Abstract
Internal ribosome entry site (IRES) elements govern protein synthesis of mRNAs that bypass cap-dependent translation inhibition under stress conditions. Picornavirus IRES are cis-acting elements, organized in modular domains that recruit the ribosome to internal mRNA sites. The aim of this study was to retrieve short RNA sequences with the capacity to adopt RNA folding patterns conserved with IRES structural subdomains, likely corresponding to RNA modules. We have applied a new program, RNAiFold, an inverse folding algorithm that determines all sequences whose minimum free energy structure is identical to that of the structural domains of interest. Sequences differing by more than 1 nt were clustered. Then, BLASTing one randomly chosen sequence from each cluster of the RNAiFold output, we retrieved viral and cellular sequences among output hits. As a proof of principle, we present the data corresponding to a coding region of Drosophila melanogaster TAF6, a transcription factor-associated protein that contains a structural motif within its coding region potentially folding into an IRES-like subdomain. This RNA region shows a biased codon usage, as predicted from structural constraints at the RNA level, it harbors conserved IRES structural motifs in loops, and interestingly, it has the capacity to confer internal initiation of translation in tissue culture cells.
Introduction
Translation initiation is a key step in the process of protein synthesis. Most cellular mRNAs initiate translation by a mechanism that depends on the recognition of the m7GpppN structure (termed cap) located at the 5′ end of most mRNAs. However, in some RNA viruses exemplified by picornaviruses and hepatitis C virus (HCV) and a subset of cellular mRNAs, which are translated under stress conditions, internal ribosome entry site (IRES) elements drive translation initiation using a cap-independent mechanism.Citation1-Citation4 IRES elements present in cellular mRNAs described so far tend to be present in genes encoding proteins required for survival under stress conditions, namely transcription factors, growth factors, apoptotic proteins, among others.Citation3 However, cellular IRES elements differ not only in nucleotide sequence but also in RNA secondary structure and trans-acting factor requirements.Citation1,Citation5 This complexity hampers the understanding of the mechanism of internal initiation and hinders progress in the prediction of novel IRES elements in mRNA sequences. Moreover, whether there are structural elements in yet-unknown cellular IRES shared with viral IRES is unknown.
RNA structure plays a fundamental role in IRES-dependent translation initiation as well as in other processes guided by RNA regulatory elements.Citation6 This is illustrated by the fact that compensatory substitutions in base-paired regions tend to conserve the secondary structure during RNA evolution.Citation7 Foot-and-mouth disease virus (FMDV) is a picornavirus characterized by a high genetic variability.Citation8 Similar to all picornavirus RNAs, protein synthesis in FMDV RNA is driven by an IRES element located at the 5′ UTR of the viral genome.Citation1 Type II IRES elements, such as the FMDV and encephalomyelitis virus (EMCV) IRES, are organized in structural domains numbered 1 to 5 in the 5′ to 3′ direction (). Each domain appears to have a specific function in which domains 1, 2, 4, and 5 consist of stem-loops that provide the binding site for RNA-binding proteins and various translation initiation factors (eIFs), with the exception of eIF4E.Citation1,Citation9
Figure 1. Secondary RNA structure of the FMDV IRES. Domains 1, 2, 3, 4, and 5 as well as the positions of the conserved GNRA, RAAA, and C-rich motifs are indicated. Nucleotide accessibility is colored according to SHAPE reactivity obtained using NMIA and 5′-radiolabeled primers.Citation7 SHAPE reactivity was calculated as describedCitation61 and represented in a colored scale in which 0 indicates an unreactive nucleotide and the average intensity at highly reactive nucleotides is set to 1.0. RNA structure was viewed with VARNA.Citation60
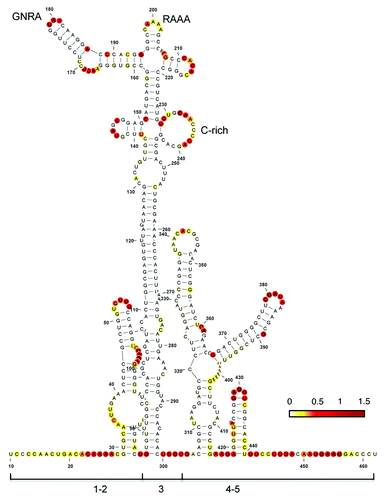
The central domain (domain 3) is a self-folding regionCitation10 with a peculiar RNA structure organization that includes conserved motifs whose disruption impairs IRES activity.Citation11-Citation13 Specifically, the GNRA (N stands for any nucleotide, and R, purine) motif of picornavirus IRES determines the RNA structural organization of the apical region of domain 3, which involves distant interactions with the also conserved RAAA stem-loop and the C-rich bulge.Citation14 On the basis of functional and structural data, these motifs have been proposed to mediate a tertiary foldingCitation7,Citation12,Citation14,Citation15 in such a way that altering one of them affects the RNA structural organization of this domain, thus leading to defective IRES elements. Given the unusual combination of motifs that constrains this unique RNA region, we hypothesized that a search for RNA sequences with the capacity to adopt a similar fold could correspond to RNA regions potentially promoting IRES activity, undetectable by other approaches.
The RNA inverse folding problem is inverse to RNA structure prediction; i.e., given a target structure, the goal is to design an RNA sequence that will fold into the target structure. Since RNA secondary structure is particularly well studied, while RNA pseudoknot and tertiary structure prediction is NP-complete, the RNA inverse folding problem usually refers to secondary structure. There is some evidence that the inverse RNA folding problem may be computationally hard, since Schnall-Levin et al.Citation16 have shown that the Inverse Viterbi Path problem is NP-complete. (Here, the latter is the problem, given a hidden Markov model [HMM] and arbitrary path π of states, of determining a sequence θ whose maximum likelihood [Viterbi] path is equal to π). Moreover, unlike the case for protein structure, it is broadly believed that RNA folds in a hierarchical fashion.Citation17 Due to this hierarchical folding, in many cases, it is possible to determine the secondary structure of an RNA molecule, without knowing its tertiary structure. Designing a sequence that can fold into a certain shape involves, at least, checking whether the proposed sequence actually folds into the target structure.
There are several algorithms for solving the RNA inverse folding problem. They all can be classified as heuristic methods, which start with an initial sequence that is iteratively modified until it either folds into the target structure or some stopping criterion is reached. The main examples are RNAinverse in the Vienna RNA Package, INFO-RNA, RNA-SSD, MODENA, and NUPACK-DESIGN.Citation18-Citation22
In contrast to the previously mentioned heuristic methods, in a very recently developed algorithm RNAiFold,Citation23 we employ Constraint Programming (CP) to solve the RNA inverse folding problem. CP has become one of the principal methodologies for solving hard combinatorial optimization problems, due to a rich modeling language and a computational model based on branch and prune. The inverse folding algorithm RNAiFold was developed using the COMET framework and RNAfold (from the Vienna RNA PackageCitation18) adapted as a plug in with COMET. COMET features a very efficient CP engine along with several global constraints that are key for the efficiency of our approach.
Here we show that a pipeline based on RNA Inverse Folding is a fast and reliable system to search for RNA regions that are predicted to adopt an RNA structure similar to conserved RNA structural subdomains of experimentally characterized IRES elements. By using this approach, we identified a coding region of Drosophila melanogaster TATA-box binding protein-associated factor (TAF6) as a candidate RNA adopting an IRES-like subdomain structure, in which conserved motifs (GNRA, RAAA, and AACCCCA) are located in loops according to RNA SHAPE structural analysis. Experimental validation of this IRES-like motif using bicistronic constructs in tissue culture cells indicated that the sense orientation had the capacity to enhance internal initiation of translation of the second cistron, relative to the activity observed with the negative orientation of the same region. As additional confirmation of this result, the immediate downstream region of TAF6 mRNA, which is not predicted to fold as an IRES-like motif, was unable to confer internal initiation of translation.
Results
Computational motif search
In order to search for RNA structural motifs with a similar folding to IRES subdomains we used conserved RNA motifs of the FMDV IRES as a model. This IRES element is 462 nt long, distributed in several domains with conserved stem-loops (). Among these stem-loops, the apical region of domain 3 adopts a unique structural organization that contains three essential RNA motifs.Citation11,Citation12 A short simplified version of this region, encompassing the RAAA and C-rich motifs (), was chosen as specific constraints in the input to RNAiFold. Thus, the structure and sequence constraints used as input to RNAiFold were the following:
Figure 2. Computational pipeline summary. (A) RNA structure and sequence constraints input for RNAiFold (I). (B) Sequences returned that fold like the input structure were then clustered (II). (C) One representative of each cluster was BLASTed (III) with match/mismatch weights of 1/-1 and gap existence/extension penalties of 1/2. (D) Returned hits were filtered (IV) using relevant keywords and genomic locations. (E) Candidate hits located in coding sequences were assessed (V) by total variation and codon usage bias determination (using BitGene http://www.bitgene.com). (F) Candidate sequences selected for experimental validation.

(((((((......)))(((((((((((.....)))))))))))(((((.......)))))))))
NNNNNNNNNNNNNNNNNNNNNNNNNNNNRAAANNNNNNNNNNNNNNNNAACCCCANNNNNNNNN
The corresponding planar representation of this structure is shown in , where our complete pipeline is represented. First, RNAiFold returned over 112 026 sequences after a time limit of 4 h (). These sequences were then clustered using a greedy algorithm in such way that two sequences in the same cluster could only differ by one nucleotide (). One sequence from each cluster was later BLASTed and the retrieved hits were filtered with an E-value limit of 3 (). A total of 3 370 unique hits were collected. It is worth noting that among the retrieved hits we identified viral genomes known to contain IRES as well as all FMDV sequences, as expected. This proves that the simplified input structure does not compromise the retrieval of the sequences corresponding to the original structure. After applying the keyword filter () we obtained around 60 candidate hits, 40 of which were discarded as they were annotated as predicted proteins. Finally, we selected six hits that were located in the appropriate position (either the 5′UTR or the coding sequence) (Table S1). In order to apply further computational validation techniques, we focused on the hits located within the coding region of the corresponding gene. and Figure S1 show the sequences of candidate hits in which putative conserved motifs are highlighted. The hit corresponding to Drosophila melanogaster TAF6 mRNA contains all relevant motifs and in a more appropriate disposition than the rest of candidates. Even though the input to RNAiFold contains both the RAAA and the C-rich motifs, so that all sequences returned by RNAiFold must contain these motifs, BLAST hits might not, since we allow non-perfect matches with a generous E-value of 3. TAF6 is a component of the core promoter-recognition complex TFIID, the RNA polymerase II general transcription factor, composed of the TATA-binding protein (TBP) and TBP-associated factors (TAFs).Citation24 Thus, we selected TAF6 as a promising case for further studies. The sequence generated by RNAiFold that allowed the identification of TAF6 by BLAST was the following:
Figure 3.Drosophila melanogaster TAF6 cDNA sequence. The sequence retrieved with RNA Inverse Folding pipeline is indicated by italics; motifs conserved with the IRES subdomain are indicated by bold letters; ATGs referred to in the text are depicted by yellow (ATG1), blue (ATG2), green (ATG3), or pink (ATG4) boxes. The region encompassing the IRES-like motif (region I) is colored in violet while the control downstream region (III) is indicated in light green.

AGAGGCCAAA AAAGGCGCGU GGCGGGGUGA AACCCCGCCG CGCCCGAAAA CCCCAUUCGG CUCU
ref|NM_079437.3| Drosophila melanogaster TBP-associated factor 6.
Computational determination of 5′ and 3′ ends
Since our candidate hit was located within the coding region of Drosophila melanogaster TAF6 mRNA, we compared codon usage between the region encompassing the predicted IRES-like motif with different 5′ and 3′ ends and the rest of the coding region. The downstream region contains three AUGs in frame with the functional start codon (). If our hit corresponds to a functional IRES-like motif, then we would expect the structural motif to be near or immediately adjacent to an alternative start codon. For that reason, we calculated codon usage for several candidate regions with variable 5′ ends (nt 25, 85, 145, 205, 265, 325) (see Materials and Methods), while the 3′ end was taken to be the nucleotide adjacent to the AUG2 or AUG3 codon (nt 522 or 747) (). shows both Codon Adaptation Index (CAI) and Total Variation (TV) for codon usage of each amino acid for all these regions. These data suggest that the region spanning nucleotides 325–522 could constitute a promising candidate for an RNA functional motif. This region shows the highest total variation, even though CAI values are similar for all regions ending in nucleotide 522. In the sequel, the candidate motif spanning nucleotides 325–522 will be designated as region I.
Figure 4. Schematic representation of the regions of Drosophila melanogaster TAF6 mRNA analyzed for internal initiation of translation. Region I was retrieved as a putative IRES-like motif by RNA Inverse Folding. Regions II, III, and IV are used as controls. Nucleotide numbers are counted relative to the A of first AUG.
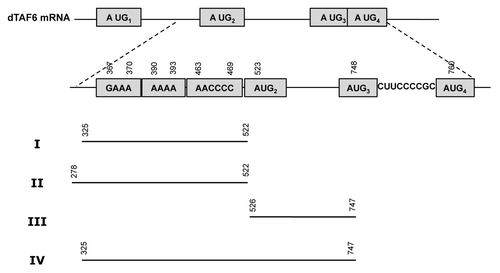
Table 1. Codon bias usage in the regions of interest
The codon usage bias for amino acids serine (S) and arginine (R) corresponding to region I are shown in Figure S2. These results indicate that the codon usage bias between the predicted IRES-like motif and the remainder of the coding sequence is significantly different and, thus, it is likely that this region has been under different selection pressure, likely to preserve RNA structure. As a control, we selected the downstream region (nucleotides 325 up to 747) ().
Experimental validation of the IRES-like subdomain
To determine whether the coding region of TAF6 mRNA encompassing an IRES-like motif could mediate internal initiation of translation, we measured the ratio of luciferase to chloramphenicol acetyl transferase (CAT) expressed from bicistronic constructs in BHK-21 cells (). In this assay, translation of CAT renders the efficiency of 5′-dependent translation initiation, while that of luciferase reflects the activity of 5′-independent translation initiation. The efficiency of internal initiation of translation of region I (nt 325–522) was 2.13 times higher in samples transfected with construct I(+), expressing the sense orientation, than the construct I(-) expressing the antisense orientation of the same region (). The higher relative internal translation initiation efficiency of region I was due to the increased luciferase expression, as the level of CAT expression remained fairly constant in all extracts (Fig. S3). This result indicated a positive capacity of this region, although weak, to mediate internal initiation of translation.
Figure 5. (A) Diagram of the bicistronic RNA. The first cistron (CAT) reports cap-dependent translation initiation, while the second cistron (luciferase) reports 5′ independent internal initiation of translation. (B) Relative internal initiation of translation of regions I, II, III, or IV in either sense (+) or antisense (-) orientation. Values (mean ± sd) observed in BHK-21 cells transfected with the corresponding bicistronic constructs were made relative to that of region I(+) that showed the highest values in all cases. Experiments were done in triplicate wells in three independent assays. (C). Schematic representation of the primary sequence of Drosophila melanogaster TAF6 protein. Numbers indicate amino acid positions. HFD stands for histone fold domain, TAF6M for middle domain, and TAF6C for the C-terminal domain. The dark gray box depicts the DNA binding region. An arrow depicts the N terminus of the polypeptide initiated at AUG2, while asterisks denote the position of methionine residues corresponding to AUG3 and AUG4.
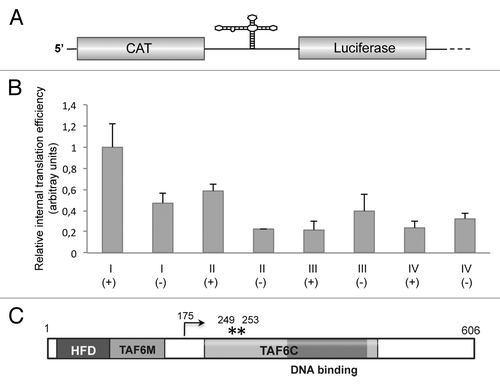
Next, to determine if upstream sequences had any effect on translation activity we generated construct II(+) encompassing nt 278 to 522 (). Transfection of construct II into BHK-21 cells yielded a relative internal initiation efficiency of 58% of construct I(+), suggesting that insertion of 50 nt belonging to the upstream region slightly reduced the capacity of region I to initiate translation. Constructs I(-) and II(-) were inactive (), in agreement with the fact the internal initiation is mediated by cis-acting elements that operate in an orientation-dependent manner.
These results suggest that region I, predicted to adopt an IRES-like motif and encompassing the GNRA, RAAA, and C-rich conserved motifs, can confer internal initiation of translation. In support of this conclusion, construct III(+), which carries the downstream region (nt 526–747) lacking the capacity to adopt the IRES-like fold and devoid of the conserved IRES motifs, was inactive (), yielding values similar to the negative orientation of regions II and III.
We then analyzed whether addition of sequences at the 3′ end of region I (up to nt 747) could modify translation initiation in bicistronic constructs. A shown in , construct IV(+) was inactive, irrespectively of the orientation analyzed. Since this construct harbors an AUG codon (, AUG2) that is out of frame with the second cistron (luciferase), the lack of activity observed in construct IV could be due to initiation at this additional AUG by scanning. In agreement with this observation, constructs V and VI, containing an extended 3′ region up to AUG3 (out of frame with the luciferase initiation codon) reduced luciferase expression (Fig. S4B). All together, we conclude that region I of TAF6 mRNA harboring an IRES-like motif conserved with domain 3 could mediate a weak but positive internal initiation of translation, validating the usefulness of the RNA Inverse Folding search to identify IRES-like structural motifs across genome sequences deposited in databases.
TAF6 is an essential protein, structurally related to histone H4.Citation25 Its primary structure () is conserved from yeast to human. It bears a histone fold domain (HFD) in its N-terminal region responsible for the interaction with TAF9.Citation26 Domain TAF6M consists of 70 residues separated from TAF6C by around 50 residues. Domain TAF6C (220 residues organized in five HEAT repeats and possessing a DNA-binding regionCitation25) modulates TAF6/TAF9 as well as other proteins interactions.Citation27 In the case that the IRES-like motif of TAF6 mRNA was active in the natural RNA context, it could promote internal initiation of translation from AUG2 resulting in a shorter polypeptide (residues 175–606, ), encompassing the TAF6C region and the C-terminal end of the protein but lacking the HFD domain and the TAF6M region. Therefore, the interaction with TBP and the DNA binding region would remain in the short TAF6 polypeptide.
SHAPE structural analysis of the IRES-like subdomain
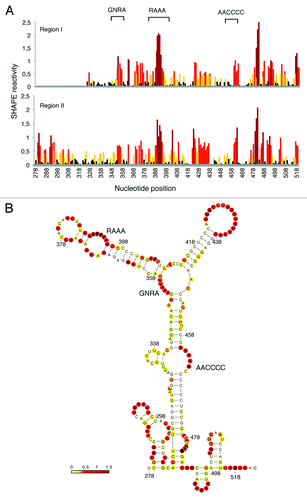
To gain information about the RNA structure of the IRES-like subdomain present in TAF6 mRNA, we performed RNA SHAPE analysisCitation7,Citation10 using transcripts produced in vitro, N-methylisatoic anhydride (NMIA) as the modifying agent and fluorescent-labeled primers. SHAPE reactivity correlates inversely with the probability that a nucleotide is base-paired, providing direct information of the local RNA flexibility in solution. The pattern of SHAPE reactivity obtained for regions I and II is shown in . Interestingly, TAF6 RNA regions I and II display very similar pattern of reactivity, confirming that a 5′end extension of RNA I does not modify its RNA structure. Furthermore, the candidate motifs (RAAA and AACCCCA, which were imposed in the input to RNAiFold) are reactive to NMIA, thus unpaired (). These results further validate the usefulness of RNA inverse folding to search for candidate IRES-like subdomains.
Figure 6. SHAPE reactivity of TAF6 regions I and II. (A) Reactivity of transcripts I and II. Values correspond to the mean SHAPE reactivity of two independent assays normalized by QuSHAPE as described.Citation58 Nucleotide positions are indicated on the x-axis. (B) RNA structure of the IRES-like subdomain. RNA structure was calculated using RNAsc (http://bioinformatics.bc.edu/clotelab/RNAsc/) and visualized with VARNA.Citation60 SHAPE reactivity was represented in a colored scale in which 0 indicates unreactive nucleotides and the average intensity at highly reactive nucleotides is set to 1.0.
Discussion
Translation control mechanisms can contribute to increase the coding capacity of the genome by generating different polypeptides from the same transcriptional unit. Although most cellular mRNAs initiate translation by a cap-dependent mechanism, alternative mechanisms are operative when cap-dependent translation is compromised, including IRES-dependent translation initiation.Citation3,Citation4,Citation28 Most known IRES elements are located in the 5′ UTR of mRNAs upstream of the initiator codon, but some exceptions exist.Citation29-Citation32 Furthermore, although cellular IRES elements described so far lack conserved structural features,Citation3,Citation4 RNA structure plays a fundamental role in viral IRES-dependent translation initiation.Citation2,Citation6,Citation7 Whether some of these structural elements can be found in yet unknown cellular IRES remains to be elucidated.
To date, the accepted strategy to identify IRES elements in mRNAs consists of functional assays testing the cap-independent capacity and the ability to resist cap-inhibitory conditions.Citation33 This is a cumbersome task to identify IRES elements in eukaryotic genomes. To facilitate this endeavor, we have made use of a unique combination of conserved structural motifs present in model viral IRES elementsCitation7,Citation12,Citation14 as a tool to search for regions putatively folding as IRES-like domains in genome sequences. Although the presence of a particular domain individually is not sufficient to define a fully functional IRES element,Citation34 the presence of one (or more) conserved motifs may provide hints to identify potential IRES in mRNAs.
Regarding the difficulties to predict functional IRES elements in genome sequences, it is worth mentioning that the detection and functional annotation of non-conding RNA (ncRNA) genes remains a task of great biological importance, since it is now understood that the human genome is pervasively transcribed, where most transcripts have no known function. Indeed, the ENCODE Consortium study reported that 93% of the human genome may be transcribed in multiple RNAsCitation35 and Clark et al.Citation36 re-affirms an earlier assertion that “given sufficient sequencing depth the whole genome may appear as transcripts.”Citation37
At the present time, there are a number of computational tools to predict the location of ncRNA genes. Specialized ncRNA gene finders exist for (1) precursor microRNA,Citation38,Citation39 (2) bacterial sRNA,Citation40 (3) rRNA genes (5S/5.8S, 16S/18S, and 23S/28S rRNA),Citation41 (4) tRNA,Citation42,Citation43 (5) H/ACA small nucleolar RNAs,Citation44,Citation45 (6) riboswitch aptamers,Citation46,Citation47 and (7) rho-independent transcription terminators.Citation48 General ncRNA gene finders that exploit comparative analysis, include the programs INFERNAL,Citation49 DARN,Citation50 and RNAz.Citation51 The algorithms employed in both specific and general ncRNA gene finders range over a variety of methods, including hidden Markov models (HMMs), generalized HMMs, stochastic context free grammars, energy minimization, support vector machines, co-variation, and so on. It is worth mentioning other recent approaches that rely on computational pipelines.Citation52,Citation53 These approaches, like ours, are not conventional machine learning methods, and thus, there is no training and cross-validation phase. For such pipelines, it makes little sense to determine measures of sensitivity and positive predictive value.
Despite this wealth of computational methods for the prediction of non-coding RNA genes, there appears to be no software for the general prediction of IRES elements, although there are INFERNAL covariance models for 27 distinct IRES families in Rfam 11.0.Citation54 Presumably, this is due to the pairwise dissimilarity of IRES elements between different species, preventing the development of a single covariance model by large groups of IRES elements. We have used cmscan from INFERNAL in order to test our TAF6 candidate against the whole Rfam covariance models and no IRES hit was returned. It follows that the sequence and structural resemblance is too low between our TAF6 candidate and existent INFERNAL covariance models. This situation highlights the usefulness of our pipeline based on RNAiFold in determining novel ncRNAs in cases for which machine learning methods appear to have limited success.
In contrast to the machine learning and dynamic programming methods used in ncRNA gene finders, our pipeline uses complete inverse folding with RNAiFold to determine those RNA sequences that fold into a target secondary structure, deemed to be of functional importance, then BLASTs these sequences and filters the hits retrieved. In cases where structure is conserved, despite no clear sequence homology, our method provides a useful approach.
Our pipeline can produce a huge number of sequences predicted to fold into the structural motif of interest. Here, we have focused on searching RNA regions folding like IRES subdomains. Some of these sequences might fold in vivo into IRES-like motifs, which have never been tried in the evolutionary history of life on earth, while others might be vestiges of previously active IRES-like motifs, and yet others might not actually fold in vivo into IRES-like motifs, due to differences between in vivo folding and in vitro folding, or due to differences between computational predictions and in vitro folding. To apply our pipeline with success, it is critical to use various computational filters and, most importantly, biological insight (IRES-elements are found in mRNAs coding proteins critical to survival, and can be located either in the 5′-UTR or upstream of AUG codons in the coding region) to prioritize the hits returned by RNAiFold.
By using a functional assay we show that the region of TAF6 adopting an IRES-like fold could mediate a weak but positive internal initiation of translation validating the usefulness of the RNA Inverse Folding search to identify IRES-like structural motifs across genome sequences deposited in databases. Our data also show that this mRNA region has a constrained RNA structure, as indicated by the differential codon usage bias relative to the remaining coding sequence, experimentally confirmed by RNA SHAPE analysis. To the best of our knowledge, our approach seems both to be new and orthogonal to existent ncRNA gene finder methods. The data presented in this paper suggests that a pipeline involving RNA Inverse Folding could complement existent methods, especially in the case of difficult ncRNA families/clans that may lack sufficient homology to be detectable using machine learning methods, as is the case for arbitrary IRES elements.
The current paper has focused on the description of a novel pipeline to identify promising candidate IRES-like subdomains, with experimental validation restricted solely to one of the hits returned by RNAiFold, as proof of principle. In future work, we plan to apply our method to identify additional potential IRES-like structural domains. Scrutiny of a number of such experimentally validated sequences could conceivably shed light on those forces in molecular evolution that could give rise to new IRES elements and related regulatory RNA molecules. Comparing unique properties of identified regulatory RNAs with those RNA sequences determined by RNAiFold to fold into the same target secondary structure could help to identify additional aspects that must be taken into account when designing novel functional RNAs in synthetic biology.
Materials and Methods
Computational pipeline
Our approach is based on using RNAiFoldCitation23 in order to calculate all (or a large number of) sequences that fold into a given characteristic secondary structure and try to find any of them in known genomes. In principle, for a given RNA family characterized primarily by structural conservation rather than sequence conservation, we can compute all those sequences that fold into a target structure, representative of the RNA family. Our pipeline is summarized in . RNAiFold optionally allows the user to stipulate certain sequence constraints, such as nucleotide identities, GC-content, etc. that may be shared by all members of an RNA family. Subsequently, we can determine whether any of the returned sequences is similar to any genomic sequences. Sequences returned by RNAiFold are clustered. Clustering was implemented in a greedy fashion by analyzing all sequences in the order in which they were returned. Briefly, the first sequence is considered the current sequence and the following ones are put in the same cluster until a sequence differs by more than 1 nt with respect to the current sequence. At this point, a new cluster with the sequence is created, the sequence becomes the new current sequence, and the approach is repeated until all the sequences have been analyzed. Next, one randomly selected representative of each cluster is BLASTed (BLAST 2.2.4 locally) using BLASTN with word size 11, match/mismatch weights of 1/-1, and gap existence/extension penalties of 1/2. Resulting hits are filtered first by gene function (following the criteria outlined in ) and then by mRNA location, either 5′UTR or coding sequence. These filters are specific to IRES-like motifs. However, general or specific filters to different RNA families can be implemented in the future without compromising the core approach of the pipeline.
The BLAST hits selected for computational validation can be found in different genomic regions within different genes. In the case of IRES elements, which are expected to be found in mRNAs encoding proteins critical for survival, IRES-like motifs might be located either in the 5′-UTR or in the coding sequence (CDS) preceding a downstream AUG codon in frame with the first functional start codon of the CDS. When the hit is found within the CDS region, we can calculate codon usage metrics for various sequence lengths encompassing the hit with a constant downstream AUG at the 3′end (nt 522 or 747) but allowing different 5′ends (nt 25, 85, 145, 205, 265, 325). If there is a functional RNA in the predicted region, which is also part of the coding region, then codon usage bias in that region would be different than in the rest of the CDS since it could have been under selective pressure to code for the active protein, as well as to maintain the functional RNA structure.
Constructs
Plasmid LD24529 (Flybase LD\_pOT2\_cDNA library of Drosophila melanogaster, Drosophila Genomic Resource Center (DGRC), Indiana University) encodes the Drosophila melanogaster TAF6 cDNA cloneCitation55 (). Insertion of the putative IRES-like motif (nt 325–522) into a bicistronic construct pBICCitation56 yielded construct I (). Briefly, the sequence of interest was amplified by PCR using the pair of oligonucleotides TAF6-1s, TAF6-1as () and inserted into the SacI restriction site of pBIC previously linearized with SacI and dephosphorylated. Colonies that carried the insert in both orientations were selected for further studies. A similar procedure was used to generate constructs II, III, IV, V, and VI (; Fig. S4A) using oligonucleotides described in . Prior to expression analysis, the nucleotide sequence of the entire length of each region under study was determined (Macrogen).
Table 2. Oligonucleotides used to generate TAF6 constructs
IRES-dependent translation activity
Bicistronic plasmids carrying the putative IRES-like motif (I, II) between the chloramphenicol acetyl transferase (CAT) and luciferase reporter genes, or the control regions (III, IV) were assayed in BHK-21 cells. Transfection of 90% confluent monolayers was performed using cationic liposomes 1 h after infection with the Vaccinia virus VT7F-3 expressing T7 RNA polymerase, as described.Citation57 This assay excludes the presence of cryptic promoters since the transfected plasmid is transcribed in the cell cytoplasm by the T7 RNA polymerase. Extracts from 105 cells were prepared 20 h after transfection in 100 µl of 50 mM Tris-HCl, pH 7.8, 120 mM NaCl, 0.5% NP40. Luciferase and CAT activities were measured as described.Citation56 Assays were performed in triplicate wells at least three times. Values correspond to the mean (± SD).
SHAPE analysis
Monocistronic constructs expressing RNA regions I and II, generated from the corresponding bicistronic plasmids by PstI digestion, were linearized with SphI prior to synthesize RNA transcripts in vitro as described.Citation10 RNAs (2 pmol) were treated with N-methylisatoic anhydride (NMIA) as described.Citation7 Prior to primer extension, 1 pmol of treated and untreated RNAs were incubated with 2 pmol of the antisense 5′-end fluorescently labeled primer 5′-TAGCCTTATG CAGTTGCTCT CC-3′ at 65 °C for 5 min, 35 °C for 5 min, and then chilled on ice for 2 min. Primer extension reactions were conducted in a final volume of 16 µl containing reverse transcriptase (RT) buffer and 1 μM each dNTP. The mix was heated at 52 °C for 1 min prior to addition of 100 U of Superscript III RT and incubated at 52 °C for 30 min. A sequencing ladder was generated using the corresponding untreated RNA in the presence of 0.1 mM ddC. NED fluorophore was used for both NMIA-treated and untreated samples while VIC fluorophore was used for the sequencing ladder. cDNA products were resolved by capillary electrophoresis. Electropherograms were analyzed using QuSHAPE software.Citation58 Quantitative SHAPE reactivity for individual data sets were normalized to a scale spanning 0 to 2, in which 0 indicates an unreactive nucleotide and the average intensity at highly reactive nucleotides is set to 1.0. Data from two independent assays were used to calculate the mean SHAPE reactivity. Secondary RNA structure was determined using RNAscCitation59 integrating values of SHAPE reactivity and visualized with VARNACitation60
Additional material
Download Zip (1.5 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The Clote Lab is funded by National Science Foundation DMS-1016618 and DBI-1262439. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. The Martinez-Salas Lab is funded by grants CSD2009-00080 and BFU2011-25437 from MINECO. We are grateful to Dr JF de Celis for the kind gift of plasmid LD24529 encoding the full sequence length of TAF6.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/rnabiology/article/26994/
References
- Martínez-Salas E, Pacheco A, Serrano P, Fernandez N. New insights into internal ribosome entry site elements relevant for viral gene expression. J Gen Virol 2008; 89:611 - 26; http://dx.doi.org/10.1099/vir.0.83426-0; PMID: 18272751
- Filbin ME, Kieft JS. Toward a structural understanding of IRES RNA function. Curr Opin Struct Biol 2009; 19:267 - 76; http://dx.doi.org/10.1016/j.sbi.2009.03.005; PMID: 19362464
- Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell 2010; 40:228 - 37; http://dx.doi.org/10.1016/j.molcel.2010.09.028; PMID: 20965418
- Liwak U, Faye MD, Holcik M. Translation control in apoptosis. Exp Oncol 2012; 34:218 - 30; PMID: 23070007
- Komar AA, Hatzoglou M. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle 2011; 10:229 - 40; http://dx.doi.org/10.4161/cc.10.2.14472; PMID: 21220943
- Martínez-Salas E. The impact of RNA structure on picornavirus IRES activity. Trends Microbiol 2008; 16:230 - 7; http://dx.doi.org/10.1016/j.tim.2008.01.013; PMID: 18420413
- Fernández N, Fernandez-Miragall O, Ramajo J, García-Sacristán A, Bellora N, Eyras E, Briones C, Martínez-Salas E. Structural basis for the biological relevance of the invariant apical stem in IRES-mediated translation. Nucleic Acids Res 2011; 39:8572 - 85; http://dx.doi.org/10.1093/nar/gkr560; PMID: 21742761
- Domingo E, Escarmis C, Martinez MA, Martinez-Salas E, Mateu MG. Foot-and-mouth disease virus populations are quasispecies. Curr Top Microbiol Immunol 1992; 176:33 - 47; http://dx.doi.org/10.1007/978-3-642-77011-1_3; PMID: 1318185
- Pacheco A, Martinez-Salas E. Insights into the biology of IRES elements through riboproteomic approaches. J Biomed Biotechnol 2010; 2010:458927; http://dx.doi.org/10.1155/2010/458927; PMID: 20150968
- Fernández N, García-Sacristán A, Ramajo J, Briones C, Martínez-Salas E. Structural analysis provides insights into the modular organization of picornavirus IRES. Virology 2011; 409:251 - 61; http://dx.doi.org/10.1016/j.virol.2010.10.013; PMID: 21056890
- López de Quinto S, Martínez-Salas E. Conserved structural motifs located in distal loops of aphthovirus internal ribosome entry site domain 3 are required for internal initiation of translation. J Virol 1997; 71:4171 - 5; PMID: 9094703
- Fernández-Miragall O, Martínez-Salas E. Structural organization of a viral IRES depends on the integrity of the GNRA motif. RNA 2003; 9:1333 - 44; http://dx.doi.org/10.1261/rna.5950603; PMID: 14561883
- Robertson ME, Seamons RA, Belsham GJ. A selection system for functional internal ribosome entry site (IRES) elements: analysis of the requirement for a conserved GNRA tetraloop in the encephalomyocarditis virus IRES. RNA 1999; 5:1167 - 79; http://dx.doi.org/10.1017/S1355838299990301; PMID: 10496218
- Fernández-Miragall O, Ramos R, Ramajo J, Martínez-Salas E. Evidence of reciprocal tertiary interactions between conserved motifs involved in organizing RNA structure essential for internal initiation of translation. RNA 2006; 12:223 - 34; http://dx.doi.org/10.1261/rna.2153206; PMID: 16373480
- Jung S, Schlick T. Candidate RNA structures for domain 3 of the foot-and-mouth-disease virus internal ribosome entry site. Nucleic Acids Res 2013; 41:1483 - 95; http://dx.doi.org/10.1093/nar/gks1302; PMID: 23275533
- Schnall-Levin M, Chindelevitch L, Berger B. In WW Cohen, A McCallum, SR Roweis (ed.) International Conference on Machine Learning 2008; volume 307. ACM International Conference Proceedings Series.
- Bailor MH, Sun X, Al-Hashimi HM. Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science 2010; 327:202 - 6; http://dx.doi.org/10.1126/science.1181085; PMID: 20056889
- Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res 2008; 36:Web Server issue W70-4; http://dx.doi.org/10.1093/nar/gkn188; PMID: 18424795
- Busch A, Backofen R. INFO-RNA--a fast approach to inverse RNA folding. Bioinformatics 2006; 22:1823 - 31; http://dx.doi.org/10.1093/bioinformatics/btl194; PMID: 16709587
- Andronescu M, Fejes AP, Hutter F, Hoos HH, Condon A. A new algorithm for RNA secondary structure design. J Mol Biol 2004; 336:607 - 24; http://dx.doi.org/10.1016/j.jmb.2003.12.041; PMID: 15095976
- Taneda A. MODENA: a multi-objective RNA inverse folding. Adv Appl Bioinform Chem 2011; 4:1 - 12; PMID: 21918633
- Zadeh JN, Wolfe BR, Pierce NA. Nucleic acid sequence design via efficient ensemble defect optimization. J Comput Chem 2011; 32:439 - 52; http://dx.doi.org/10.1002/jcc.21633; PMID: 20717905
- Garcia-Martin JA, Clote P, Dotu I. RNAiFOLD: a constraint programming algorithm for rna inverse folding and molecular design. J Bioinform Comput Biol 2013; 11:1350001; http://dx.doi.org/10.1142/S0219720013500017; PMID: 23600819
- Hisatake K, Ohta T, Takada R, Guermah M, Horikoshi M, Nakatani Y, Roeder RG. Evolutionary conservation of human TATA-binding-polypeptide-associated factors TAFII31 and TAFII80 and interactions of TAFII80 with other TAFs and with general transcription factors. Proc Natl Acad Sci U S A 1995; 92:8195 - 9; http://dx.doi.org/10.1073/pnas.92.18.8195; PMID: 7667268
- Shao H, Revach M, Moshonov S, Tzuman Y, Gazit K, Albeck S, Unger T, Dikstein R. Core promoter binding by histone-like TAF complexes. Mol Cell Biol 2005; 25:206 - 19; http://dx.doi.org/10.1128/MCB.25.1.206-219.2005; PMID: 15601843
- Scheer E, Delbac F, Tora L, Moras D, Romier C. TFIID TAF6-TAF9 complex formation involves the HEAT repeat-containing C-terminal domain of TAF6 and is modulated by TAF5 protein. J Biol Chem 2012; 287:27580 - 92; http://dx.doi.org/10.1074/jbc.M112.379206; PMID: 22696218
- Wright KJ, Marr MT 2nd, Tjian R. TAF4 nucleates a core subcomplex of TFIID and mediates activated transcription from a TATA-less promoter. Proc Natl Acad Sci U S A 2006; 103:12347 - 52; http://dx.doi.org/10.1073/pnas.0605499103; PMID: 16895980
- Martínez-Salas E, Piñeiro D, Fernández N. Alternative Mechanisms to Initiate Translation in Eukaryotic mRNAs. Comp Funct Genomics 2012; 2012:391546; http://dx.doi.org/10.1155/2012/391546; PMID: 22536116
- Herbreteau CH, Weill L, Décimo D, Prévôt D, Darlix JL, Sargueil B, Ohlmann T. HIV-2 genomic RNA contains a novel type of IRES located downstream of its initiation codon. Nat Struct Mol Biol 2005; 12:1001 - 7; http://dx.doi.org/10.1038/nsmb1011; PMID: 16244661
- Henis-Korenblit S, Shani G, Sines T, Marash L, Shohat G, Kimchi A. The caspase-cleaved DAP5 protein supports internal ribosome entry site-mediated translation of death proteins. Proc Natl Acad Sci U S A 2002; 99:5400 - 5; http://dx.doi.org/10.1073/pnas.082102499; PMID: 11943866
- Du X, Wang J, Zhu H, Rinaldo L, Lamar KM, Palmenberg AC, Hansel C, Gomez CM. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 2013; 154:118 - 33; http://dx.doi.org/10.1016/j.cell.2013.05.059; PMID: 23827678
- Burkart C, Fan JB, Zhang DE. Two independent mechanisms promote expression of an N-terminal truncated USP18 isoform with higher DeISGylation activity in the nucleus. J Biol Chem 2012; 287:4883 - 93; http://dx.doi.org/10.1074/jbc.M111.255570; PMID: 22170061
- Martínez-Salas E. Internal ribosome entry site biology and its use in expression vectors. Curr Opin Biotechnol 1999; 10:458 - 64; http://dx.doi.org/10.1016/S0958-1669(99)00010-5; PMID: 10508627
- Fernández-Miragall O, López de Quinto S, Martínez-Salas E. Relevance of RNA structure for the activity of picornavirus IRES elements. Virus Res 2009; 139:172 - 82; http://dx.doi.org/10.1016/j.virusres.2008.07.009; PMID: 18692097
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al, ENCODE Project Consortium, NISC Comparative Sequencing Program, Baylor College of Medicine Human Genome Sequencing Center, Washington University Genome Sequencing Center, Broad Institute, Children’s Hospital Oakland Research Institute. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007; 447:799 - 816; http://dx.doi.org/10.1038/nature05874; PMID: 17571346
- Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KV, Morillon A, et al. The reality of pervasive transcription. PLoS Biol 2011; 9:e1000625 - , discussion e1001102; http://dx.doi.org/10.1371/journal.pbio.1000625; PMID: 21765801
- van Bakel H, Nislow C, Blencowe BJ, Hughes TR. Most “dark matter” transcripts are associated with known genes. PLoS Biol 2010; 8:e1000371; http://dx.doi.org/10.1371/journal.pbio.1000371; PMID: 20502517
- Xue C, Li F, He T, Liu GP, Li Y, Zhang X. Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinformatics 2005; 6:310; http://dx.doi.org/10.1186/1471-2105-6-310; PMID: 16381612
- Ng KL, Mishra SK. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics 2007; 23:1321 - 30; http://dx.doi.org/10.1093/bioinformatics/btm026; PMID: 17267435
- Tjaden B. Prediction of small, noncoding RNAs in bacteria using heterogeneous data. J Math Biol 2008; 56:183 - 200; http://dx.doi.org/10.1007/s00285-007-0079-5; PMID: 17354017
- Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 2007; 35:3100 - 8; http://dx.doi.org/10.1093/nar/gkm160; PMID: 17452365
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997; 25:955 - 64; PMID: 9023104
- Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 2005; 33:Web Server issue W686-9; http://dx.doi.org/10.1093/nar/gki366; PMID: 15980563
- Freyhult E, Edvardsson S, Tamas I, Moulton V, Poole AM. Fisher: a program for the detection of H/ACA snoRNAs using MFE secondary structure prediction and comparative genomics - assessment and update. BMC Res Notes 2008; 1:49; http://dx.doi.org/10.1186/1756-0500-1-49; PMID: 18710502
- Tafer H, Kehr S, Hertel J, Hofacker IL, Stadler PF. RNAsnoop: efficient target prediction for H/ACA snoRNAs. Bioinformatics 2010; 26:610 - 6; http://dx.doi.org/10.1093/bioinformatics/btp680; PMID: 20015949
- Chang TH, Huang HD, Wu LC, Yeh CT, Liu BJ, Horng JT. Computational identification of riboswitches based on RNA conserved functional sequences and conformations. RNA 2009; 15:1426 - 30; http://dx.doi.org/10.1261/rna.1623809; PMID: 19460868
- Singh P, Bandyopadhyay P, Bhattacharya S, Krishnamachari A, Sengupta S. Riboswitch detection using profile hidden Markov models. BMC Bioinformatics 2009; 10:325; http://dx.doi.org/10.1186/1471-2105-10-325; PMID: 19814811
- Naville M, Ghuillot-Gaudeffroy A, Marchais A, Gautheret D. ARNold: a web tool for the prediction of Rho-independent transcription terminators. RNA Biol 2011; 8:11 - 3; http://dx.doi.org/10.4161/rna.8.1.13346; PMID: 21282983
- Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics 2009; 25:1335 - 7; http://dx.doi.org/10.1093/bioinformatics/btp157; PMID: 19307242
- Zytnicki M, Gaspin C, Schiex T. Darn: a weighted constraint solver for RNA motif localization. Constraints 2008; 13:91 - 109; http://dx.doi.org/10.1007/s10601-007-9033-9
- Gruber AR, Neuböck R, Hofacker IL, Washietl S. The RNAz web server: prediction of thermodynamically stable and evolutionarily conserved RNA structures. Nucleic Acids Res 2007; 35:Web Server issue W335-8; http://dx.doi.org/10.1093/nar/gkm222; PMID: 17452347
- Parker BJ, Moltke I, Roth A, Washietl S, Wen J, Kellis M, Breaker R, Pedersen JS. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res 2011; 21:1929 - 43; http://dx.doi.org/10.1101/gr.112516.110; PMID: 21994249
- Hoeppner MP, Gardner PP, Poole AM. Comparative analysis of RNA families reveals distinct repertoires for each domain of life. PLoS Comput Biol 2012; 8:e1002752; http://dx.doi.org/10.1371/journal.pcbi.1002752; PMID: 23133357
- Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 2013; 41:Database issue D226 - 32; http://dx.doi.org/10.1093/nar/gks1005; PMID: 23125362
- Rubin GM, Hong L, Brokstein P, Evans-Holm M, Frise E, Stapleton M, Harvey DA. A Drosophila complementary DNA resource. Science 2000; 287:2222 - 4; http://dx.doi.org/10.1126/science.287.5461.2222; PMID: 10731138
- Martínez-Salas E, Sáiz JC, Dávila M, Belsham GJ, Domingo E. A single nucleotide substitution in the internal ribosome entry site of foot-and-mouth disease virus leads to enhanced cap-independent translation in vivo. J Virol 1993; 67:3748 - 55; PMID: 8389904
- López de Quinto S, Sáiz M, de la Morena D, Sobrino F, Martínez-Salas E. IRES-driven translation is stimulated separately by the FMDV 3′-NCR and poly(A) sequences. Nucleic Acids Res 2002; 30:4398 - 405; http://dx.doi.org/10.1093/nar/gkf569; PMID: 12384586
- Karabiber F, McGinnis JL, Favorov OV, Weeks KM. QuShape: rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA 2013; 19:63 - 73; http://dx.doi.org/10.1261/rna.036327.112; PMID: 23188808
- Zarringhalam K, Meyer MM, Dotu I, Chuang JH, Clote P. Integrating chemical footprinting data into RNA secondary structure prediction. PLoS One 2012; 7:e45160; http://dx.doi.org/10.1371/journal.pone.0045160; PMID: 23091593
- Darty K, Denise A, Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009; 25:1974 - 5; http://dx.doi.org/10.1093/bioinformatics/btp250; PMID: 19398448
- McGinnis JL, Duncan CD, Weeks KM. High-throughput SHAPE and hydroxyl radical analysis of RNA structure and ribonucleoprotein assembly. Methods Enzymol 2009; 468:67 - 89; http://dx.doi.org/10.1016/S0076-6879(09)68004-6; PMID: 20946765