
Abstract
Massive parallel sequencing (MPS) technologies have paved the way into new areas of research including individualized medicine. However, sequencing of trace amounts of DNA or RNA still remains a major challenge, especially for degraded nucleic acids like circulating DNA. This together with high cost and time requirements impedes many important applications of MPS in medicine and fundamental science. We have established a fast, cheap and highly efficient protocol called ‘Capture and Amplification by Tailing and Switching’ (CATS) to directly generate ready-to-sequence libraries for MPS from nanogram and picogram quantities of both DNA and RNA. Furthermore, those DNA libraries are strand-specific, can be prepared within 2–3 h and do not require preliminary sample amplification steps. To exemplify the capacity of the technique, we have generated and sequenced DNA libraries from hundred-picogram amounts of circulating nucleic acids isolated from human blood plasma, one nanogram of mRNA-enriched total RNA from cultured cells and few nanograms of bisulfite-converted DNA. The approach for DNA library preparation from minimal and fragmented input described here will find broad application in diverse research areas such as translational medicine including therapy monitoring, prediction, prognosis and early detection of various human disorders and will permit high-throughput DNA sequencing from previously inaccessible material such as minute forensic and archeological samples.
Introduction
Massive parallel sequencing (MPS) of nucleic acids requires the preparation of amplified libraries where the DNA region of interest is located between known 5′- and 3′- terminal sequences. Current methods for MPS libraries construction utilize either RNA or DNA adaptors ligation to the 5′- and 3′- ends of the target RNA or DNA molecules.Citation1,Citation2 Ligation of adaptors is not only time consuming but also a low efficiency process that requires micrograms of nucleic acid inputs. In addition, the resulting cDNA libraries are contaminated with cross- and self-ligation adaptor by-products and require additional purification steps both before and after pre-amplification.Citation3
More than a decade ago, a method was described that harnesses the template switching activity of the Moloney murine leukemia virus reverse transcriptase (MMLV-RT) to attach adaptors of choice to the 5′-end of cDNA generated from poly(A) tailed mRNA molecules.Citation4,Citation5 At the same time, a 3′-adaptor sequence was incorporated into a poly(dT) reverse transcription primer. This principle, named SMART (switching mechanism at the 5′ end of the RNA transcript), is currently used in an Illumina Ultra Low RNA sequencing kit (Clontech) to generate full-length cDNA copies of mRNA molecules from a single cell. However, the method subsequently still requires (1) fragmentation of the amplified cDNA, (2) ligation of platform-specific 5′/3′-end adaptors and (3) pre-amplification of the adaptors-ligated DNA fragments.Citation6,Citation7 Although the SMART method is capable of preparing cDNA for sequencing from single-cell amounts of RNA, it is time consuming, expensive and restricted to mRNA sequencing. To our knowledge, the approach of using template switching activity of MMLV-RT has not been yet applied to sequence (1) any DNA molecules and (2) RNA molecules other than long RNAs.
In this article we describe the “Capture and Amplification by Tailing and Switching” (CATS) method to generate ready-to-sequence DNA libraries from picogram amounts of either DNA or RNA molecules in a time frame of few hours. Small (< 150 bp) DNAs or RNAs (e.g., miRNAs, piRNAs, degraded or bisulfite-converted DNA for methylation analysis of trace amounts of DNA) can be used as an input directly, while long RNA or DNA molecules have to be at first fragmented by a corresponding approach (e.g., sonication for DNA or Mg2+ incubation for RNA). The procedure we describe is drastically cheaper when compared with any commercial kit for cDNA generation for deep sequencing available on the market to date. We believe that our protocol will become “a method of choice” for DNA and RNA next-generation sequencing experiments. Most importantly, this approach will permit sequencing of nucleic acids from sources from which sequencing was hitherto impossible due to the minimal requirements of the input. Examples of those may include: DNA and RNA from small (diagnostic) amounts of liquid biopsies, or microsomes, targeted compartments of the cells (e.g., micronuclei, endoplasmic reticulum), fossils, remnants of extinct organisms, and forensics samples containing minute and highly fragmented DNA molecules.
Results
Principle of DNA Library Construction
The strategy used in this study for cDNA library construction is illustrated in . Briefly, short single stranded DNA or RNA fragments are polyadenylated or polydeoxyadenylated with either poly(A) polymerase or terminal deoxytransferase. Subsequently, a cDNA strand synthesis is performed in the presence of the anchored poly(dT) oligonucleotide containing a custom 3′-adaptor sequence. When the reverse transcriptase reaches the 5′ end of the DNA (or RNA) template, the enzyme’s terminal transferase activity adds additional nucleotides (predominantly dC) that are not encoded by the template. On the next step, the template switching oligonucleotide (TSO) containing three 3′-terminal rG nucleotides and a custom 5′-adaptor sequence is added to the RT reaction product, which serves as a second template for the reverse transcriptase. The complementary interaction of the three consecutive rG nucleotides at the 3′-end of the TSO and the dC-rich extended sequence of the cDNA are thought to promote template switching. The second cDNA strand is generated during the first cycle of the standard PCR reaction from a forward primer which is either fully or partially complementary to the 3′-terminus of the first cDNA strand. Furthermore, the reverse primer used for the PCR amplification of the cDNA (together with forward primer) is either fully or partially complementary to the 3′-terminus of the second cDNA strand. Since the PCR forward primer does not share complementarity with the TSO and the PCR reverse primer is not complementary to the poly(dT) primer, the excess of both TSO and poly(dT) primers does not interfere with the PCR amplification that follows cDNA synthesis. It has to be mentioned that during adaptor ligation-based cDNA synthesis, one of the ligated adaptors is always complementary to (1) RT primer used for first strand cDNA synthesis and (2) one of the PCR amplification primers. As a result, besides their time and labor intensiveness, adaptor-ligation methods demand additional purification steps before pre-amplification of the cDNA libraries and have a limited number of possible pre-amplification cycles.
Figure 1. Schematic representation of cDNA preparation methods using a combination of poly(A) or poly(dA) tailing and template switching capacity of MMLV-RT.
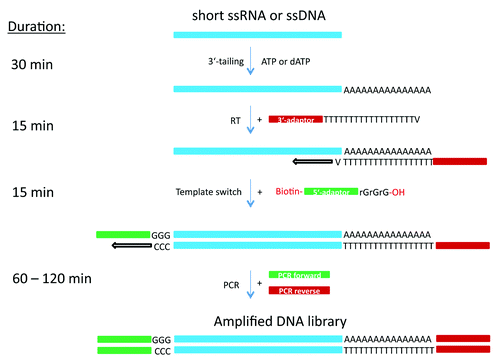
Several well-known by-products may occur during preparation of DNA libraries using the template switching approach ().Citation8-Citation10 Primarily, poly(dT) reverse transcription primer together with TSO theoretically can yield a certain amount of “empty” cDNA libraries. However, by using a similar molar ratio of poly(dT) primer and poly(A) tail the incidence of “empty” cDNA molecules is decreased to undetectable levels (). Although the average length of the poly(A) tail is hard to control, and thus, to calculate the exact poly(dT):primer/poly(A) tail ratio, no detectable “empty” cDNA molecules appeared after 17 PCR cycles when using 1 µM TSO together with 100 nM poly(dT) primer and after 26 PCR cycles when using 1 µM TSO together with 1 nM poly(dT) primer (). Another possible by-product of the RT and template switching reactions are 5′-terminal sequence concatemers resulting from secondary template switching events occurring when the reverse transcriptase reaches the end of the TSO. However, under our experimental conditions (1 μM of TSO; 100 units SmartScribe RT polymerase per reaction) the occurrence of the secondary template switching was hardly detectable (Supplementary material ). In addition, blocking the 5′-OH group of TSO either with three consecutive abasic sites or with biotin reduced the incidence of secondary template switches at the end of TSO to undetectable levels (Supplementary material ). Consequently, blocking the sample DNA with 5′-biotin dramatically prevented the template switching reaction (Supplementary material ). Finally, preliminary template switching events were described to occur by some researchers, especially when the TSO is added before the reverse transcriptase reaches the 5′-end of the RNA.Citation10 However, we did not observe a significant percentage of any of the three by-products when analyzing synthetic small RNA and DNA (cel-miR-39) libraries either using Sanger sequencing or next generation sequencing.
Table 1. Known by-products of cDNA preparation using template switching activity of MMLV-RT.
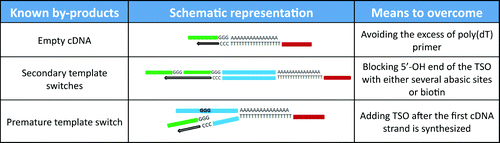
Figure 2. (A) The structure of cDNA prepared using adaptor sequences for the Illumina sequencing platform. Note, the absence of sequence complementarity between PCR primers and terminal adaptors allowing pre-amplification of the cDNA library without prior purification of the first cDNA strand. (B) Electropherogram obtained after 3% agarose gel electrophoresis of amplified DNA libraries obtained from 1 ng (or 5 pg) of either synthetic cel-miR-39 RNA or DNA molecules. The number of PCR amplification cycles and the concentration of reverse poly(dT) primer are indicated below each electropherogram. In addition, Agilent Bioanalyser (High Sensitivity DNA chips) and automated Sanger sequencing were used to estimate the purity of DNA libraries after the column purification step (upper chromatogram) and additional gel extraction step (lower chromatogram). Agilent Bioanalyser data and Sanger chromatograms shown only for 5 pg RNA and DNA inputs.
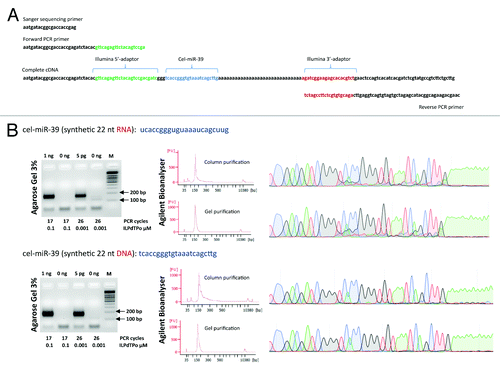
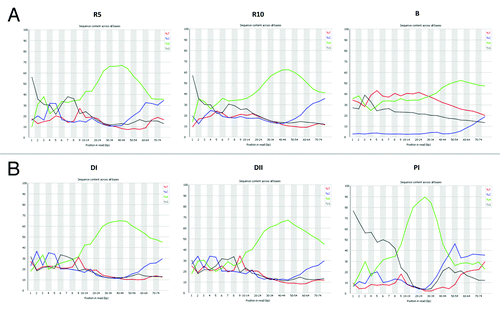
Figure 3. Nucleotides distribution of reads obtained after MiSeq Illumina sequencing of the libraries generated from: (A) Poly(A) enriched RNA (R10 and R5) and bisulfite converted DNA (B) from U2OS cells; (B) Human blood plasma circulating DNA (DI and DII) and circulating RNA (RI). Note, the significant bias toward reads containing 5′-terminal G nucleotides in the RNA samples. In addition, RNA molecules starting with A nucleotide are incorporated into the DNA library with lower efficiency. Note, no bias toward a 5′-terminal nucleotide was observed when DNA was taken as input. Note, the significantly lower content of dC nucleotide in the libraries generated from bisulfite-converted DNA.
DNA library construction for Illumina MiSeq (HiSeq) platform
To construct DNA libraries suitable for Illumina MiSeq or HiSeq platforms we have used adaptor sequences from the NEBnext Small RNA Sequencing Kit (New England Biolabs). The sequence corresponding to the 5′-adaptor was incorporated into the TSO and the 3′-adaptor sequences was used to design a terminal tag of the poly(dT) primer (). We have used either 1 ng or 5 pg of 22 nt RNA and DNA as inputs for the DNA library preparation (). The efficience of cDNA synthesis was equal for DNA and RNA. When using 1 ng of nucleic acids, a single PCR product was strongly visible after 17 PCR pre-amplification cycles (1/100 cDNA to PCR dilution). When 5 pg of nucleic acids were used as input, the amount of PCR cycles required to pre-amplify cDNA to similar levels increased to 26. When a 10/100 cDNA to PCR dilution was used, the amount of cycles necessary to generate DNA libraries decreased proportionally (Figure S3C). The amplified cDNAs were subsequently purified either by simple column purification or by extraction from agarose gel (). The only contaminating by-product in the reaction was an excess of PCR primers, most of which can be removed by column purification. Sanger sequencing further confirmed that cDNA prepared from synthetic short DNA was pure (). However, when RNA was used as a template, a certain percentage of truncated fragments was indeed observed. The occurrence of shorter fragments in the libraries cannot be explained by premature template switching, but rather by the presence of synthetic cel-miR-39 RNA templates that were truncated by 5 bases (5′-GGGTGTAAATCAGCTTG-3′), probably as by-products of synthesis or due to degradation in storage, since the synthetic cel-miR-39 RNA oligonucleotide had been stored for several years. Indeed, those shorter by-products were no longer visible on Sanger electropherogram when a newly synthesized HPLC purified cel-miR-39 RNA oligonucleotide was taken as template (Supplementary materials ). Furthermore, DNA libraries generated from cel-miR-39 RNA molecules, but not from cel-miR-39 DNA, contained frequent truncation of the last nucleotide, what could be explained by a trace 3′-exoribonuclease activity in the solution. Interestingly, in RNA derived libraries the second T nucleotide from 3′-end was frequently substituted by C – a phenomenon that has yet to be explained.
Despite the principal simplicity of the protocol, several critical points have to be taken into account when preparing cDNA library using this method. Primarily, the poly(A) tailing reaction is critical for the optimal yield of cDNA. Thus, too long poly(A) tails would eventually decrease the effective concentration of poly(dT) primer, which would not only decrease the amount of cDNA but also produce a smear of larger by-products on the gel (Supplementary material , ). In our hands, 10 min poly(A)-tailing time and the 0.1 mM of final ATP gave decent results for the 22 nt RNA input. Second, the supplier and the brand of MMLV-RT appeared to be critical for the sensitivity of the approach. Thus, out of 6 tested commercial MMLV-RTs only SuperScribe II (Invitrogen), SMARTScribe RT (Clontech) and SMART RT (Clontech) yielded detectable amounts of cDNA after pre-amplification with current protocol, while SuperScribe III (Invitrogen), Multiscribe RT (Applied Biosystems) and M-MLV from NEB required 4 more cycles of pre-amplification for a DNA library to be visible on agarose gel (Supplementary material ). This phenomenon could be explained by the fact that different MMLV-RT variants might possess different RNase H and terminal transferase activities (the latter is thought to facilitate the template switching reaction). Finally, the structure of TSO appears to be critical for the sensitivity and the performance of the method. Both pure DNA and pure RNA TSO failed to yield any adequate amount of the targeted cDNA after 17 cycles of pre-amplification PCR (Supplementary material ). This could be explained by the fact that a sequence of three riboG has much stronger affinity for the template switching than three deoxyriboG, while the pure RNA oligonucleotide is prone to forming significant secondary structures that decrease the availability of the 3′-terminus. Furthermore, when a TSO with four instead of three terminal riboG nucleotides was used, the yield of the cDNA was dramatically reduced (Supplementary material ), presumably due to the ability of four consecutive G to form quadruplex structures.Citation11
We also tested an option of blocking the terminal 3′-OH group of the TSO to prevent its poly(A) tailing which might occur when poly(A) polymerase is not completely deactivated. Although, in our hands, thermal deactivation of E.coli poly(A) polymerase for 20 min at 65 °C before the RT reaction was complete, the usage of 3′-OH blocked TSO would be mandatory in case of (1) poly(A) tailing and the RT are performed simultaneously or (2) no heat inactivation is possible after poly(A) tailing of RNA. Surprisingly, blocking the 3′-OH terminal of TSO with either monophosphate or biotin abrogated the efficacy of cDNA synthesis under the conditions used (Supplementary material ). Nevertheless, when the 3′-OH group of TSO was blocked with phosphate or dideoxycytidine (ddC), similar amounts of cDNA product appeared four PCR cycles later.
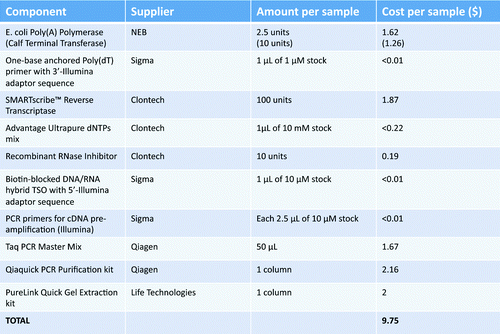
Current commercial kits for cDNA library preparation for next generation sequencing of RNA and DNA are priced between $200 and $500 per sample depending on the application, type of the kit and manufacturer. The rough estimates of the costs required for a single DNA library preparation using our method is listed in . According to our estimation, a researcher or clinician would need to spend less than $10 per sample to obtain ready-to-be-sequenced DNA library.
Table 2. Cost estimation of the consumables per sample of cDNA preparation according to the here presented protocol. Calculations include prices in US dollars without special discounts (as by end of 2013). Common consumables (e.g., laboratory plastic, gel electrophoresis reagents etc.) have not been included in the calculations.
DNA libraries prepared from poly(A)-enriched RNA and bisulfite-converted DNA from U2OS cell line
Having optimised the technique of cDNA preparation using short single stranded synthetic DNA and RNA of a given control sequence (cel-miR-39), we have applied this method to biological samples. At first, we have tested the feasibility of our protocol to obtain DNA libraries from one nanogram of poly(A)-enriched cellular transcriptome and few nanograms of bisulfite-converted DNA. In our hands, one well of a 24-well plate containing fully confluent U2OS cells yielded approximately 100 ng/µL of total RNA in a 50 µL eluate. After enrichment for poly(A)-tailed mRNAs, fragmentation with Mg2+ and purification (see Material and Methods), the final yield constituted a few nanograms per microliter of RNA in a volume of 50 µL. One microliter of poly(A) tailed RNA (1 ng/µL) was used for single DNA library preparation according to the protocol described for the synthetic cel-miR-39 RNA (Figure S5A). Pre-treatment of RNA with T4 PNK before poly(A) tailing was mandatory to achieve adequate yields of DNA library after 19 amplification cycles (Figure S5A). The DNA libraries obtained from either 10 min or 5 min Mg2+ fragmented RNA (samples R10 and R5) consisted of single peaks of the mean size 171 bp and 179 bp, respectively, according to the Agilent Bioanalyser. One microliter of poly(dA)-tailed bisulfite-converted DNA from U2OS cells (approx. Three ng/µL) was used for a single DNA library preparation reaction according to the protocol described for synthetic cel-miR-39 DNA (Figure S5B). To obtain a ready-to-sequence sample, bisulfite-converted DNA library was cut from the agarose gel roughly between 200 and 400 bp.
DNA libraries prepared from human circulating RNA and DNA
Additionally, we have tested the feasibility of DNA library preparation from circulating nucleic acids. Previously, we have optimised the protocol for RNA isolation from biological fluids, including protein-rich blood plasma. Our extraction procedure allows almost complete recovery of small RNAs as judged by the recovery of pre-mixed synthetic cel-miR-39 from C.elegans.Citation12 Concentration of both RNA isolated from 400 µL and DNA isolated from 800 µL of human blood plasma constituted approximately 150–200 pg/µL in the final eluates of 50 µL (as analyzed by Agilent Bioanalyser). One microliter of the circulating RNA poly(A)-tailing reaction (containing approx. 150–200 pg/µL of RNA input) was used for library preparation reaction according to the protocol optimised for the synthetic cel-miR-39 RNA (Figure S5D). Importantly, pre-treatment with T4 polynucleotide kinase (T4 PNK) was mandatory to obtain the cDNA library from circulating plasma RNA, indicating that most of the RNA in blood plasma carries either phosphate or cyclo-phosphate at 3′-end (common by-products of RNase cleavage). One microliter of poly(dA)-tailed circulating DNA (approx. 150 pg) was used for a single library preparation reaction according to the protocol described for synthetic cel-miR-39 DNA (Figure S5C). Amplified libraries obtained from plasma DNA differed significantly from the RNA-derived libraries in terms of the fragments lengths distribution (Figure S5C, S5D). To obtain ready-to-sequence samples, circulating DNA libraries were cut from the agarose gel roughly between 200 and 500 bp. According to the Bioanalyser data, libraries prepared from circulating RNA consisted of single peak with a mean length 7–8 nt shorter than the library generated from control 22 nt cel-miR-39 RNA. This indicates that most of the circulating RNAs in human blood plasma consist of RNA fragments that are considerably shorter than microRNAs (Bioanalyser data; Supplementary material 5D). In contrast, DNA libraries prepared from plasma DNA consisted of two broad peaks of distinct sizes ranging from 150 to 400 bp, the larger peak presumably representing the nucleosome-protected DNA that is approximately 140 bp long.
Initial deep sequencing data preparation
The cDNA library prepared from cel-miR-39 DNA was sequenced together with the circulating DNA libraries (mixed 10%:90%). The cDNA library prepared from cel-miR-39 RNA was sequenced together with one of the poly(A)-enriched RNA libraries (mixed 10%:90%) and the circulating RNA library (mixed 5%:95%). The MiSeq data were exported as FASTQ file and the reads were subjected to adaptor and poly(A) tail removal as well as quality trimming. The presence of the 5′ sequencing adaptor due to secondary template switches was found in a negligible fraction of the reads, from which it was removed (Table S1). Reads retaining a minimum length of 20 nt were used for alignment.
Next generation sequencing of poly(A)-enriched RNA and bisulfite-converted DNA from human cancer cell line U2OS
The usable fraction of reads obtained from Mg2+ fragmented poly(A)-enriched RNA libraries was 69% for R10 (10 min of fragmentation) and 77% for R5 (5 min of fragmentation) (Table S1). Library R10 was mixed with 10% cel-miR-39 control library, which was well represented among the usable reads. Of the usable reads not corresponding to the cel-mir-39 control, > 90% could be mapped to the human genome and transcriptome. In each case, fragments from approximately 30.000 known gene loci could be detected (FPKM > 1, Table S1), as well as from 683 (R10) and 1087 (R5) potential unannotated loci. We have also observed that in both the R10 and R5 library the sequencing reads contained a significant bias toward reads starting with G nucleotides (). The bias to the first G nucleotides was also profound in the circulating RNA sample, but was absent in circulating DNA and bisulfite-treated DNA samples ().
As anticipated, in the bisulfite-converted DNA library the frequency of C nucleotides was drastically lower as compared with the incidence of A, T and G nucleotides (). The usable fraction of reads was 87.16%. Out of these, 55.49% were uniquely mapped to the human genome prepared for bisulfite-converted read mapping, while further 28.70% of reads mapped more than once. The amount of reads which could not be mapped was only 15.82%. The single NGS experiment with bisulfite-treated DNA was aimed merely to demonstrate the proof-of-principle that such DNA can be sequenced directly without additional pre-amplification steps. However, the amount of reads generated with the MiSeq (15 – 20 millions) platform is not sufficient to generate an informative map of CpG methylated regions throughout the genome. Higher output platforms (such as HiSeq) have to be used to obtain adequate coverage.
Next generation sequencing of circulating DNA and RNA
Approximately 70% of the reads obtained from human plasma circulating DNA samples were usable for mapping to the cel-miR-39 control DNA sequence or the human genome (Table S1). The percentage of control cel-miR-39 library input (10%) was reflected in the raw read output. The relative representation of cel-miR-39 was increased in the fraction of usable reads, pointing to the fact that the fraction of reads that are shorter than 20 nt after adaptor removal corresponds to short DNA fragments in the source material. The percentage of usable reads from circulating DNA that did not correspond to the cel-miR-39 library and that could be mapped to the human genome was 35% for DI and 54% for DII. A very small fraction of the unaligned reads is constituted by fragments of cel-miR-39 that were missed during sequence alignment due to multiple sequence errors or severe truncation. Additional mapping of the unaligned reads to human repetitive DNA elements from RepBaseCitation13 yielded negligible fractions (83 reads in DI and 32 reads in DII).
The number of circulating RNA reads usable for mapping constituted only 21.86% due to the large fraction of the reads which were shorter than 20 nt (76.39%) and empty reads (1.75%) (Table S1). The percentage of control cel-miR-39 library input (5%) was recovered in the raw read output (4.24%), but strongly enriched in the fraction of usable reads with at least 20 nt (19.4%). Allowing no mismatches and gaps during mapping to the human genome and transcriptome, the fraction of usable non-control reads which were mapped only once in the human genome was 3.38%, while non-uniquely mapped reads constituted further 19.97%. Nevertheless, 231 known gene loci and 41 potential unannotated loci could be detected (FPKM > 1), among them several micro-RNAs and other small RNA species. The proportion of unmapped reads was > 75%. We did not sequence a second library generated from circulating small RNA, since it has become evident that additional means are required to purify “informative” RNAs suitable for short read alignment, such as circulating miRNAs and other nuclease resistant RNAs > 20 nt from short (< 19 nt) RNA fragments which are mainly the final by-products of RNase degradation, and possibly parts of tRNA.
Of the large fraction of circulating RNA reads that did not align to the human genome or transcriptome, one third (35%) mapped with 0–2 mismatches to microbial reference sequences from the Human Microbiome Project database, indicating a considerable presence of exogenous RNA sequences in the human plasma (Table S1). In contrast, the amount of unaligned reads from circulating DNA mapping to microbial sequences was only around 5% for both DI and DII. The origin of the further unaligned reads remains yet to be elucidated.
C. elegans control library sequence data
Among the libraries generated from either DNA or RNA cel-miR-39 and which were premixed into certain samples (Table S1), minor fractions of by-products were observed. Cel-miR-39 RNA libraries contained 2.1% (library sequenced together with F10) and 4.1% (library sequenced together with RI) of 5 nt truncated fragments, while both libraries generated from cel-miR-39 DNA showed only 0.05% of those. Since the cel-miR-39 sequence contains a GGG triplet, the reads with preliminary template switching events would have a sequence of TGTAAATCAGCTTG instead of TCACCGGGTGTAAATCAGCTTG. These fragments composed 0.95% of the Cel-miR39 RNA reads from the library sequenced together with RI. However, virtually no premature template switch events were observed in both cel-miR-39 DNA libraries (0.02% of Cel-miR-39 reads in DI and 0.03% in DII) or the second RNA (0.03% in F10) library. In addition, the percentage of secondary template switch events was negligible (between 0.029–0.034% for DNA libraries and between 0.0002–0.0008% for libraries generated from RNA). Furthermore, we estimated the error rate per nucleotide among cel-miR-39 molecules that were mapped in full-length. The error rate estimated to be generally low at 0.0–0.5% for DNA and 0.0–0.7% for RNA. An exception is the thymine base at position nine, directly after the GGG triplet, which showed an error rate of 2.96% in DI and 4.63% in DII, 2.54% in RI and 3.41% in F10, of which the majority (82 – 91%) were substations to adenine (A). RNA cel-miR-39 reads also showed slightly increased error rates at positions 11 (thymine, 1.22–1.44%) and 18 (guanine, 0.89–0.94%), again mostly substituted to adenine (82–91%). However, this data cannot further elucidate whether the observed errors arose during library generation and the sequencing process or they represent errors in the actual cel-miR-39 DNA or RNA oligonucleotides that were created during oligonucleotide synthesis or storage.
Analysis of Exon 2 in the highly covered GAPDH in the data from poly(A)-enriched RNA libraries from the U2OS cell lines reflected the error rates observed in the control library from synthetic cel-mir-39 (Figures S6). In both experiments (R10 and R5), the error rates were < 0.7% for most bases. An exception is a thymine base (chr12:6644005), which shows an error rate of approximately 4% in both R10 and R5, with a majority of substitutions (55 and 58%) to adenine. The respective sequence contexts of the bases with the highest error rates are identical for the cel-mir-39 control libraries and GAPDH Exon2 (5′-GG|T|G-3′ in top strand) from the poly(A) enriched RNA libraries. It is unlikely that these errors were introduced during the reverse transcription step, since the error rates of MMLV RT enzymes are usually much lower. For instance, the error rate of SuperScript™ II RT (Life Technologies) is 0,006% (1 to 15000 bases) according to the manufacturer’s manual, what is much lower than the error rate of the Illumina MiSeq Sequencing platform (observed to be 0.8% on averageCitation14). However, the error rate introduced by the sequencer may not be uniform over the different base contexts. Therefore, control libraries from a wide range of oligonucleotides that specifically contain many different possible base contexts would be necessary to investigate the error rates observed not only in our protocol, but also in other well-established RNA or DNA library generation methods.
Discussion
Current technologies for next generation sequencing utilize a limited length of the fragments to be sequenced in one cluster. Thus, Illumina MiSeq allows 50 nt, 150 nt, 300 nt, 500 nt or 600 nt rounds of sequencing-by-synthesis by standard settings. Ion torrent PGM platform is able to sequence either approximately 200 nt or 400 nt fragments downstream the 3′-end of the sequencing primer. As a result, a ready-to-be-sequenced DNA library must at first be prepared from crude nucleic acids material and contain the sample DNA between adaptors of a given sequence. Importantly, current commercially available kits for cDNA generation rely solely on the consequent ligation of 5′- and 3′-adaptors to the fragmented RNA and DNA molecules of interest. We have developed an approach which does not require adaptor ligation and instead utilizes a combination of polyadenylation reaction to capture 3′-end and template switching activity of MMLV-RT to capture the 5′-end of nucleic acids during the first strand cDNA synthesis reaction. We have originally used synthetic 22 nt single stranded DNA and RNA molecules to develop and attune this method of cDNA library genaration; however, larger fragments can be efficiently incorporated into the ready-to-be-sequenced cDNA library as was demonstrated by preparing cDNA libraries from circulating DNA, bisulfite-converted DNA and cellular transcriptomes.
The first step in the analysis of cellular transcriptomes is the conversion of RNA species into cDNA. Many current methods for transcriptome analysis involve (1) primary synthesis of long cDNAs by either random or mRNA-specific priming, (2) pre-amplification of cDNA, (3) subsequent fragmentation of pre-amplified cDNA, (4) adaptors ligation and (5) a final pre-amplification step.Citation1,Citation2 However, a cDNA pre-amplification step may not be required with those commercial kits which use micrograms of input material.
Obviously, fractionation of total RNA, poly(A)-enriched RNA, or DNA and its direct use as input material for DNA library generation is a less time consuming procedure. Furthermore, small RNA and DNA molecules cannot be efficiently pre-amplified using random priming and their capture relies solely on adaptor ligations to their 5′ and 3′ ends – a fundamentally inefficient process. Unlike adaptor ligation, the efficiency of poly(A) tailing does not depend on the concentration of the free 3′-ends of the templates. In addition, primers carrying poly(dT) stretches (e.g., 30 consecutive dTs) capture poly(A) tailed molecules from very diluted solutions due to the low dissociation constant (Kd) of the poly(A)/poly(T) dimers. In contrast, the 3′-end ligation rate is (1) heavily dependent on the relative concentration of adaptors and free 3′-ends and (2) requires threshold amounts of nucleic acids in the sample. The efficiency of template switching can be approximately evaluated by analyzing the frequency of secondary template switching events. In our hands, when using a 5′-unblocked TSO, the percent of observed concatemers was between 10–20%. The efficency of adaptor ligation in any experimental setup is never as high. Moreover, similar to other commercial assays that rely on adaptor ligation, our method is strand-specific and thus retains the strandeness of RNAs. Finally, similar to most currently used techniques, our method allows multiplexing. Different barcodes can be incorporated into the 3′-adaptor sequence after the poly(A) tail and sequenced using the Illumina index read primer; in addition, secondary barcodes could be incorporated into the sequence of the template switching oligonucleotide.
One drawback of this technique is that template switching efficiency is apparently higher for RNA molecules having G nucleotide in their 5′-terminal end. Furthermore, RNA molecules ending with A incorporated the TSO with lower efficiency. Interestingly, such differences in template switching were not observed with DNA templates. Apparently, MMLV-RT mediates template switching more frequently when the terminal cDNA nucleotide is dC (occurring when 5′-end of RNA is occupied with G) – complementary to the last rG in the TSO. It remains to be tested whether the observed bias can be amended by using TSO with different or modified 3′-terminal nucleotides. However, methods which rely on adaptor ligation exhibit similar biases. Recent data uncovered severe biases in the sequencing of small non-protein coding RNA (small RNA-seq or sRNA-seq), such that the expression levels of some RNAs appeared to be artificially enhanced and others diminished or even undetectable.Citation3 While the large poly(dA) tail between the captured DNA sequence and the 3′-adaptor provides an obstacle to paired-end sequencing with a conventional second read primer due to the missing library complexity at this end, this could likely be amended by using a custom second read sequencing primer complimentary to the poly(dA) tail.
We did not compare our template switching-based method with the methods which are currently on the market for cDNA library generation directly as such comparison would not be possible. Current techniques require 10–100 fold higher inputs of DNA and RNA and are much more time consuming and expensive. Even those methods which allow DNA library generation from a single cell's RNA rely on (1) long RNAs to be first randomly or poly(A) primed and (2) cDNA to be pre-amplified before fractionation, adaptor ligation and sequencing.Citation1,Citation2 Furthermore, small circulating RNAs, miRNAs, piRNAs, endogenous siRNAs are only 20–30 nt long and, thus, cannot be pre-amplified before generating cDNA using random priming. Finally, the costs to generate ready-to-be-sequenced cDNA libraries using our method are comparable with conventional cDNA generation protocols for simple RT-qPCR, a fact that makes this method broadly affordable for researchers worldwide. In addition, the performance of the method described here in terms of mapping efficiency is similar to approaches published previously. Thus, after mRNA sequencing from single cell amounts using the SMART-seq approach, the mapping efficiency to the human reference genome constituted 52.5–70.3% unique mapping and 18.8–25.5% multimapping depending on the cell line sequenced.Citation6,Citation7 Likewise, we could map more than 90% of the reads obtained from poly(A) enriched RNA to human genome; moreover, uniquely mapped reads summed up to 59.72% and 62.56% (sample R10 and R5 respectively), while 28.55 and 30.56% reads mapped more than once. However, both the R10 and R5 libraries still contained a remarkable percentage of rRNAs (about 25%) which are highly repetitive in the genome (Agilent Bioanalyser QC, data not shown). The mapping efficiency of our bisulfite-converted DNA data (84.18% mapped to human genome) was comparable to other reports which used conventional methods for library preparation.Citation15,Citation16 Circulating DNA sequencing of plasma samples from two individuals yielded 35.42% and 53.91% of usable reads that could be mapped to the human reference genome; however, we could not find a proper benchmark in the scientific literature. It is feasible that some unmapped reads could derive from cells with significant genomic rearrangements or microorganisms. Finally, the relatively small percentage of mappable reads found in circulating RNA sequencing data set (only 23.35% can be mapped to human reference genome) and the considerable fraction of remaining reads that map to sequences of the human microbiome indicate the presence of circulating RNA from other species. Indeed in one previous report, a significant fraction of the circulating RNA appeared to originate from exogenous species.Citation17 We did not perform a detailed analysis of reads mapping to microbial sequences, as it was not the focus of this paper. At this stage, we merely aimed to demonstrate the technical feasibility of the approach itself to prepare DNA libraries from ultra-low amounts of nucleic acids.
We have chosen nanogram-amounts of bisulfite-treated DNA and poly(A)-enriched RNA as well as hundred-picogram amounts of circulating nucleic acids, because this represents the amount of nucleic acids per microliter of eluate which can typically be obtained from one confluent 24-well cell culture plate (mRNA and bisulfite-converted DNA) or several hundred microliters of blood plasma (circulating nucleic acids), thus allowing an easy integration into existing experimental or diagnostic workflows. To our knowledge, there were no reports so far describing next generation sequencing of (1) cellular bisulfitome (sequencing of bisulfite converted genomic DNA in order to determine its methylation state) from few nanograms of bisulfite-converted DNA without preliminary pre-amplification and (2) strand-specific mRNA transcriptome from one nanogram of poly(A) enriched RNA. Finally, there were no reports demonstrating the preparation of DNA library for deep sequencing from several picograms of short (22 nt) RNA and DNA.
Massive parallel sequencing of circulating RNA from blood plasma has been described before using adaptors ligation approaches for library generationCitation17,18.19; however, the amount of RNA input used was several nanograms per reaction. Unlike previous reports, we prepared cDNA library from two hundred picograms of the circulating plasma RNA - the amount of RNA per microliter of eluate which was obtained from as little as 400 microliters of plasma that was high-speed centrifuged to remove contaminating nucleic acids from cells or cell debris.
Likewise, to our knowledge, library generation for deep sequencing from only hundred-picogram of total circulating plasma DNA (in our case corresponding to the amount per microliter of eluate obtained from 800 microliters of plasma after high-speed centrifugation) has never been reported. However, recently some research groups described targeted amplicon sequencing (TAm-Seq) of several nanogram-scales of blood plasma DNA.Citation20,Citation21 Their work has demonstrated the proof-of-principle that circulating DNA from tumor is an informative and highly sensitive biomarker of metastatic breast cancer. However, TAm-Seq technique can only sequence circulating DNA from specific DNA loci (amplifiable by primers of known sequence) and not the whole circulating DNA. In another report, Murtaza and co-authors used 2.3 - 40 nanograms of circulating plasma DNA for library generation from whole circulating DNA using the commercial TruPLEX-FD kit.Citation22 However, the capacity of the TruPLEX-FD technology to generate libraries from lower quantity of circulating DNA has not been shown. Moreover, the exact mechanism of the method behind TruPLEX-FD technique is not disclosed, and thus, cannot be reproduced without purchasing the highly priced kit from the commercial supplier. Nevertheless, the results of those studies have demonstrated that mutated DNA which is found in the primary tumors can be subsequently detected in the blood plasma. They have further indicated the possibility to monitor the evolution of tumors in response to therapy in circulating DNA of serial plasma samples.Citation21 The development of new techniques for circulating DNA detection is therefore of particular interest.Citation23
In comparison to the previous studies our DNA sequencing approach could overcome many obstacles to circulating DNA-based approaches. While currently used techniques allowed the detection of selected DNA loci frequently mutated in tumors in the plasma of cancer patients, a standard panel of genes is unlikely to work for all patients.Citation23 Our method provides a comprehensive and significantly more sensitive, cheaper and faster way of library generation from whole circulating plasma DNA than any method published so far. The high sensitivity of the presented technique can enable regular screening for mutations in the plasma DNA to improve early detection of cancer as well as detection of recurrences/metastases in asymptomatic patients with previously diagnosed non-metastatic disease. Further, identification of new mutations in circulating tumor DNA over time, i.e., during the course of cancer therapy, might indicate tumor evolution and give rise to new treatment targets not indicated in the primary tumor. Finally, as changes in DNA methylation occur early in cancer developmentCitation24 and can give rise to therapy resistanceCitation25,Citation26 sensitive methods for deep sequencing are supposed to enable comprehensive methylation analysis of circulating DNA for early cancer detection and estimation of therapy response and prognosis.
One recent report described a method of generating strand-specific DNA libraries from small amounts of RNA molecules.Citation27 Similar to our method, this approach, named ‘Peregrine’ also utilizes a template switching oligonucleotide and was shown to generate ready-to-sequence DNA libraries from randomly fragmented RNA molecules. However, the Peregrine approach relies on a much higher input of RNA material (10–200 ng) and requires a purification step using magnetic beads after the first cDNA strand synthesis. Furthermore, unlike the method described here, Peregrine is based on random priming and was demonstrated to generate libraries from RNA molecules only larger than 200 nt. However, small RNA molecules such as miRNA and piRNA cannot be efficiently captured by random priming. Finally, we demonstrate for the first time the possibility to generate DNA libraries from DNA fragments using the template switch approach.
In another report the Weissman group described two novel protocols for the amplification and deep sequencing of very small amounts of mRNA. Both of them imply cDNA generation with either oligo(dT) or random oligonucleotide primers and subsequent semi-randomly primed PCR or phi29 DNA polymerase-based cDNA amplification.Citation28 Nevertheless, similarly to the SMART-seq technique, both methods were restricted to long mRNA sequencing and still involves adaptors-ligation after pre-amplification of the cDNA. As a result both procedures are significantly more time consuming and expensive than the method described in this paper. Several other research groups also performed mRNA deep sequencing form single cell amounts of mRNA; however all those approaches are based on cDNA pre-amplification and subsequent adaptor ligation.Citation29,Citation30 Finally, an elegant tagmentation approach (which implies simultaneous fragmentation and ligation of adaptors) has been reported to permit DNA library preparation from small quantities of genomic DNA.Citation31 This principle has been included into the Nextera DNA sample preparation kits, but requires at least 50 ng of DNA input and, apparently, is restricted to the long DNA molecules. The full capacity of the tagmentation technique for DNA library preparation is yet to be tested and compared with other methods.
It has to be indicated that CATS is not limited to library generation for sequencing but could also be used in other applications including microarray analyses. The ability of CATS to generate sequencing data directly from traces of circulating plasma DNA or RNA without the need for ligation steps and without prior selection, fractionation or amplification provides new experimental possibilities. This technique will be particularly valuable for the expanding field of individualised medicine such as early detection, therapy monitoring, prediction and prognosis of various human diseases using high-throughput sequencing. Forensic and archeological sciences will also benefit from this method as it allows deep sequencing of highly degraded and small amounts of DNA and RNA. Finally, the cost-efficient, easy to automate “one-tube process” and the short hands-on and turnaround time would also enable its application for routine diagnostics.
Materials and Methods
RNA and DNA samples
Synthetic cel-miR-39 (Sigma-Aldrich), a 22 nt microRNA from C.elegans was used as an input for small RNA sequencing control. Synthetic 22 nt (Sigma-Aldrich) DNA version of cel-miR-39 was used as an input for DNA sequencing control. Circulating DNA was isolated from the plasma fraction of blood samples from two voluntary healthy donors (DI, female and DII, male). The circulating RNA was isolated from the blood plasma of two voluntary female healthy donors (RI and RII). The study has been approved by the Ethical Committee of the Medical Faculty in Heidelberg. Circulating DNA and RNA isolated from human blood plasma, bisulfite-converted DNA from U2OS cells and Mg2+-fractionated poly(A)-enriched total RNA from U2OS cells were used as inputs for cDNA library preparation and sequenced on MiSeq sequencer (Illumina). Enrichment of poly(A) mRNA from total RNA was performed using NEBNext® Poly(A) mRNA Magnetic Isolation Module (New England Biolabs). Fragmentation of poly(A) mRNA was done using NEBNext® Magnesium RNA Fragmentation Module (New England Biolabs). Circulating RNA from 400 µl of human blood plasma was extracted as described earlier.Citation12 Circulating DNA samples were prepared using the QIAamp DNA Blood Mini Kit (Qiagen) from 800 µl of human blood plasma according to the manufacturer’s instructions but with minor modifications (addition of linear acrylamide to a final concentration of 20 mg/ml and increased volume of AL buffer and ethanol to 800 µL). Genomic DNA was isolated from cultured U2OS cells using QIAamp DNA mini kit (Qiagen) and bisulfite-treated with Epitect Bisulfite kit (Qiagen) using manufacturer’s recommendations.
Oligonucleotides for cDNA Synthesis
The sequences of all primers used in this work are provided in the Supplementary materials. Several template switch oligonucleotides (TSO) of different structures were tested during the development of the method. All oligonucleotides were synthesized by Eurofins Operon or Sigma Aldrich.
First-Strand cDNA Synthesis and Template Switching
Synthetic small RNA or DNA was diluted in water to achieve concentrations of 1 ng/μl and 5 pg/μl and was used as starting material to synthesize first-strand cDNA. The optimized protocol to generate the ready-to-sequence DNA library was as follows. The RNA was polyadenylated using E.coli poly(A) polymerase (New England Biolabs) in 1x PAP buffer containing 10 units Recombinant RNase inhibitor (Clontech) and 0.1 mM ATP for 10 min at 37 °C and the reaction was terminated by heating at 65 °C for 20 min. The DNA was poly(dA) tailed using terminal deoxynucleotide transferase (New England Biolabs) in 1x TdT buffer and 0.1 mM dATP for 30 min at 37 °C and the enzyme was heat-inactivated for 10 min at 70 °C. Before poly(dA) tailing, circulating DNA and bisulfite-converted DNA samples were denatured by heating at 95 °C for 5 min and fast cooling on ice. In some experiments (indicated in the figures) RNA and DNA templates were pre-treated with T4 Polynucleotide Kinase (New England Biolabs) for 10 min in 1xPAP/TdT buffer before poly(A/dA) tailing. In case of blood plasma RNA and Mg2+-fractionated poly(A)-enriched total RNA templates pre-treatment with T4 Polynucleotide Kinase dramatically increased the efficiently of DNA libraries preparation. For the reverse transcription, 1 μl of poly(A) tailed RNA or poly(dA) tailed DNA was mixed with 2.5 μl of 1x First-Strand RT buffer containing 20% DMSO and 1 μl of the one-base anchored Illumina poly(dT) primer AGA CGT GTG CTC TTC CGA TCT (T)x30V (final concentration 0.1 μM for 1 ng and 0.001 μM for 5 pg of RNA or DNA). The entire solution was incubated at 72 °C for 2 min and then cooled to 42 °C for 1 min. In the following step a master mix containing 2 μl 5x First-Strand RT buffer (Clontech), 1 μl dNTP (10 mM each), 1 μl SmartScribe RT polymerase (Clontech), 0.25 μl DTT (100 mM) and 0.25 μl of Recombinant RNase Inhibitor (Clontech) was added to the DNA(RNA)/primer solution and incubated for 15 min at 42 °C. Next, 1 μl of 10 μM 5′-biotin blocked template switch oligonucleotide (TSO) GTT CAG AGT TCT ACA GTC CGA CGA TC rGrGrG was added to the RT reaction and incubated for another 15 min at 42 °C. The RT reaction was terminated by heating at 70 °C for 10 min. Either 1 μl or 10 μl of RT reaction was used for cDNA amplification in a total volume of 100 μl. The amplification of cDNA was performed in 2xTaq polymerase master mix (Qiagen) using cDNA amplification primers () at a final concentration of 250 nM. The primers were as follows: CAA GCA GAA GAC GGC ATA CGA GAT CGT GAT GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATCT (forward) and AAT GAT ACG GCG ACC ACC GAG ATC TAC ACG TTC AGA GTT CTA CAG TCC GA (reverse). The amplified cDNAs were column purified using Qiaquick PCR Purification kit (Qiagen) and sequenced using Sanger automated sequencing (GATC GmbH, Konstanz, Germany). For next generation sequencing, the DNA fragments were additionally purified from 4% agarose gel using PureLink Gel Extraction kit (Life Technologies) and analyzed with Agilent Bioanalyser High Sensitivity DNA chips.
Deep Sequencing
Illumina MiSeq platform was used to sequence DNA libraries prepared by the method described above. A custom sequencing primer (Figure S4) was used for sequencing to resolve the problem with required complexity of the first six bases to ensure proper image registration and clusters identification. DNA libraries were diluted to a concentration of 5 nM, denatured with 0.2 N NaOH for 5 min and further diluted to 11 pM shortly before loading into the MiSeq cassette. The MiSeq run was performed using MiSeq Reagent Kit (50-cycles) for 77 cycles.
Data Analysis
Initial check of the FASTQ was done with FastQC (Babraham Bioinformatics). Cutadapt version 1.3 was applied for removal 5′- and 3′-adaptor sequences and of 3′ Poly(A) tails as well as for read length selection. Quality trimming (Q20) was performed using the FastX toolkit (version 0.0–13, http://hannonlab.cshl.edu/fastx_toolkit/index.html). Short read alignment to the cel-mir-39 sequencing control was performed with Bowtie2 (version 2.10) in local very sensitive alignment mode.Citation32 Short reads not corresponding to cel-mir-39 were selected using Samtools (version 0.1.19) and converted from SAM format to FASTQ for downstream alignment compatibility using the bam2fastx script included in TopHat2 (version 2.0.10).Citation33,Citation34 The 1000 Genomes Project reference genome (GRCh37 with decoy 5) was used for alignment of short reads to the human genome. Bowtie2 with local alignment was used for mapping of human circulating DNA sequencing reads. Tophat2 was used to align circulating and cell-line RNA reads to the reference genome and to the Gencode human reference transcriptome (version 19),Citation35 applying mapping with Bowtie2 in the very sensitive mode with reduced segment length for spliced mapping from 25 to 20 nt and performing search for microexons and potential transcript fusions. The relative transcript abundance was calculated as fragments per kilobase of transcript per megabase of genome sequence (FPKM) using Cufflinks (version 2.1.1) with masked rRNA, mt_rRNA and mt_tRNA genes from GencodeV19 and correction of multimapping and fragment bias.Citation36-Citation38 The included Cuffcompare script was used for annotation against known GencodeV19 transcripts. Circulating RNA reads not aligned to human sequences were mapped to the reference genome database of the NIH Human Microbiome ProjectCitation39 using Bowtie2 (version 2.11). Bismark (version 0.10.0) was used to align short reads from DNA bisulfite sequencing of U2OS cells to normal as well as bisulfite-converted versions (C > T and G > A) of the human genome and methylation analysis.Citation16
Additional material
Download Zip (1.1 MB)Acknowledgment
We thank Prof. Alexander Tonevitsky from Moscow State University (Russia) for support.
Funding
This work was supported by the University Clinic Heidelberg and the German Cancer Research Center
Contributions
A.T. developed the protocol, performed all experiments and wrote this manuscript; H.S performed all bioinformatics analysis, participated in study design and the protocol optimisation and edited the manuscript. A.S. and M.Z. P.L advised and participated in Illumina MiSeq DNA sequencing and reviewed the manuscript. B.B. coordinated the study, participated in study design and edited the manuscript.
References
- Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, Sivachenko A, Thompson DA, Wysoker A, Fennell T, et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods 2013; 10:623 - 9; http://dx.doi.org/10.1038/nmeth.2483; PMID: 23685885
- Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods 2014; 11:41 - 6; http://dx.doi.org/10.1038/nmeth.2694; PMID: 24141493
- Raabe CA, Tang TH, Brosius J, Rozhdestvensky TS. Biases in small RNA deep sequencing data. Nucleic Acids Res 2014; 42:1414 - 26; PMID: 24198247
- Matz M, Shagin D, Bogdanova E, Britanova O, Lukyanov S, Diatchenko L, Chenchik A. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res 1999; 27:1558 - 60; http://dx.doi.org/10.1093/nar/27.6.1558; PMID: 10037822
- Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques 2001; 30:892 - 7; PMID: 11314272
- Picelli S, Björklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 2013; 10:1096 - 8; http://dx.doi.org/10.1038/nmeth.2639; PMID: 24056875
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 2014; 9:171 - 81; http://dx.doi.org/10.1038/nprot.2014.006; PMID: 24385147
- Cocquet J, Chong A, Zhang G, Veitia RA. Reverse transcriptase template switching and false alternative transcripts. Genomics 2006; 88:127 - 31; http://dx.doi.org/10.1016/j.ygeno.2005.12.013; PMID: 16457984
- Kapteyn J, He R, McDowell ET, Gang DR. Incorporation of non-natural nucleotides into template-switching oligonucleotides reduces background and improves cDNA synthesis from very small RNA samples. BMC Genomics 2010; 11:413; http://dx.doi.org/10.1186/1471-2164-11-413; PMID: 20598146
- Tang DT, Plessy C, Salimullah M, Suzuki AM, Calligaris R, Gustincich S, Carninci P. Suppression of artifacts and barcode bias in high-throughput transcriptome analyses utilizing template switching. Nucleic Acids Res 2013; 41:e44; http://dx.doi.org/10.1093/nar/gks1128; PMID: 23180801
- Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res 2006; 34:5402 - 15; http://dx.doi.org/10.1093/nar/gkl655; PMID: 17012276
- Turchinovich A, Weiz L, Burwinkel B. Isolation of circulating microRNA associated with RNA-binding protein. Methods Mol Biol 2013; 1024:97 - 107; http://dx.doi.org/10.1007/978-1-62703-453-1_8; PMID: 23719945
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110:462 - 7; http://dx.doi.org/10.1159/000084979; PMID: 16093699
- Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 2012; 13:341; http://dx.doi.org/10.1186/1471-2164-13-341; PMID: 22827831
- Krueger F, Kreck B, Franke A, Andrews SR. DNA methylome analysis using short bisulfite sequencing data. Nat Methods 2012; 9:145 - 51; http://dx.doi.org/10.1038/nmeth.1828; PMID: 22290186
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011; 27:1571 - 2; http://dx.doi.org/10.1093/bioinformatics/btr167; PMID: 21493656
- Wang K, Li H, Yuan Y, Etheridge A, Zhou Y, Huang D, Wilmes P, Galas D. The complex exogenous RNA spectra in human plasma: an interface with human gut biota?. PLoS One 2012; 7:e51009; http://dx.doi.org/10.1371/journal.pone.0051009; PMID: 23251414
- Semenov DV, Baryakin DN, Brenner EV, Kurilshikov AM, Vasiliev GV, Bryzgalov LA, Chikova ED, Filippova JA, Kuligina EV, Richter VA. Unbiased approach to profile the variety of small non-coding RNA of human blood plasma with massively parallel sequencing technology. Expert Opin Biol Ther 2012; 12:Suppl 1 S43 - 51; http://dx.doi.org/10.1517/14712598.2012.679653; PMID: 22509727
- Williams Z, Ben-Dov IZ, Elias R, Mihailovic A, Brown M, Rosenwaks Z, Tuschl T. Comprehensive profiling of circulating microRNA via small RNA sequencing of cDNA libraries reveals biomarker potential and limitations. Proc Natl Acad Sci U S A 2013; 110:4255 - 60; http://dx.doi.org/10.1073/pnas.1214046110; PMID: 23440203
- Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012; 4:36ra68; http://dx.doi.org/10.1126/scitranslmed.3003726; PMID: 22649089
- Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013; 368:1199 - 209; http://dx.doi.org/10.1056/NEJMoa1213261; PMID: 23484797
- Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013; 497:108 - 12; http://dx.doi.org/10.1038/nature12065; PMID: 23563269
- Lippman M, Osborne CK. Circulating tumor DNA--ready for prime time?. N Engl J Med 2013; 368:1249 - 50; http://dx.doi.org/10.1056/NEJMe1301249; PMID: 23484798
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415 - 28; PMID: 12042769
- Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res 1994; 54:2552 - 5; PMID: 8168078
- Das PM, Singal R. DNA methylation and cancer. J Clin Oncol 2004; 22:4632 - 42; http://dx.doi.org/10.1200/JCO.2004.07.151; PMID: 15542813
- Langevin SA, Bent ZW, Solberg OD, Curtis DJ, Lane PD, Williams KP, Schoeniger JS, Sinha A, Lane TW, Branda SS. Peregrine: A rapid and unbiased method to produce strand-specific RNA-Seq libraries from small quantities of starting material. RNA Biol 2013; 10:502 - 15; http://dx.doi.org/10.4161/rna.24284; PMID: 23558773
- Pan X, Durrett RE, Zhu H, Tanaka Y, Li Y, Zi X, Marjani SL, Euskirchen G, Ma C, Lamotte RH, et al. Two methods for full-length RNA sequencing for low quantities of cells and single cells. Proc Natl Acad Sci U S A 2013; 110:594 - 9; http://dx.doi.org/10.1073/pnas.1217322109; PMID: 23267071
- Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2012; 2:666 - 73; http://dx.doi.org/10.1016/j.celrep.2012.08.003; PMID: 22939981
- Ramsköld D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 2012; 30:777 - 82; http://dx.doi.org/10.1038/nbt.2282; PMID: 22820318
- Parkinson NJ, Maslau S, Ferneyhough B, Zhang G, Gregory L, Buck D, Ragoussis J, Ponting CP, Fischer MD. Preparation of high-quality next-generation sequencing libraries from picogram quantities of target DNA. Genome Res 2012; 22:125 - 33; http://dx.doi.org/10.1101/gr.124016.111; PMID: 22090378
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012; 9:357 - 9; http://dx.doi.org/10.1038/nmeth.1923; PMID: 22388286
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078 - 9; http://dx.doi.org/10.1093/bioinformatics/btp352; PMID: 19505943
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14:R36; http://dx.doi.org/10.1186/gb-2013-14-4-r36; PMID: 23618408
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 2012; 22:1760 - 74; http://dx.doi.org/10.1101/gr.135350.111; PMID: 22955987
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28:511 - 5; http://dx.doi.org/10.1038/nbt.1621; PMID: 20436464
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7:562 - 78; http://dx.doi.org/10.1038/nprot.2012.016; PMID: 22383036
- Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol 2011; 12:R22; http://dx.doi.org/10.1186/gb-2011-12-3-r22; PMID: 21410973
- Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, et al, NIH HMP Working Group. The NIH Human Microbiome Project. Genome Res 2009; 19:2317 - 23; http://dx.doi.org/10.1101/gr.096651.109; PMID: 19819907