Abstract
Recently, we have determined the secondary structure of the 5′-terminal region of p53 mRNA that starts from the P1 transcription initiation site and includes two major translation initiation codons responsible for the synthesis of p53 and ΔNp53 isoform. Here, we showed that when this region was extended into 5′ direction to the P0 transcription start site, the two characteristic hairpin motifs found in this region were preserved. Moreover, the presence of alternatively spliced intron 2 did not interfere with the formation of the larger hairpin in which the initiation codon for p53 was embedded. The impact of the different variants of p53 5′-terminal region, which start at P0 or P1 site and end with the initiation codon for p53 or ΔNp53, on the translation of luciferase reporter protein was compared. Strikingly, the efficiency of translation performed in rabbit reticulocyte lysate differed by two orders of magnitude. The toe-printing analysis was also applied to investigate the formation of the ribosomal complex on the model mRNA constructs. The relative translation efficiencies in HeLa and MCF-7 cells were similar to those observed in the cell lysate, although some differences were noted in comparison with cell-free conditions. The results were discussed in terms of the role of secondary structure folding of the 5′-terminal region of p53 mRNA in translation and possible modes of p53 and ΔNp53 translation initiation.
Introduction
Recent genome-wide studies have identified multiple transcriptional start sites in human genes.Citation1-Citation3 Other studies have uncovered a variety of potential translation initiation codons upstream of the coding sequences. Global mapping of such positions in mammalian cells has shown that nearly half of the transcripts are potentially translated from alternative initiation sites.Citation4 Since many genetic disorders seem to be attributed to malfunctions in transcriptional and translational regulations a better understanding of expression mechanisms of the genes, which are transcribed and/or translated from multiple start sites is of high importance.
One of the most intensively studied genes is tumor suppressor p53, which makes use of several transcription promoters, undergoes alternative splicing, and uses alternative translation initiation sites to generate multiple protein isoforms.Citation5-Citation7 Transcription of p53 gene is initiated mainly at P1 and P0 start sites separated by ca. 110 nucleotides, which are located within exon 1. The full-length p53 protein is synthesized from the initiation codon AUG1 localized in exon 2. One of the p53 isoforms is ∆Np53 that lacks the N-terminal transactivation domain. This form is translated from the alternative initiation codon AUG2 present in exon 4, 114 nucleotides downstream of the AUG1 codon. It has also been proposed that the ∆Np53 isoform may originate from alternatively spliced pre-mRNA in which intron 2 is retained.Citation8 The ΔNp53 isoform can bind to the full-length p53 protein playing the role of a negative regulator of the p53 level, thus inhibiting its activity as a transcription factor and growth suppressor.Citation9,Citation10
The major regulator of the p53 level in normally functioning cells is the Mdm2 protein.Citation5-Citation7 The protein attaches ubiquitin residues that drive p53 to proteasome, where it is degraded. However, it has been observed that p53 expression is also regulated at the translational level and the 5′-untranslated region of p53 mRNA is involved in this regulation.Citation11,Citation12 This region exerts properties of IRES elements, which enable internal initiation of translation of full-length p53 as well as ΔN-p53 isoform.Citation10,Citation13 The differentiated activity of the IRES elements involved in p53 translation may play an important function in the mechanism describing the role of both forms of p53 in the regulation of the cellular cycle.Citation13 On the other hand, at the transcriptional level, a switch between promoter sites for p53 mRNA from primary P0 to P1 has been observed for normal to tumor tissues.Citation14 The region between these two promoters has been suggested to form a stable secondary structure reducing translation efficiency.Citation15 It seems, therefore, that the secondary structure folding of the 5′-terminal region of p53 mRNA plays an important role in p53 expression at both the transcriptional and translational levels. This assumption requires detailed structural studies of different variants of the p53 5′-terminal region and an analysis of their impact on translation of the full-length p53 protein and ΔN-p53 isoform.
Recently, we have determined the secondary structure of the 5′-terminal region of p53 mRNA that starts from the P1 transcription initiation site and includes two translation initiation codons responsible for the synthesis of p53 and ΔNp53 isoform. Alternative mechanisms of p53 mRNA translation initiation have also been investigated in vitro using mRNA fragments.Citation16 Here, we characterized the secondary structure of the 5′-terminal region of p53 mRNA that started from the P0 transcription initiation site. Moreover, the intron 2 sequence was also retained in one transcript to see how its presence influences folding of this mRNA region. In parallel with the functional studies, we evaluated the impact of different variants of the p53 5′-terminal region beginning at the P0 and P1 transcription start sites on translation efficiency of a reporter protein. Relative efficiencies of translation initiation from the sites corresponding to full-length p53 and its ΔNp53 isoform were compared in rabbit reticulocyte lysate as well as in HeLa and MCF-7 cell lines. The results were discussed in terms of the role of the secondary structure folding of the 5′-terminal region of p53 mRNA in the translation process and the possible modes of p53 and ΔNp53 translation initiation.
Results
Folding of the 5'-terminal region of p53 mRNA variants that begin at the P0 transcription start site
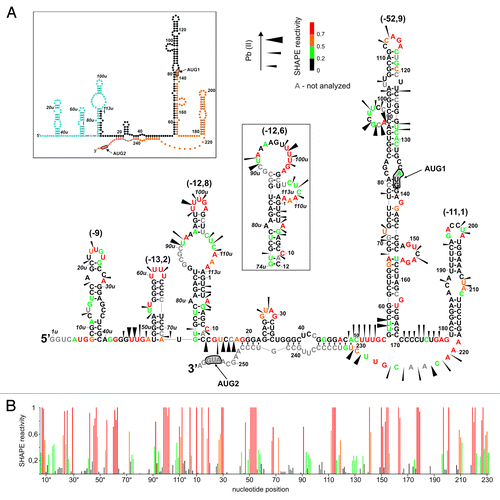
Two 5′-terminal fragments of p53 mRNA were synthesized by in vitro transcription. The fragments began at the transcription start site P0. RNA P0-p53 (253 nt) was terminated with the p53 initiation codon AUG1 and RNA P0-ΔNp53 (370 nt) with AUG2 for ΔNp53 isoform ( and ). In order to simplify the comparison of the variants of the 5′-terminal region of p53 mRNA, the two regions were individually numbered. Nucleotides located between promoters P0 and P1 were denoted with numbers with superscript “u” (upstream; residues 1u–113u), while nucleotides of intron 2 with numbers with superscript “i” (intronic; residues 1i–117i). In that way, the numbering of other nucleotides was consistent with that used earlier for variants beginning at the transcription start site P1.Citation16 Secondary structures of the P0-p53 and P0-ΔNp53 RNAs were analyzed by means of Pb2+ cleavage and SHAPE probing methods.Citation17-Citation19 The experimental results of the SHAPE approach were used in RNAStructure v.5.4 computer program as structural constraints to predict secondary structure models of the investigated regions of p53 mRNA.
Figure 1. Structural probing of P0-p53 RNA by Pb2+-induced cleavage and SHAPE methods. (A) The secondary structure model of P0-p53 RNA. Relative intensities of Pb2+ cleavages correlate with the size of black triangles. Nucleotide symbols are colored (black, green, orange, red) according to their SHAPE reactivities. Nucleotides that were not analyzed are in gray. The predicted ΔG values (kcal/mol) of structural motifs are in brackets. In the inset schematic representation of predicted secondary structure for the P0-p53, RNA is shown. Colors denote parts of the 5′-terminal region of p53 mRNA: blue, region between P0-P1 transcription promoters; black, 5′ untranslated region downstream P1 promoter. (B) Normalized SHAPE reactivity as a function of nucleotide position. Bars are colored using the scheme shown in (A).
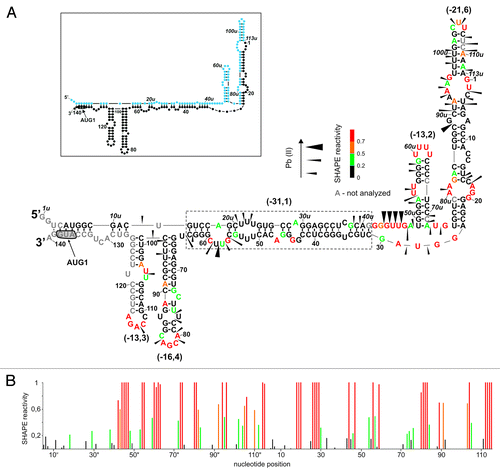
Figure 2. Secondary structure of P0-ΔNp53 RNA analyzed by Pb2+-induced cleavage and SHAPE. (A) The secondary structure model of P0-ΔNp53 RNA. Cleavages induced by Pb2+ ions are displayed on the secondary structure model of P0-ΔNp53 RNA that is most consistent with experimental data. Alternative arrangement of G74u-C12 region is also shown in the inset (details in the text). Nucleotide symbols are colored to reflect their SHAPE reactivity. The predicted ΔG values (kcal/mol) of structural motifs are in brackets. In the inset schematic representation of predicted secondary structure for the P0-ΔNp53 RNA is shown. Colors denote parts of the 5′-terminal region of p53 mRNA: blue, region between P0-P1 transcription promoters; black, 5′ untranslated region downstream P1 promoter; orange, p53 open reading frame (ORF). (B) Normalized SHAPE reactivity as a function of nucleotide position.
Secondary structure of RNA P0-p53
shows a secondary structure model of RNA P0-p53 with the lowest free energy of ΔG = -133 kcal/mol. In this structure stretches U3u-G41u and C31–G139 interact with each other forming a large double-stranded region with several bulges, internal loops, and two hairpin motifs G65-C98 and C99-G126. A part of this region (boxed in ) is characterized by a relatively large thermodynamic stability (ΔG = -31.1 kcal/mol). The other nucleotides of this RNA are arranged into two hairpins U49u-A72u and U75u-A25, which are separated from the stable double-stranded region by two single-stranded stretches G42u-A48u and G26-G30. The hairpin U49u-A72u with ΔG = -13.2 kcal/mol consists of nine base pairs out of which seven pairs are G-C type. The hairpin U75u-A25 contains two asymmetric internal loops, a two-nucleotide bulge, non-standard G-A interaction, and a three-nucleotide-membered apical loop.
The proposed secondary structure of RNA P0-p53 is well supported by the results of experimental probing with Pb2+-induced cleavage and SHAPE methods (; Fig. S1). Strong Pb2+ cleavages are observed in the G43u-G47u and G56-C59 stretches. A strong cleavage also occurs in hairpin U75u-A25 between C88u and G89u from the G-A non-standard base pair. Weak cleavages also take place in most apical loops of the hairpin motifs, which are present in this RNA. Some cleavages occur in selected double-stranded stretches, for example, in the upper part of hairpin U75u-A25 (nucleotides U98u-A112u). They are likely to be a consequence of several less stable A-U pairs present in the stem and of the local breathing of the RNA structure.
The results of the SHAPE approach map the apical loop regions of all hairpins in RNA P0-p53, larger single-stranded regions and most bulged residues or located in internal loops (). A comparison of Pb2+ and NMIA probing data also reveals some discrepancies. Most of these concern weaker recognition of purine-rich stretches in single-stranded RNA regions by Pb2+ ions.Citation17 An exception is the apical loop of hairpin U49u-A72u, which consists of four U residues. The loop is not cleaved by Pb2+ but is strongly modified by NMIA.
Structural probing of RNA P0-ΔNp53
In the secondary structure of RNA P0-ΔNp53 with the free energy of -172.3 kcal/mol, the region between G19 and C247 is arranged identically as in RNA P1-ΔNp53, which has been studied earlier in our laboratory (; Fig. 2 in ref. Citation16). The most characteristic structural elements of this region are two hairpin motifs. In one of them, the initiation codon AUG1 is embedded while the other hairpin has been shown to bind the Mdm2 and hnRNPC1/C2 proteins.Citation20,Citation21 The patterns of Pb2+ cleavages and NMIA modification sites in the corresponding regions of both RNAs are very similar (; Fig. S2). However, in RNA P0-ΔNp53, stronger Pb2+ cleavages occur at uridine residues in positions 57 and 58, at the bottom of hairpin G56-C169. Stronger cleavages also take place in bulge region A91-C98, as well as at U122, G127, and U128. On the other hand, weak cleavages, which were present in RNA P1-ΔNp53 (see ref. Citation16) disappear in the pyrimidine-rich region C188-U193 in hairpin U180-A218. As regards the SHAPE data, the only difference in the modification patterns is the increase of reactivity of C116, G118, C119, and the downstream U128-C131 region in RNA P0-ΔNp53 (; Fig. 2 in ref. Citation16).
The region G1u-A118, which spans the transcription start sites P0 and P1, folds into a secondary structure different than the one determined for RNA P0-p53. The only common motif present in both RNAs is hairpin U49u-A72u, and its presence in RNA P0-ΔNp53 is confirmed by the results of Pb2+ cleavage and SHAPE mapping (). The stretch G74u-C12 folds into a hairpin motif, similarly as in RNA P0-p53 but the hairpin is arranged differently. It has a large asymmetric purine-rich internal loop, and a five-nucleotide-membered apical loop. Moreover, the hairpin has one non-standard A-A base pair with NMIA reactive A5 residue, and bulged C10 nucleotide that is also strongly modified by this reagent. Several suboptimal structures proposed for hairpin G74u-C12 by the RNAstructure program suggest that it is a relatively unstable structural element, which may show a tendency to rearrange. This region may fold into an alternative hairpin in which the apical loop is enlarged to 14 nucleotides and the U106u-A112u region is bulged out (). The mapping results of RNA P0-ΔNp53 support both these hairpins, which have almost identical ΔG values of -12.8 and -12.6 kcal/mol.
Importantly, the 5′-proximal region of RNA P0-ΔNp53 is arranged differently compared with RNA P0-p53. It folds into hairpin structure G8u-C39u of low stability with ΔG of -9 kcal/mol. This hairpin is separated from hairpin U49u-A72u by a single-stranded stretch, which is clearly recognized by Pb2+ ions and NMIA.
The role of intron 2 in folding of the 5'-terminal region of p53 mRNA
The 5′-terminal fragment of p53 mRNA with intron 2 retained, RNA P1-ΔNp53(int2) (376 nt), started from transcription promoter P1 and ended with the initiation codon AUG2 for translation of ΔNp53 isoform. The secondary structure of this RNA was probed with Pb2+ cleavage and SHAPE methods. shows a secondary structure model proposed for RNA P1-ΔNp53(int2) by the RNAstructure program with the use of experimentally determined structural constrains.
Figure 3. Secondary structure arrangement of the P1-ΔNp53(int2) RNA. (A) The secondary structure model of P1-ΔNp53(int2) RNA. Cleavages induced by Pb2+ ions as well as SHAPE data are displayed in the figure. Numbers in brackets denote predicted ΔG values (kcal/mol) for selected structural motifs. In the inset, schematic representation of predicted secondary structure for the P1-ΔNp53(int2) RNA is shown. Colors denote parts of the 5′-terminal region of p53 mRNA: black, 5′ untranslated region downstream P1 promoter; orange, p53 open reading frame (ORF); green, intron 2. (B) Normalized SHAPE reactivity as a function of nucleotide position. Bars are colored using the scheme shown in (A).
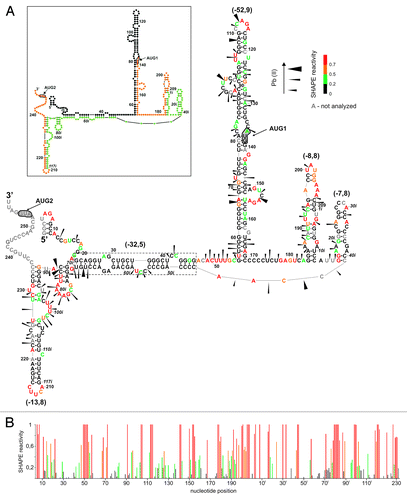
In the secondary structure of RNA P1-ΔNp53(int2), the G56-C169 hairpin motif is formed, despite the presence of the intervening intron 2 sequence. This is supported by the characteristic NMIA modification of single-nucleotide bulges U72 and G141, nucleotides in the apical loop, and in two multi-nucleotide bulges A91-C98 and A149-A155. Nucleotides of the internal loop containing the p53 initiation codon are also weakly modified. A similar modification has been earlier observed in RNAs P1-554 and H56-169 in which the same hairpin motif was present.Citation16 The pattern of Pb2+ cleavages also supports the presence of hairpin G56-C169. Characteristic cleavages occur in multi-nucleotide bulges A91-C98 and A149-A155, in the apical loop, and at sites located in the vicinity of AUG1 initiation codon.
The presence of intron 2 in RNA P1-int2 precludes the formation of hairpin U180-A218 and results in four new structural motifs of the hairpin type: G183-C12i, G16i-C41i, G72i-C86i, and G89i-C235. The 5′-terminal nucleotides of intron 2, G1i-C12i, interact with the p53-coding sequence G183-C194 forming the G183-C12i hairpin. High reactivity of nucleotides of the upper part of this hairpin, A197-A207, suggests that this region may exist in a single-stranded form. Hairpin G183-C12i has a relatively low thermodynamic stability with ΔG = -8.8 kcal/mol and its several nucleotides undergo NMIA modification or Pb2+ cleavage. Hairpin G16i-C41i is also relatively low stable (ΔG = -7.8 kcal/mol) and strong modification of G16i and G17i by NMIA may suggest disruption of two base pairs at the hairpin base. Downstream nucleotides of the intron, C46i-U70i, interact with nucleotides G23-G46 of the untranslated region of p53 mRNA. A thermodynamically very stable double-stranded region is formed with ΔG of the isolated duplex of -32.5 kcal/mol. Asymmetric internal loops within the helix are mapped by Pb2+ ions. The other nucleotides of intron 2 are arranged into two hairpin motifs G72i-C86i and G89i-C235. Hairpin G72i-C86i is mapped by both the SHAPE and Pb2+ cleavage methods. Hairpin G89i-C235 consists of a distal part of intron 2 (nucleotides G89i-G117i) and a stretch of the coding region of p53 mRNA (nucleotides A215-C235). NMIA modifies nucleotides of six-membered bulge C97i-U102i and five-membered apical loop. Pb2+ ions also recognize the bulge C97i-U102i but do not induce cleavages within the apical loop region despite the presence of four pyrimidine residues. Several Pb2+ cleavages occur in region C222-C229. This is most likely a consequence of weak base pairs A-U and G-U in the stem and a multi-nucleotide bulge in the opposite strand of the hairpin. Finally, the hairpin motifs of RNA P1-ΔNp53(int2) are separated by three long single-stranded stretches A47-C55, C170-A182, and C42i-A45i. These regions are well mapped by the SHAPE and Pb2+ cleavage methods. Exceptionally, the C170-U176 region is resistant to modification but these nucleotides have also been unreactive in other earlier studied variants of p53 mRNA.Citation16
Translation efficiency of model mRNAs is influenced by both the regions spanning transcription start sites P0 and P1 and those located between two initiation codons
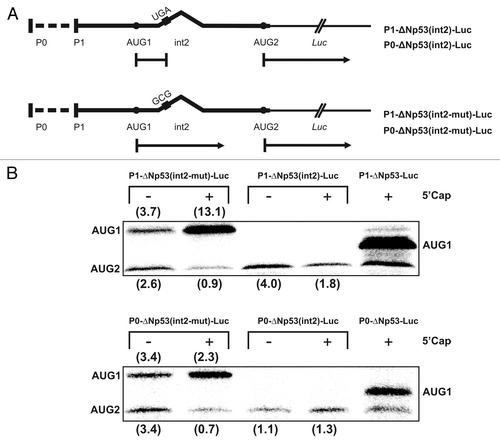
In order to evaluate the role of variants of the 5′-terminal region of p53 mRNA in translation several model mRNA constructs were synthesized (). In the expression plasmids the variant sequences preceded the sequence encoding the Renilla luciferase reporter protein. Before performing translation assays, it was important to show that the major structural elements of the 5′-terminal region of p53 mRNA—two characteristic hairpin motifs G56-C169 and U180-A218 were preserved in these conditions. To this end, the P1-ΔNp53-Luc construct was used. Folding of the 5′-terminal region in this model, mRNA was analyzed in RRL by DMS modification. It turned out that the G56-C169 and U180-A218 hairpins were preserved and their structure was not disturbed by the downstream luciferase sequence (data not shown).
Figure 4. Translation of model mRNA constructs with the sequence encoding reporter luciferase protein proceeded by variants of the p53 5′-terminal region in cell-free conditions in RRL. (A) Schematic representation of model mRNA constructs. (B) Model mRNAs were translated in RRL in the presence of 35S-methionine. The autoradiograms in the insets show the translation products and the bar charts display quantitatively the translation efficiency of model mRNAs, uncapped and capped, from two initiation codons AUG1 and AUG2. The logarithmic scale was used for data representation. All values are averages of at least three independent experiments and were normalized to the translation efficiency of luciferase mRNA. The values calculated for the longer protein products, which were synthetized with the (C and D) templates were corrected for the presence of an additional 35S-methionine residue.
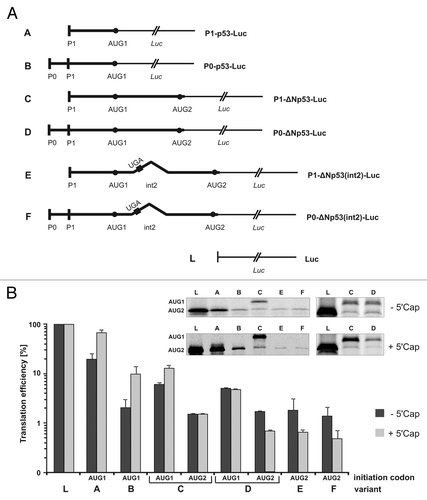
The model mRNA constructs with various 5′-terminal regions of p53 mRNA were translated in RRL in the presence of 35S-methionine. Panel B of shows protein products obtained with capped and uncapped templates (the insets) and the bar chart illustrates quantitatively the effects of variants on translation. The translation efficiency is expressed in percentages in relation to the translation efficiency of the control capped and uncapped mRNAs encoding luciferase. In the mRNA constructs in which the luciferase open reading frame was preceded by the coding sequence located between AUG1 and AUG2, the N-extended protein product contained an additional methionine residue. This was taken into account while calculating the relative amounts of 35S-labeled translation products.
A comparison of the translation efficiency of P1-p53-Luc and P0-p53-Luc (, transcripts A and B) shows that the region located between two transcription start sites P0 and P1 has a strong inhibitory effect. Translation efficiency is reduced by the factor of 6.9 and 9.3, for capped and uncapped mRNAs, respectively. For the capped P1-ΔNp53-Luc and P0-ΔNp53-Luc (, transcripts C and D) with both the initiation codons AUG1 and AUG2, translation efficiency is further reduced. Thus, the extension of P1-p53-Luc sequence in the 3′ direction to AUG2 results in a 5.9-fold decrease of the translation efficiency from AUG1, and the addition of region P0-P1 to its 5′ end reduces the efficiency by an additional factor of 2.7. In total, 16-fold inhibition is observed. The presence of region P0-P1 also reduces the translation from AUG2 by the factor of 2.2, as determined for capped P1-ΔNp53-Luc and P0-ΔNp53-Luc (, transcripts C and D). These differences are smaller when translation of uncapped templates is compared. The extension of the P1-p53-Luc sequence to AUG2 results in a 3.5-fold decrease of the translation efficiency from AUG1, but unexpectedly, further addition of region P0-P1 has almost no effect on translation from either AUG1 or AUG2.
Presence of the intron 2 sequence does not substantially influence translation efficiency from AUG2, and AUG1 codon is likely to be active in the initiation of translation
The presence of intron 2 in the P1-ΔNp53(int2)-Luc and P0-ΔNp53(int2)-Luc mRNAs (, transcripts E and F) results in the lack of translation of the reporter protein from AUG1. It is obvious since the intron has a termination codon UGA within the reading frame of the p53 coding sequence. Translation initiation at AUG1 potentially leads to a 25-aminoacid-long polypeptide. Both mRNAs containing intron 2 are translated from AUG2 with ca. 1% efficiency, similar to that observed with the templates with no intron embedded (). Thus, the presence of the P0-P1 region does not affect translation efficiency. Also, the 5′ cap structure does not have a stimulatory effect, and upon its addition the translation, is even reduced from ca. 1.5 to 0.5%.
In order to find out whether translation of not completely spliced, p53 mRNAs with intron 2 may also be initiated at AUG1, mutated P1-ΔNp53(int2)-Luc and P0-ΔNp53(int2)-Luc were synthesized (). The UGA stop codon in the intron sequence was replaced by the GCG triplet for alanine. The original and mutated mRNAs, capped and uncapped, were translated in RRL and the resulting protein products are shown in . The translation efficiency is expressed in percentages in relation to the translation efficiency of the capped and uncapped control mRNAs encoding luciferase protein. With the P1-ΔNp53(int2-mut)-Luc 4-fold stimulation of translation from AUG1 and a 2-fold decrease of the translation efficiency from AUG2 occur upon cap addition (). With capped P0-ΔNp53(int2-mut)-Luc almost no change of translation from AUG1 is observed whereas the translation efficiency from AUG2 decreases 5-fold. For these templates but uncapped the addition of region P0-P1 has almost no effect on translation from either AUG1 or AUG2, similarly as for the same templates without the intron sequence (, transcripts C and D). Interestingly, both the uncapped P1-ΔNp53(int2-mut)-Luc and P0-ΔNp53(int2-mut)-Luc constructs gave the translation products in a ratio of approximately 1:1.
Figure 5. The effect of mutation of the termination codon present in intron 2 on translation efficiency. (A) The STOP codon UGA present in P1-ΔNp53(int2)-Luc and P0-ΔNp53(int2)-Luc constructs was mutated to GCG codon for alanine. (B) The mutated and wild-type mRNA constructs were translated in RRL in the presence of 35S-methionine and the relative amounts of translation products initiated from AUG1 and AUG2 were determined. All values are averages of at least three independent experiments and were normalized to the translation efficiency of luciferase mRNA (not shown). The 5′-capped P1-ΔNp53-Luc construct was translated as a reference.
The toe-printing analysis reveals pausing of the initiation complexes at the initiation codons directly preceding the luciferase sequence while those embedded in hairpin G56-C169 are hardly detected
The toe-printing analysisCitation22,Citation23 was performed with P1-p53-Luc, P1-ΔNp53-Luc, and P1-ΔNp53(int2)-Luc mRNAs. In the first template, translation was initiated from AUG1, in the second from AUG1 and AUG2, and in the third case AUG1 and AUG2 were separated by the termination codon UGA present in intron 2. The reactions of primer extension inhibition by 48S and 80S initiation complexes in RRL were performed with 32P-end-labeled primers in the presence of GMP-PNP and cycloheximide (Cx). GMP-PNP is a non-hydrolysable GTP analog, an inhibitor of ribosomal subunit joining. The formation of polysomes is blocked by Cx, a translocation inhibitor, which stalls ribosomes. Interestingly, when AMV reverse transcriptase was used, the toe-prints were generated only by pausing the complexes at the initiation codons directly preceding the sequence of the reporter luciferase protein (). It was AUG1 in P1-p53-Luc, and AUG2 in P1-ΔNp53-Luc and P1-ΔNp53(int2)-Luc. The toe-prints were located +15–17 nucleotides downstream the adenosine residue in AUG codons and the bands distribution was very similar in the presence of GMP-PNP and cycloheximide, Cx. Moreover, the toe-print bands were detected in the control lines when the blocking reagents were omitted. An analogous effect has been earlier observed by others and explained by suggesting a delay in the conversion of the 48S intermediate into the 80S complex.Citation22
Figure 6. The analysis of ribosomal complexes paused on model mRNAs containing different variants of the p53 5′-terminal region. The toe-printing of 48S and 80S initiation complexes formation in RRL with P1-p53-Luc (A), P1-ΔNp53-Luc (B and D) and P1-ΔNp53(int2)-Luc (C) mRNAs. For (A–C), toe-printing reactions were performed with the 5′-end-radiolabeled primer annealed to the luciferase coding sequence downstream of the AUG2 codon and the primer was elongated by AMV reverse transcriptase. For (D), the toe-print for AUG1 was generated with Superscript III reverse transcriptase and with the primer annealed to nucleotides 232–257 within the p53 5′-terminal region. Lane - Cap demonstrates inhibition of initiation complex assembling at the final concentration of 2 mM m7GpppG cap analog. The products of primer extension inhibition were resolved on 8% polyacrylamide gels in denaturing conditions. Positions of the AUG initiation codons and toe-print bands are marked on the autoradiograms. C and CL denote control reactions in water and RRL respectively, when GMP-PNP and Cx were omitted. A dideoxynucleotide sequence ladder was generated with the same primers as those used in the toe-printing reactions.
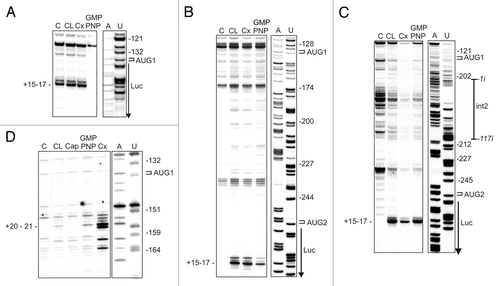
Surprisingly, in P1-ΔNp53-Luc and P1-ΔNp53(int2)-Luc mRNAs the toe-prints were observed for AUG2 and no signals were detected for AUG1. In both these cases, AUG1 is located in the stable G56-C169 hairpin and this seems to preclude the toe-print occurrence. The expected signal for AUG1 was detected when the experimental protocol was changed and Superscript III instead of AMV reverse transcriptase was applied which seems to penetrate double-stranded RNA structures.Citation24 In contrast, it has been described that in these conditions the toe-printing analysis of an mRNA with two initiation codons in strong Kozak’s context has revealed the toe-print almost exclusively for the 5′-proximal initiation codon. The second codon gave the expected signal when the first codon was in unfavorable Kozak’s context.Citation24
Translation efficiency of model mRNA changes differently for products initiated at AUG1 and AUG2 upon increasing concentration of the cap analog
The P1-ΔNp53-Luc mRNA was translated in RRL in the presence of 35S-methionine and increasing concentration of the cap analog (m7GpppG) to inhibit cap-dependent translation. The amounts of both protein products, initiated at AUG1 and AUG2, were determined, and following quantification to the values with no cap analog added, they were displayed in , in panel A.
Figure 7. The influence of increasing concentration of the cap analog on translation and stability of translation products in RRL. (A) The capped P1-ΔNp53-Luc mRNA was translated in RRL in the presence of 35S-methionine and increasing concentration of the cap analog (m7GpppG) to inhibit cap-dependent translation. The amounts of both protein products were determined, and following quantification and normalization to the values with no cap analog added, they were displayed on the graph. (B) The protein products generated from AUG1 and AUG2 during prolonged incubation in RRL are shown.
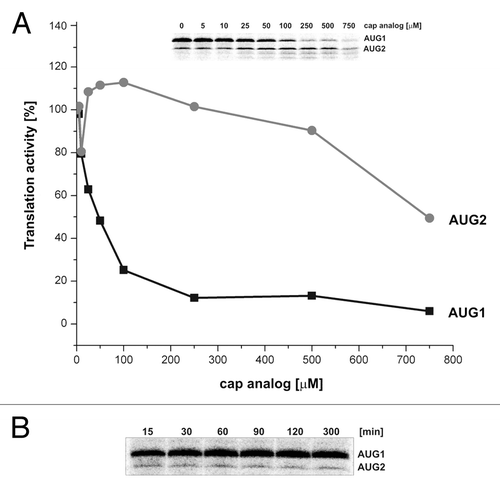
Translation from AUG1 is strongly inhibited already at low concentrations of the cap analog (). At its 250 μM concentration, the amount of translation product is on the level of 10% of the initial value with no inhibitor. In contrast, translation from AUG2 slightly increases up to 100 μM of the cap analog but at its higher concentration, translation activity gradually diminishes. At a concentration of 250 μM, translation activity from AUG2 codon is still equal to that observed with no inhibitor ().
In order to find out whether during translation in RRL protein products may be degraded upon prolonged incubation time, the P1-ΔNp53-Luc mRNA was translated in the presence of 35S-methionine for up to 5 h. The protein products initiated at AUG1 and AUG2 codons were separated by gel electrophoresis (). Product quantification shows that the amount of protein synthesized from AUG1 increases over time and reaches its maximum after approximately 1 h incubation. Subsequently, its amount slightly decreases but still more than 85% is present in the mixture after 5 h incubation in RRL (data not shown).
Translation of model mRNA constructs in HeLa and MCF-7 cells
HeLa and MCF-7 cells were co-transfected with equi-molar concentration of 5′-capped model mRNA constructs containing Renilla luciferase coding sequence along with 5′-capped and poly(A)-tailed firefly luciferase RNA to control transfection efficiency. After 4 h, cells were lysed and the luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega). The quantitative results of this assay are displayed as bars charts in .
Figure 8. Translation of model mRNA constructs with the sequence encoding reporter luciferase protein proceeded by variants of the p53 5′-terminal region in MCF-7 and HeLa cell lines. Model mRNAs were introduced to HeLa and MCF-7 cells along with Firefly luciferase mRNA as a transfection control. After transfection Dual Luciferase Assay was performed and Renilla luciferase activity was normalized for each construct. All transfections were repeated at least three times in different cell passages.
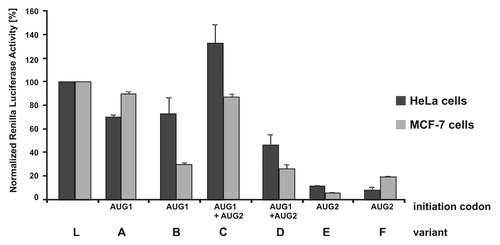
The data shows that translation efficiency of P1-p53-Luc and P0-p53-Luc (, transcripts A and B) is lower than that of the control Luc message (L) in both cell lines. Although in HeLa cells the constructs are translated with a similar efficiency as in MCF-7 cells, P0-p53-Luc, which contains the additional P0-P1 region, is translated almost three times less efficiently. In the case of P1-ΔNp53-Luc and P0-ΔNp53-Luc (, transcripts C and D), it was not possible to quantify the translation products initiated from AUG1 and AUG2 separately using the Dual-Luciferase Reporter Assay. Therefore, the summarized efficiencies of translation from both these AUG codons were determined. It turns out that in both cell lines the presence of the P0-P1 region strongly (2–3-fold) reduces the amounts of protein products (, transcripts C and D). Finally, the lowest translation level is observed in the case of P1-ΔNp53(int2)-Luc and P0-ΔNp53(int2)-Luc mRNAs (, transcripts E and F). In HeLa cells, their translation activity is ca. 10% of that determined for the control Luc template. In MCF-7 cells the relative amounts of translation products are 6% and 20% for P1-ΔNp53(int2)-Luc and P0-ΔNp53(int2)-Luc, respectively.
Discussion
Compared with transcriptional regulation, translational control of existing mRNAs allows for more rapid changes in the cellular concentrations of the encoded proteins.Citation25-Citation28 The most highly regulated phase of translation is initiation. The underlying principle of the translation initiation mechanism of eukaryotic mRNAs has been proposed by Kozak as so-called scanning model.Citation29 Recent elegant investigations are still adding important information to this widely accepted model.Citation30-Citation32 The 5′ untranslated regions of mRNAs from which the initiation complex begins scanning have been proposed to modulate translation efficiency and together with protein factors they may participate in translation regulation. In particular, several lines of evidence have suggested that the 5′-terminal region of p53 mRNA plays an important role in the regulation of p53 expression and its major isoform ΔNp53 at the translational level.Citation11,Citation33 In order to better understand the functioning of this region we have undertaken comprehensive studies of variants of the 5′-terminal region of p53 mRNA.
Recently, we have characterized the secondary structure of the 5′-terminal region of p53 mRNA that begins at P1 transcription site.Citation16 Here, we show that when this region is extended into 5′ direction to the P0 site its major structural features are preserved (–; Fig. S4). In transcripts that end with AUG1 initiation codon, hairpins G65-C98 and C99-G126 are present in both RNAs P0-p53 and P1-p53. Moreover, the C99-G126 hairpin also exists in the 3′-extended transcripts ending with AUG2, in which this motif constitutes the apical part of hairpin G56-C169. Importantly, in these longer transcripts, two characteristic hairpin elements are present. The larger hairpin G56-C169 embeds the initiation codon for the full-length p53 while the smaller hairpin U180-A218 has earlier been shown to interact with the Mdm2 protein.Citation20 Thus, in mRNA fragments that are extended to the AUG2 codon, the two characteristic hairpins are formed regardless of whether the RNA synthesis begins from transcription promoter P0 or P1.
It turns out that the region spanning the two transcription initiation sites P0 and P1 does not disturb the secondary structure motifs of p53 mRNA found downstream ( and ; Fig. S4). However, the arrangement of the P0-P1 stretch differs depending on whether the transcripts end with the AUG1 or AUG2 codon. In the shorter transcript, RNA P0-p53 nucleotides G1u to G41u interact with the downstream sequence forming a long, imperfect helix, relatively stable thermodynamically, which is closed with the U49u-A72u hairpin (). This hairpin is also present in the longer transcript RNA P0-ΔNp53 (), which suggests that it folds independently and may be a common structural element of region P0-P1 in p53 mRNA. The other nucleotides of this longer transcript are arranged into hairpin motifs of modest stability separated by single-stranded RNA stretches.
The presence of intron 2 leads to the destruction of some crucial structural elements and the formation of new interactions (; Fig. S4). Obviously, intron 2 disrupts the U180-A218 hairpin. The intron nucleotides form two new hairpins and interact with some non-coding and coding stretches of the 5′-terminal region. Most importantly, these interactions do not interfere with the formation of the characteristic hairpin G56-C169.
In order to get some idea on how the secondary structure of the 5′-terminal region of p53 mRNA may impact the scanning of this region by translating ribosome, we performed computer simulation of this process (Fig. S5). The ribosome movement and opening of the structure were mimicked by hybridization of a 30-nucleotide long oligomer sliding along sequences of variants of the 5′-terminal regions.Citation34 For RNAs P0-ΔNp53 and P1-ΔNp53, which begin at P0 or P1 promoters and extend to AUG2, the free energy change profiles for the target structure opening are closely superimposed on each other. This suggests that the secondary structure of the P0-P1 stretch is responsible for the differences in the translation efficiency of mRNAs controlled by these 5′-terminal regions. In contrast, for RNAs P0-p53 and P1-p53, which end with AUG1, the corresponding free energy change profiles for the target structure opening are clearly distinct. In that case, the possible impact of the RNA secondary structure on translation may depend on both the region spanning the P0 and P1 promoters and the downstream nucleotide sequence.
Having the suggestion that the 5′-terminal regions of p53 mRNA may have an impact on the scanning of this region by translating ribosome, we decided to evaluate this effect in vitro in RRL. It seems that cell lysates are deprived of most regulatory proteins,Citation35 therefore such conditions are particularly well suited to evaluate the effect of RNA secondary structure on translation. The efficiency of translation of model mRNAs with various 5′-terminal regions of p53 mRNA differed by two orders of magnitude (; note that translation efficiency values are expressed in the logarithmic scale). In the case of the compared capped and uncapped constructs that end with AUG1 ca. 4-fold stimulation of the translation is observed (, transcripts A and B). This is consistent with some earlier observations on cap-dependent translation in RRL.Citation35 For 3′-extended constructs with both initiation codons, the translation efficiency from AUG1 increases only 2-fold for the transcript that begins at P1 and no stimulation is observed for the transcript initiated at P0 (, transcripts C and D). Unexpectedly, in the case of the protein products synthesized from AUG2 no impact or even slight inhibition of translation takes place upon cap addition. This suggests that translation from this initiation site occurs via a cap-independent mechanism. Such a mechanism is also consistent with the impact of the cap analog. At low analog concentrations of up to 250 µM, translation of P1-ΔNp53-Luc mRNA from AUG2 is stimulated or remains unchanged (), similar to our earlier observations for RNA-554 with 1/3 coding sequence of p53 mRNA.Citation16 In contrast, translation initiated from AUG1 strongly decreases upon addition of the cap analog and this effect is even more pronounced for P1-ΔNp53-Luc mRNA than for RNA-554.Citation16 In the 5′-terminal region of p53 mRNA, the presence of IRES elements has been suggested and our data, particularly those concerning translation initiated from AUG2, support this suggestion.Citation12,Citation13
It turns out that the region located between the P0 and P1 promoters strongly (7–9-fold) reduces the translation efficiency initiated from AUG1 (, compare transcripts A and B). Translation of mRNAs with long 5′ UTRs has been believed to result in lower productivity of the translation systems. This could be the effect of a significant off-rate movement along extended 5′ UTRs.Citation32 However, this does not explain the observed impact of region P0-P1 on translation, since with the transcripts extended to AUG2 much smaller (1–3-fold) effects are noticed upon P0-P1 addition (, compare transcripts C and D). Thus, the differences in the translation activity seem to be rather a consequence of secondary structure folding of cap-proximal regions. The 5′-terminal region of P0-p53-Luc is likely to be involved in strong secondary interactions just at the beginning of the template as in RNA P1-p53 (). Such a structural arrangement may interfere either with loading of the 43S complex onto the mRNA or the scanning process.Citation36,Citation37 In contrast, at least the first seven nucleotides of the P0-P1 region in P0-ΔNp53-Luc are single-stranded and the downstream nucleotides form relatively unstable hairpin structures as in RNA P0-ΔNp53. Therefore, secondary structure folding of this region does not interfere with loading of the 43S complex or moving the complex along the 5′-proximal region of mRNA. A highly stable hairpin G56-C169 with ΔG of -52.9 kcal/mole is present downstream in this transcript, which can be a substantial hindrance to the scanning process. It has been shown that stable hairpins with ΔG lower than -30 to -40 kcal/mole severely inhibit translation even when located distantly from the 5′ end of mRNA.Citation38 Moreover, the AUG1 codon is embedded within hairpin G56-C169 and its accessibility to initiator tRNA might be restricted. Limited accessibility of AUG initiation codons seems to contribute to low translation efficiency.Citation39 Recent studies have shown that the expression level of GFP reporter protein in E. coli cells can vary 250-fold in the library of model mRNAs. The stability of mRNA secondary structure near the start codon explained more than half of the variations in the expression level and mRNA with a more stable local structure had reduced protein expression.Citation40
In case of two model mRNAs containing intron 2 translation of the reporter protein, sequence is observed only from AUG2 codon. Moreover, neither the presence of the P0-P1 region nor 5′ capping change the translation efficiency from this site. This may suggest that these templates are translated via a cap-independent mechanism and possibly IRES elements are involved in initiation similarly as proposed for p53 mRNA.Citation12,Citation13 Interestingly, our data suggest that in mRNAs containing intron 2 also AUG1 is active in translation. This is shown for the templates transcribed either from P1 or P0 sites (). Mutation UGA/GCG (codon stop/alanine) in the intron sequence results in the appearance of a protein longer than the product synthetized from the templates without the intron. This strongly suggests that during translation of not completely spliced p53 mRNAs containing intron 2 also a short, 25-aminoacid-long polypeptide is synthetized from AUG1.
Importantly, in all the variants of the 5′-terminal region of p53 mRNA extended to AUG2 the first initiation codon AUG1 is embedded in the G56-C169 hairpin (this work and ref. Citation16). Even in the mRNA construct with intron 2 the structural surrounding of this codon is the same. The G56-C169 hairpin seems to strongly reduce p53 translation efficiency despite favorable Kozak’s context of AUG1. A stable hairpin structure is probably responsible for the lack of the toe-printing signal for AUG1 when analyzed with AMV reverse transcriptase, although in P1-p53-Luc mRNA in which there is no G56-C169 hairpin a clear signal of ribosome pausing at AUG1 is detected (). Pausing of the initiation complex at this codon but embedded in the G56-C169 hairpin in P1-ΔNp53-Luc mRNA is revealed with SuperScript III reverse transcriptase that gives stronger toe-prints and seems to more easily unfold the double-stranded RNA structures.Citation24 However, it remains unclear how the ribosome unfolds and moves through the G56-C169 hairpin. Recently, a way of ribosome moving through the stable hairpin motifs has been proposed.Citation30 The most important role in this process is played by the helicase activity of the initiation factors and some proteins recruited by the scanning complex as well as the activity of the ribosome itself as a helicase.Citation25,Citation41,Citation42
The hairpin G56-C169 contributes at least partially to the determination of the ratio of translation products initiated from AUG1 and AUG2. Both these codons are located in optimal Kozak’s context. It has been observed that if two AUG codons were in optimal Kozak’s context but were separated by 78 nucleotides of an unstructured nucleotide sequence the ratio of translation products was ca. 1:1.Citation43 For translation of p53 mRNA, the ratio of ca. 5:1 for p53 and ΔNp53 is usually determined. One may speculate that the hairpin G56-C169 causes pausing of the moving ribosome, thus increasing the translation efficiency from AUG1. Similarly, as it has been observed for stable hairpins located just downstream of initiation codons.Citation44 Alternatively, only a small fraction of scanning ribosomes can go through the hairpin and initiate translation from the AUG2 codon. However, translation from AUG2 is more likely to be initiated predominantly or exclusively via the IRES-directed mechanism.
Finally, the assumption that protein factors may contribute to translation regulation of p53 is supported by the translation results of our model mRNAs in HeLa and MCF-7 cells (). Some differences in translation efficiencies are observed in these two cell lines and in cell-free conditions. In MCF-7 cells, P1-p53-Luc and P0-p53-Luc (transcripts A and B in ) are translated with different efficiencies but similar to those observed in RRL. However, these mRNAs are translated similarly to each other in HeLa cells. This may suggest a higher involvement of helicases in translation in this cell line. Although the relative translation efficiencies in HeLa and MCF-7 cells are similar to those observed in RRL, substantial differences are noted in some cases in comparison with cell-free conditions. For example, P1-ΔNp53-Luc is translated more effectively than P0-ΔNp53-Luc (transcripts C and D in ) suggesting that the P0-P1 region may bind some inhibiting proteins. Several proteins have been identified that bind to the 5′ UTR of p53 mRNA, for example, the full-length p53, Mdm2, ribosomal protein L26, nucleolin, the polypyrimidine tract-binding protein PTB, and the tumor-suppressor protein Pdcd4.Citation33,Citation45 It has to be established whether any of these proteins (or any others which have not been identified yet) bind specifically to the P0-P1 region. As the human genome is predicted to encode ca. 1000 RNA-binding proteins with a large percentage of them implicated in translation,Citation46 such proteins are likely to exist.
Materials and Methods
DNA template constructs
All variants of the 5′ non-coding region of p53 mRNA described in this study (except P1-ΔNp53[int2] and P0-ΔNp53[int2]) were amplified from human liver cDNA (Ambion) using the following forward and reverse primers: for P1-p53: FS, 5′-CTAGAGCCAC CGTCCAGGGA GC-3′ and RS, 5′-TCCATGGCAG TGACCCGGAA GG-3′; for P1-ΔNp53: FS and RL, 5′-TCCATTGCTT GGGACGGCAA GG-3′; for P0-p53: FP0, 5′-GTCATGGCGA CTGTCCAGCT TTG-3′ and RS; for P0-ΔNp53: FP0 and RL.
PCR reaction mixtures contained 0.5 ng cDNA, 0.25 μM forward and reverse primers, 75 mM Tris-HCl, pH 8.8, 20 mM (NH4)2SO4, 0.01% Tween 20, 1.5 mM MgCl2, 200 μM each dNTP, and 1.25 units of Taq polymerase (Thermo Scientific). The reactions were performed on a Biometra T Gradient thermocycler for 30 cycles of initially 5 min at 95 °C, then 30 s at 95 °C, 30 s at 58 °C, 2 min at 72 °C, and finally 5 min at 72 °C. The reaction products were purified by phenol/chloroform (1:1, v/v) extraction and precipitated with ethanol. The obtained dsDNA templates were dissolved in TE buffer. Variants of the 5′ non-coding region of p53 mRNA were cloned into pRL-CMV vector (Promega) and transformed into E. coli DH5α competent cells (Invitrogen). The sequence of each construct was confirmed by sequencing.
In the case of P1-ΔNp53(int2) and P0-ΔNp53(int2) DNA, in the first PCR reaction sequences upstream and downstream of intron 2 were amplified from human liver cDNA while the intron 2 sequence from human genomic DNA (European Collection of Cell Cultures). In the next PCR reaction, all DNA fragments were assembled to obtain P1-ΔNp53(int2) and P0-ΔNp53(int2) DNAs. The P1-ΔNp53(int2-mut)-Luc and P0-ΔNp53(int2-mut)-Luc templates with UGA stop codon in intron 2 mutated to GCG were constructed by site-directed mutagenesis.
In vitro transcription
For the purpose of in vitro transcription all plasmid constructs were linearized using NotI restriction enzyme (Thermo Scientific). The in vitro transcription reaction with AmpliScribe T7, T3, and SP6 High Yield Transcription Kit (Epicenter Biotechnologies) or RiboMax Large Scale RNA Production System-T7 (Promega) was performed as recommended by the manufacturers. 5′-capped RNAs were synthesized in a reaction containing Anti-Reverse Cap Analog (ARCA, Epicenter Biotechnologies) in a final concentration of 3 mM, 1.5 mM GTP, and 7.5 mM of the remaining nucleotides. After the transcription reaction, one unit of DNaseI was added and the reaction was incubated 15 min at 37 °C. All RNAs were purified using RNeasy MinElute Cleanup kit (Qiagen) or RNA Clean-up and Concentration kit (Norgen). The size and integrity of each transcript were verified using agarose gel electrophoresis and ethidium bromide staining.
SHAPE analysis
The reaction mixture containing 20 pmol of RNA (0.4 μM final concentration) in renaturation buffer (10 mM Tris pH 8.0, 100 mM KCl, and 0.1 mM EDTA), and in final volume of 20 μl, was heated at 90 °C for 3 min and slowly cooled (0.1 °C/s) to 4 °C. The folding buffer (40 mM Tris pH 8.0, 5 mM MgCl2, 130 mM KCl, and 0.1 mM EDTA) was then added followed by water, to final volume of 146 μl. The sample was incubated at 37 °C for 10 min and separated into two reactions. The RNA solution was mixed with 7.3 μl 180 mM NMIA (Invitrogen) in DMSO (8–16 mM final concentration). The control reaction contained DMSO without NMIA. Both reactions were incubated for 50 min at 37 °C and RNA was precipitated with 0.3 M sodium acetate, pH 5.2, 1 μl glycogen (20 mg/ml), and 3 vol of ethanol. After centrifugation, RNA was resuspended in 10 mM Tris pH 8.0 and 0.1 mM EDTA.
Pb2+-induced cleavage
Prior to the cleavage reaction with Pb2+ ions, 50 pmol of unlabeled RNA was renatured in the buffer: 40 mM NaCl, 10 mM Tris-HCl pH 7.2, 10 mM MgCl2 by heating for 5 min at 65 °C and slowly cooling to 37 °C.Citation17,Citation18 Then, the RNA solution was divided into four samples and the lead acetate solution was added to the final concentration of 0.25 mM, 0.5 mM, 1 mM, and an equal volume of water to the control reaction. After incubation at 37 °C for 5 min, the reactions were terminated by mixing their aliquots with 20 mM EDTA solution. All samples were ethanol precipitated and resuspended in 10 mM Tris pH 8.0 and 0.1 mM EDTA.
Analysis of modifications and cleavage sites by primer extension
The primer extension reaction was performed by mixing modified RNA (2 pmol), 5′-end-32P-labeled DNA primer (2 pmol), and water in final volume of 12 μl. The reaction mixture was incubated at 95 °C for 1 min, at 60 °C for 5 min and at 52 °C for 2 min, and then 8 μl of the reverse transcription mix was added (final reaction conditions: 50 mM Tris HCl pH 8.3, 75 mM KCl, 3 mM MgCl2, 6 mM DTT, 500 μM each dNTP, and five units of SuperScipt III reverse transcriptase - Invitrogen). The reactions were performed at 52 °C for 10 min. cDNA samples were then treated with 1 μl of 4 M NaOH, placed on ice, heated at 95 °C for 5 min, placed on ice and treated with 160 mM Trizma (Sigma). Dideoxy sequencing markers were generated in the same way using unmodified RNA and dideoxy terminating nucleotides (0.2 mM). In the next step cDNAs were precipitated with 0.3 M sodium acetate, pH 5.2, 1 μl glycogen (20 mg/ml), and 3 vol of ethanol. After centrifugation cDNAs were resuspended in water. Following addition of 8 M urea/dyes/20 mM EDTA solution the samples were denatured for 2 min at 95 °C and loaded on 8% polyacrylamide, 0.75% bis-acrylamide, 7 M urea gels. Electrophoresis was performed at 2000 V for 2–3 h, followed by phosphorimaging with Typhoon 8600 analyzer (Molecular Dynamics).
Computer analysis of SHAPE data and RNA structure prediction
Bands that corresponded to NMIA-modified nucleotides and those present in the control lane were integrated using Semi-Automated Footprinting Analysis (SAFA) software to obtain numerical output of the band intensities.Citation47 Raw intensities were normalized using noRNAlize program.Citation48 The program works under Matlab environment and performs statistical analyses of the weak signals from NMIA modifications, subsequently using that information for signal normalization. All secondary structure predictions were performed using the RNAstructure 5.4 program.Citation49 Normalized SHAPE intensities were entered as pseudo-free energy constrains to obtain secondary structures for all studied RNA molecules.
In vitro translation
In vitro translation reactions were performed in nuclease-treated rabbit reticulocyte lysate (RRL) as described by the manufacturer (Promega). Briefly, the translation mixture containing 17.5 μl of RRL, 20 μM amino acid mixture without methionine, 1 μl of 35S-methionine (1000 Ci/mol) (Hartman Analytic), 20 units of recombinant ribonuclease inhibitor (Promega) and 2.5 pmole of 5′-capped or uncapped reporter RNA construct in the final volume of 25 μl was incubated for 90 min at 30 °C. For the purpose of translation inhibition experiments, RRL was preincubated for 15 min at 30 °C with increasing concentration of the m7GpppG cap analog (0–750 μM; Epicenter Biotechnologies) as an inhibitor and equimolar amounts of magnesium acetate. Aliquots of translation reactions were resolved on 15% SDS-polyacrylamide gels, followed by radioisotope imaging with FLA 5100 image analyzer (Fuji). Band intensities were analyzed using MultiGauge software (Fuji).
Toe-printing of 48S and 80S complexes in rabbit reticulocyte lysate (RRL)
The primer-extension analysis by AMV reverse transcriptase (Promega) in nuclease-treated RRL (Promega) was prepared as described previously.Citation22 Primer 5′-TAATAAATGA ATCAAGAACA TCC-3′ complementary to nt 81–103 in Renilla luciferase coding sequence was employed to produce toe-prints for P1-p53-Luc, P1-ΔNp53-Luc and P1-ΔNp53(int2)-Luc transcripts. Primers 5′-TCCATTGCTT GGGACGGCAA GG-3′ complementary to 5′ untranslated region (nt 232–257) and 5′-CTTCCCACAG GTCTCTGCTA GG-3′ complementary to nt 63–84 in the intron 2 sequence were used to obtain toe-prints for AUG1 codon in P1-ΔNp53-Luc and P1-ΔNp53(int2)-Luc, respectively. The translation mixture contained 7.14 μl of RRL, 10 units of recombinant ribonuclease inhibitor (Promega) and 13 mM Mg(OAc)2. In the control reaction, 7.14 μl water was added instead of RRL. To assemble 48S and 80S ribosomal complexes, the mixture was pretreated with 2 mM GMP-PNP (Sigma) or 1 mg/ml cycloheximide (Sigma), respectively. The mixtures were incubated for 5 min at 30 °C, followed by addition of 1 pmol of 5′-capped reporter RNA construct and incubated for another 5 min at 30 °C. Then, about 5 pmol of 32P-labeled oligonucleotide primer and 8 μl of RT-Mix (0.6 μl of 320 mM Mg(OAc)2, 1 μl of 10 mM dNTPs, 1 μl of 10 x Reconstitution buffer (200 mM Tris-HCl pH 7.5, 25 mM Mg(OAc)2, 10 mM DTT, 1 M KCl), 4.9 μl water, and five units of AMV reverse transcriptase (Promega) were added to each sample and incubated for 15 min at 30 °C. Additionally, the 48S and 80S reconstitution mixtures were supplemented with 1 μl of water or 1 μl of cycloheximide. cDNA was purified by phenol extraction and ethanol precipitation. Samples were separated on 8% denaturing polyacrylamide gel and analyzed by phosphorimaging using FLA 5100 image analyzer (Fuji). Migration of cDNAs products was compared with a dideoxynucleotide sequence ladder obtained in the reverse transcription reaction using the same primer.
Cell culture, transfection, and dual-luciferase assay
HeLa and MCF-7 cells were maintained in RPMI (Sigma Aldrich) or high-glucose DMEM (CytoGen), respectively, both supplemented with 10% fetal bovine serum (FBS; Sigma Aldrich), 1 x non-essential amino acids mixture (Gibco) and 1 x antibiotic-antimycotic solution (Sigma Aldrich). Twenty-four hours before transfection, 2 × 10Citation5 cells were seeded per well in a 12-well plate in 2 ml of complete growth medium. On the day of transfection, when cell density reached 80–90%, transfection with 5′-capped monocistronic RNAs was performed using the TransMessenger transfection reagent (Qiagen). Cells were co-transfected with equimolar concentration of 5′-capped RNA constructs containing Renilla luciferase coding sequence along with 5′-capped and poly(A)-tailed Firefly luciferase RNA to control the transfection efficiency (1.6 μg in total). Both reporters were mixed with 6 μl of TransMessenger reagent and added drop-wise onto the cells in 300 μl of appropriate medium without serum and antibiotics. After 3 h, the transfection complex was removed from the cells and the fresh complete medium was added. Four hours post-transfection, the cells were lysed with 250 μl of 1 x Passive Lysis Buffer (Promega) for 20 min at room temperature with rocking. The luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega) according to the instruction manual. Briefly, first 20 μl of prepared lysate was added to 100 μl of Luciferase Assay Reagent II (LARII) and Firefly luciferase activity was measured for 10 s (with a 2 s pre-read delay). This reaction was quenched by adding 100 μl of Stop&Glo Reagent to the same well and the Renilla luciferase activity was assayed. Both luciferase activities were measured in white, flat-bottomed 96-well plates (Perkin Elmer) using 2030 Multilabel Reader VICTOR X4 (Perkin Elmer). All the transfections were repeated at least three times in different cell passages.
Additional material
Download Zip (1.3 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of our laboratory for valuable comments on the manuscript. Dr Michal Legiewicz is thanked for helpful suggestions on SHAPE approach and Dorota Kowalczykiewicz for assistance in preparation of some variants of the 5′-terminal region of p53 mRNA. The authors also thank Dr Edward Darzynkiewicz for providing us the m7GpppG cap analog. This work was supported by the Polish Ministry of Science and Higher Education (grant number NN301272037 to JC).
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/rnabiology/article/26562
References
- Davuluri RV, Suzuki Y, Sugano S, Plass C, Huang TH. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet 2008; 24:167 - 77; http://dx.doi.org/10.1016/j.tig.2008.01.008; PMID: 18329129
- Yamashita R, Sathira NP, Kanai A, Tanimoto K, Arauchi T, Tanaka Y, Hashimoto S, Sugano S, Nakai K, Suzuki Y. Genome-wide characterization of transcriptional start sites in humans by integrative transcriptome analysis. Genome Res 2011; 21:775 - 89; http://dx.doi.org/10.1101/gr.110254.110; PMID: 21372179
- Yamashita R, Sugano S, Suzuki Y, Nakai K. DBTSS: DataBase of Transcriptional Start Sites progress report in 2012. Nucleic Acids Res 2012; 40:Database issue D150 - 4; http://dx.doi.org/10.1093/nar/gkr1005; PMID: 22086958
- Lee S, Liu B, Lee S, Huang SX, Shen B, Qian SB. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci U S A 2012; 109:E2424 - 32; http://dx.doi.org/10.1073/pnas.1207846109; PMID: 22927429
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9:749 - 58; http://dx.doi.org/10.1038/nrc2723; PMID: 19776744
- Olivier M, Petitjean A, Marcel V, Pétré A, Mounawar M, Plymoth A, de Fromentel CC, Hainaut P. Recent advances in p53 research: an interdisciplinary perspective. Cancer Gene Ther 2009; 16:1 - 12; http://dx.doi.org/10.1038/cgt.2008.69; PMID: 18802452
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413 - 31; http://dx.doi.org/10.1016/j.cell.2009.04.037; PMID: 19410540
- Ghosh A, Stewart D, Matlashewski G. Regulation of human p53 activity and cell localization by alternative splicing. Mol Cell Biol 2004; 24:7987 - 97; http://dx.doi.org/10.1128/MCB.24.18.7987-7997.2004; PMID: 15340061
- Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, Oren M, Hainaut P. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 2002; 21:6722 - 8; http://dx.doi.org/10.1038/sj.onc.1205874; PMID: 12360399
- Grover R, Candeias MM, Fåhraeus R, Das S. p53 and little brother p53/47: linking IRES activities with protein functions. Oncogene 2009; 28:2766 - 72; http://dx.doi.org/10.1038/onc.2009.138; PMID: 19483723
- Halaby MJ, Yang DQ. p53 translational control: a new facet of p53 regulation and its implication for tumorigenesis and cancer therapeutics. Gene 2007; 395:1 - 7; http://dx.doi.org/10.1016/j.gene.2007.01.029; PMID: 17395405
- Yang DQ, Halaby MJ, Zhang Y. The identification of an internal ribosomal entry site in the 5′-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene 2006; 25:4613 - 9; http://dx.doi.org/10.1038/sj.onc.1209483; PMID: 16607284
- Ray PS, Grover R, Das S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep 2006; 7:404 - 10; PMID: 16440000
- Strudwick S, Carastro LM, Stagg T, Lazarus P. Differential transcription-coupled translational inhibition of human p53 expression: a potentially important mechanism of regulating p53 expression in normal versus tumor tissue. Mol Cancer Res 2003; 1:463 - 74; PMID: 12692266
- Bienz-Tadmor B, Zakut-Houri R, Libresco S, Givol D, Oren M. The 5′ region of the p53 gene: evolutionary conservation and evidence for a negative regulatory element. EMBO J 1985; 4:3209 - 13; PMID: 3004941
- Błaszczyk L, Ciesiołka J. Secondary structure and the role in translation initiation of the 5′-terminal region of p53 mRNA. Biochemistry 2011; 50:7080 - 92; http://dx.doi.org/10.1021/bi200659b; PMID: 21770379
- Ciesiołka J, Michałowski D, Wrzesinski J, Krajewski J, Krzyzosiak WJ. Patterns of cleavages induced by lead ions in defined RNA secondary structure motifs. J Mol Biol 1998; 275:211 - 20; http://dx.doi.org/10.1006/jmbi.1997.1462; PMID: 9466904
- Kirsebom LA, Ciesiołka J. Pb+2–induced cleavage of RNA. In Hartmann RK, Bindereif A, Schön A, Westhof E, eds. Handbook of RNA Biochemistry. WILEY-VCH Verlag, 2013:ISBN: 978-3-527-32764-5.
- Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc 2006; 1:1610 - 6; http://dx.doi.org/10.1038/nprot.2006.249; PMID: 17406453
- Candeias MM, Malbert-Colas L, Powell DJ, Daskalogianni C, Maslon MM, Naski N, Bourougaa K, Calvo F, Fåhraeus R. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol 2008; 10:1098 - 105; http://dx.doi.org/10.1038/ncb1770; PMID: 19160491
- Christian KJ, Lang MA, Raffalli-Mathieu F. Interaction of heterogeneous nuclear ribonucleoprotein C1/C2 with a novel cis-regulatory element within p53 mRNA as a response to cytostatic drug treatment. Mol Pharmacol 2008; 73:1558 - 67; http://dx.doi.org/10.1124/mol.107.042507; PMID: 18296503
- Dmitriev SE, Pisarev AV, Rubtsova MP, Dunaevsky YE, Shatsky IN. Conversion of 48S translation preinitiation complexes into 80S initiation complexes as revealed by toeprinting. FEBS Lett 2003; 533:99 - 104; http://dx.doi.org/10.1016/S0014-5793(02)03776-6; PMID: 12505166
- Gould PS, Bird H, Easton AJ. Translation toeprinting assays using fluorescently labeled primers and capillary electrophoresis. Biotechniques 2005; 38:397 - 400; http://dx.doi.org/10.2144/05383ST02; PMID: 15786806
- Kozak M. Primer extension analysis of eukaryotic ribosome-mRNA complexes. Nucleic Acids Res 1998; 26:4853 - 9; http://dx.doi.org/10.1093/nar/26.21.4853; PMID: 9776744
- Aitken CE, Lorsch JR. A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol 2012; 19:568 - 76; http://dx.doi.org/10.1038/nsmb.2303; PMID: 22664984
- Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 2010; 11:113 - 27; http://dx.doi.org/10.1038/nrm2838; PMID: 20094052
- Liwak U, Faye MD, Holcik M. Translation control in apoptosis. Exp Oncol 2012; 34:218 - 30; PMID: 23070007
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009; 136:731 - 45; http://dx.doi.org/10.1016/j.cell.2009.01.042; PMID: 19239892
- Kozak M, Shatkin AJ. Migration of 40 S ribosomal subunits on messenger RNA in the presence of edeine. J Biol Chem 1978; 253:6568 - 77; PMID: 681367
- Abaeva IS, Marintchev A, Pisareva VP, Hellen CU, Pestova TV. Bypassing of stems versus linear base-by-base inspection of mammalian mRNAs during ribosomal scanning. EMBO J 2011; 30:115 - 29; http://dx.doi.org/10.1038/emboj.2010.302; PMID: 21113134
- Paek KY, Park SM, Hong KY, Jang SK. Cap-dependent translation without base-by-base scanning of an messenger ribonucleic acid. Nucleic Acids Res 2012; 40:7541 - 51; http://dx.doi.org/10.1093/nar/gks471; PMID: 22638585
- Vassilenko KS, Alekhina OM, Dmitriev SE, Shatsky IN, Spirin AS. Unidirectional constant rate motion of the ribosomal scanning particle during eukaryotic translation initiation. Nucleic Acids Res 2011; 39:5555 - 67; http://dx.doi.org/10.1093/nar/gkr147; PMID: 21415006
- Vilborg A, Wilhelm MT, Wiman KG. Regulation of tumor suppressor p53 at the RNA level. J Mol Med (Berl) 2010; 88:645 - 52; http://dx.doi.org/10.1007/s00109-010-0609-2; PMID: 20306257
- Tsao D, Shabalina SA, Gauthier J, Dokholyan NV, Diatchenko L. Disruptive mRNA folding increases translational efficiency of catechol-O-methyltransferase variant. Nucleic Acids Res 2011; 39:6201 - 12; http://dx.doi.org/10.1093/nar/gkr165; PMID: 21486747
- Andreev DE, Dmitriev SE, Terenin IM, Prassolov VS, Merrick WC, Shatsky IN. Differential contribution of the m7G-cap to the 5′ end-dependent translation initiation of mammalian mRNAs. Nucleic Acids Res 2009; 37:6135 - 47; http://dx.doi.org/10.1093/nar/gkp665; PMID: 19696074
- Alekhina OM, Vassilenko KS. Translation initiation in eukaryotes: versatility of the scanning model. Biochemistry (Mosc) 2012; 77:1465 - 77; http://dx.doi.org/10.1134/S0006297912130056; PMID: 23379522
- Malys N, McCarthy JE. Translation initiation: variations in the mechanism can be anticipated. Cell Mol Life Sci 2011; 68:991 - 1003; http://dx.doi.org/10.1007/s00018-010-0588-z; PMID: 21076851
- Babendure JR, Babendure JL, Ding JH, Tsien RY. Control of mammalian translation by mRNA structure near caps. RNA 2006; 12:851 - 61; http://dx.doi.org/10.1261/rna.2309906; PMID: 16540693
- Hinnebusch AG. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol Mol Biol Rev 2011; 75:434 - 67; http://dx.doi.org/10.1128/MMBR.00008-11; PMID: 21885680
- Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009; 324:255 - 8; http://dx.doi.org/10.1126/science.1170160; PMID: 19359587
- Hinnebusch AG, Lorsch JR. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol 2012; 4:a011544; http://dx.doi.org/10.1101/cshperspect.a011544; PMID: 22815232
- Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 2005; 361:13 - 37; http://dx.doi.org/10.1016/j.gene.2005.06.037; PMID: 16213112
- Matsuda D, Mauro VP. Determinants of initiation codon selection during translation in mammalian cells. PLoS One 2010; 5:e15057; http://dx.doi.org/10.1371/journal.pone.0015057; PMID: 21124832
- Kozak M. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc Natl Acad Sci U S A 1990; 87:8301 - 5; http://dx.doi.org/10.1073/pnas.87.21.8301; PMID: 2236042
- Wedeken L, Singh P, Klempnauer KH. Tumor suppressor protein Pdcd4 inhibits translation of p53 mRNA. J Biol Chem 2011; 286:42855 - 62; http://dx.doi.org/10.1074/jbc.M111.269456; PMID: 22033922
- Araujo PR, Yoon K, Ko D, Smith AD, Qiao M, Suresh U, Burns SC, Penalva LO. Before It Gets Started: Regulating Translation at the 5′ UTR. Comp Funct Genomics 2012; 2012:475731; http://dx.doi.org/10.1155/2012/475731; PMID: 22693426
- Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA 2005; 11:344 - 54; http://dx.doi.org/10.1261/rna.7214405; PMID: 15701734
- Vicens Q, Gooding AR, Laederach A, Cech TR. Local RNA structural changes induced by crystallization are revealed by SHAPE. RNA 2007; 13:536 - 48; http://dx.doi.org/10.1261/rna.400207; PMID: 17299128
- Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 2010; 11:129; http://dx.doi.org/10.1186/1471-2105-11-129; PMID: 20230624