002; 277:1099 - 106; http://dx.doi.org/10.1074/jbc.M108340200; PMID: 11696543
- Fred RG, Tillmar L, Welsh N. The role of PTB in insulin mRNA stability control. Curr Diabetes Rev 2006; 2:363 - 6; http://dx.doi.org/10.2174/157339906777950570; PMID: 18220641
- Roush SF, Slack FJ. Micromanagement: a role for microRNAs in mRNA stability. ACS Chem Biol 2006; 1:132 - 4; http://dx.doi.org/10.1021/cb600138j; PMID: 17163657
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 2010; 79:351 - 79; http://dx.doi.org/10.1146/annurev-biochem-060308-103103; PMID: 20533884
- Tang X, Muniappan L, Tang G, Ozcan S. Identification of glucose-regulated miRNAs from pancreatic beta cells reveals a role for miR-30d in insulin transcription. RNA 2009; 15:287 - 93; http://dx.doi.org/10.1261/rna.1211209; PMID: 19096044
- Melkman-Zehavi T, Oren R, Kredo-Russo S, Shapira T, Mandelbaum AD, Rivkin N, et al. miRNAs control insulin content in pancreatic β-cells via downregulation of transcriptional repressors. EMBO J 2011; 30:835 - 45; http://dx.doi.org/10.1038/emboj.2010.361; PMID: 21285947
- Zhao X, Mohan R, Özcan S, Tang X. MicroRNA-30d induces insulin transcription factor MafA and insulin production by targeting mitogen-activated protein 4 kinase 4 (MAP4K4) in pancreatic β-cells. J Biol Chem 2012; 287:31155 - 64; http://dx.doi.org/10.1074/jbc.M112.362632; PMID: 22733810
- Jacovetti C, Abderrahmani A, Parnaud G, Jonas JC, Peyot ML, Cornu M, et al. MicroRNAs contribute to compensatory β cell expansion during pregnancy and obesity. J Clin Invest 2012; 122:3541 - 51; http://dx.doi.org/10.1172/JCI64151; PMID: 22996663
- Plaisance V, Abderrahmani A, Perret-Menoud V, Jacquemin P, Lemaigre F, Regazzi R. MicroRNA-9 controls the expression of Granuphilin/Slp4 and the secretory response of insulin-producing cells. J Biol Chem 2006; 281:26932 - 42; http://dx.doi.org/10.1074/jbc.M601225200; PMID: 16831872
- Giddings SJ, Chirgwin J, Permutt MA. Effects of glucose on proinsulin messenger RNA in rats in vivo. Diabetes 1982; 31:624 - 9; http://dx.doi.org/10.2337/diabetes.31.7.624; PMID: 6761201
- Karolina DS, Armugam A, Tavintharan S, Wong MT, Lim SC, Sum CF, et al. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS One 2011; 6:e22839; http://dx.doi.org/10.1371/journal.pone.0022839; PMID: 21829658
- Reed MJ, Meszaros K, Entes LJ, Claypool MD, Pinkett JG, Gadbois TM, et al. A new rat model of type 2 diabetes: the fat-fed, streptozotocin-treated rat. Metabolism 2000; 49:1390 - 4; http://dx.doi.org/10.1053/meta.2000.17721; PMID: 11092499
- Fred RG, Bang-Berthelsen CH, Mandrup-Poulsen T, Grunnet LG, Welsh N. High glucose suppresses human islet insulin biosynthesis by inducing miR-133a leading to decreased polypyrimidine tract binding protein-expression. PLoS One 2010; 5:e10843; http://dx.doi.org/10.1371/journal.pone.0010843; PMID: 20520763
- Altirriba J, Barbera A, Del Zotto H, Nadal B, Piquer S, Sánchez-Pla A, et al. Molecular mechanisms of tungstate-induced pancreatic plasticity: a transcriptomics approach. BMC Genomics 2009; 10:406; http://dx.doi.org/10.1186/1471-2164-10-406; PMID: 19715561
- McClenaghan NH, Barnett CR, Ah-Sing E, Abdel-Wahab YH, O’Harte FP, Yoon TW, et al. Characterization of a novel glucose-responsive insulin-secreting cell line, BRIN-BD11, produced by electrofusion. Diabetes 1996; 45:1132 - 40; http://dx.doi.org/10.2337/diabetes.45.8.1132; PMID: 8690162
- Efrat S. Development of engineered pancreatic beta-cell lines for cell therapy of diabetes. Adv Drug Deliv Rev 1998; 33:45 - 52; http://dx.doi.org/10.1016/S0169-409X(98)00019-2; PMID: 10837652
- Limbert C, Päth G, Jakob F, Seufert J. Beta-cell replacement and regeneration: Strategies of cell-based therapy for type 1 diabetes mellitus. Diabetes Res Clin Pract 2008; 79:389 - 99; http://dx.doi.org/10.1016/j.diabres.2007.06.016; PMID: 17854943
- Sun LL, Jiang BG, Li WT, Zou JJ, Shi YQ, Liu ZM. MicroRNA-15a positively regulates insulin synthesis by inhibiting uncoupling protein-2 expression. Diabetes Res Clin Pract 2011; 91:94 - 100; http://dx.doi.org/10.1016/j.diabres.2010.11.006; PMID: 21146880
- Narushima M, Kobayashi N, Okitsu T, Tanaka Y, Li SA, Chen Y, et al. A human beta-cell line for transplantation therapy to control type 1 diabetes. Nat Biotechnol 2005; 23:1274 - 82; http://dx.doi.org/10.1038/nbt1145; PMID: 16186810
- Gartner W, Koc F, Nabokikh A, Daneva T, Niederle B, Luger A, et al. Long-term in vitro growth of human insulin-secreting insulinoma cells. Neuroendocrinology 2006; 83:123 - 30; http://dx.doi.org/10.1159/000094875; PMID: 16888402
- Labriola L, Peters MG, Krogh K, Stigliano I, Terra LF, Buchanan C, et al. Generation and characterization of human insulin-releasing cell lines. BMC Cell Biol 2009; 10:49; http://dx.doi.org/10.1186/1471-2121-10-49; PMID: 19545371
- Ravassard P, Hazhouz Y, Pechberty S, Bricout-Neveu E, Armanet M, Czernichow P, et al. A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J Clin Invest 2011; 121:3589 - 97; http://dx.doi.org/10.1172/JCI58447; PMID: 21865645
- Guo-Parke H, McCluskey JT, Kelly C, Hamid M, McClenaghan NH, Flatt PR. Configuration of electrofusion-derived human insulin-secreting cell line as pseudoislets enhances functionality and therapeutic utility. J Endocrinol 2012; 214:257 - 65; http://dx.doi.org/10.1530/JOE-12-0188; PMID: 22685334
- Rosero S, Bravo-Egana V, Jiang Z, Khuri S, Tsinoremas N, Klein D, et al. MicroRNA signature of the human developing pancreas. BMC Genomics 2010; 11:509; http://dx.doi.org/10.1186/1471-2164-11-509; PMID: 20860821
- Chu K, Tsai MJ. Neuronatin, a downstream target of BETA2/NeuroD1 in the pancreas, is involved in glucose-mediated insulin secretion. Diabetes 2005; 54:1064 - 73; http://dx.doi.org/10.2337/diabetes.54.4.1064; PMID: 15793245
- Gu C, Stein GH, Pan N, Goebbels S, Hörnberg H, Nave KA, et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab 2010; 11:298 - 310; http://dx.doi.org/10.1016/j.cmet.2010.03.006; PMID: 20374962
- Sharma A, Moore M, Marcora E, Lee JE, Qiu Y, Samaras S, et al. The NeuroD1/BETA2 sequences essential for insulin gene transcription colocalize with those necessary for neurogenesis and p300/CREB binding protein binding. Mol Cell Biol 1999; 19:704 - 13; PMID: 9858593
- Kaneto H, Nakatani Y, Miyatsuka T, Matsuoka TA, Matsuhisa M, Hori M, et al. PDX-1/VP16 fusion protein, together with NeuroD or Ngn3, markedly induces insulin gene transcription and ameliorates glucose tolerance. Diabetes 2005; 54:1009 - 22; http://dx.doi.org/10.2337/diabetes.54.4.1009; PMID: 15793239
- Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 2009; 324:1710 - 3; http://dx.doi.org/10.1126/science.1174381; PMID: 19460962
- Martin A, Komada MR, Sane DC. Abnormal angiogenesis in diabetes mellitus. Med Res Rev 2003; 23:117 - 45; http://dx.doi.org/10.1002/med.10024; PMID: 12500286
- Kolluru GK, Bir SC, Kevil CG. Endothelial dysfunction and diabetes: effects on angiogenesis, vascular remodeling, and wound healing. Int J Vasc Med 2012; 2012:918267; http://dx.doi.org/10.1155/2012/918267; PMID: 22611498
- Razumilava N, Bronk SF, Smoot RL, Fingas CD, Werneburg NW, Roberts LR, et al. miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology 2012; 55:465 - 75; http://dx.doi.org/10.1002/hep.24698; PMID: 21953056
- Zhang H, Zuo Z, Lu X, Wang L, Wang H, Zhu Z. MiR-25 regulates apoptosis by targeting Bim in human ovarian cancer. Oncol Rep 2012; 27:594 - 8; PMID: 22076535
- Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004; 432:226 - 30; http://dx.doi.org/10.1038/nature03076; PMID: 15538371
- Ehehalt F, Knoch K, Erdmann K, Krautz C, Jäger M, Steffen A, et al. Impaired insulin turnover in islets from type 2 diabetic patients. Islets 2010; 2:30 - 6; http://dx.doi.org/10.4161/isl.2.1.10098; PMID: 21099291
- Støy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al, Neonatal Diabetes International Collaborative Group. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA 2007; 104:15040 - 4; http://dx.doi.org/10.1073/pnas.0707291104; PMID: 17855560
- Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J, et al. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab 2010; 21:652 - 9; http://dx.doi.org/10.1016/j.tem.2010.07.001; PMID: 20724178
- Liu M, Lara-Lemus R, Shan SO, Wright J, Haataja L, Barbetti F, et al. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes 2012; 61:828 - 37; http://dx.doi.org/10.2337/db11-0878; PMID: 22357960
- Hausser J, Syed AP, Bilen B, Zavolan M. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Res 2013; 23:604 - 15; http://dx.doi.org/10.1101/gr.139758.112; PMID: 23335364
- van de Bunt M, Gaulton KJ, Parts L, Moran I, Johnson PR, Lindgren CM, et al. The miRNA profile of human pancreatic islets and beta-cells and relationship to type 2 diabetes pathogenesis. PLoS One 2013; 8:e55272; http://dx.doi.org/10.1371/journal.pone.0055272; PMID: 23372846
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, et al, MAGIC investigators, GIANT Consortium. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2010; 42:579 - 89; http://dx.doi.org/10.1038/ng.609; PMID: 20581827
- Wang H, Lin YZ, Zhou Y, Lu HM, Lu SH, Luo C, et al. Circulatory level of microRNA 92a in patients of stable angina pectoris with diabetes mellitus. China Journal of Modern Medicine 2011; 23:718 - 22
- Sepramaniam S, Armugam A, Lim KY, Karolina DS, Swaminathan P, Tan JR, et al. MicroRNA 320a functions as a novel endogenous modulator of aquaporins 1 and 4 as well as a potential therapeutic target in cerebral ischemia. J Biol Chem 2010; 285:29223 - 30; http://dx.doi.org/10.1074/jbc.M110.144576; PMID: 20628061
- Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY) 2009; 1:425 - 37; PMID: 20157526
- Su JL, Kilpatrick KE, Champion BR, Morris DC, Lehmann JM, Kost TA. Fluorescent microtiter screening assay for immunocytochemically reactive antibodies. Biotechniques 1997; 22:320 - 4; PMID: 9043705