Abstract
Humans have two nearly identical copies of the Survival Motor Neuron (SMN) gene: SMN1 and SMN2. The two SMN genes code for identical proteins; however, SMN2 predominantly generates a shorter transcript due to skipping of exon 7, the last coding exon. Skipping of SMN2 exon 7 leads to production of a truncated SMN protein that is highly unstable. The inability of SMN2 to compensate for the loss of SMN1 results in spinal muscular atrophy (SMA), the second most prevalent genetic cause of infant mortality. Since SMN2 is almost universally present in SMA patients, correction of SMN2 exon 7 splicing holds the promise for cure. Consistently, SMN2 exon 7 splicing has emerged as one of the best studied splicing systems in humans. The vast amount of recent literature provides a clue that SMN2 exon 7 splicing is regulated by an intron definition mechanism, which does not require cross-exon communication as prerequisite for exon inclusion. Our conclusion is based on the prominent role of intronic cis-elements, some of them have emerged as the frontrunners among potential therapeutic targets of SMA. Further, the widely expressed T-cell-restricted intracellular antigen-1 (TIA1), a member of the Q-rich domain containing RNA-binding proteins, has recently been found to regulate SMN exon 7 splicing by binding to intron 7 sequences away from the 5′ ss. These findings make a strong argument for an "intron definition model", according to which regulatory sequences within a downstream intron are capable of enforcing exon inclusion even in the absence of a defined upstream 3′ ss of an alternatively spliced exon.
Introduction
Spinal muscular atrophy (SMA) is a leading genetic cause of infant mortality.Citation1–Citation3 The disease is attributed to deletion or mutation within Survival Motor Neuron 1 (SMN1) gene.Citation4 SMN2, a nearly identical copy of SMN1, fails to prevent SMA due to a critical C-to-T mutation at the 6th position (C6U mutation in RNA) of exon 7.Citation5–Citation7 C6U mutation leads to a predominant exon 7 skipping from SMN2, producing mostly truncated protein SMNΔ7, which is unstable.Citation8 High SMN2 copies capable of producing cumulative high levels of SMNΔ7 as well as full-length SMN fully rescue SMA mice, suggesting that SMNΔ7 does not play a dominant negative role.Citation9 Full-length SMN protein (here after referred to as “SMN”) has multiple functions that include snRNP biogenesis, transcription, splicing, cell signaling, macromolecular trafficking and stress granule formation.Citation10–Citation18 Consistent with the essential role of SMN in cellular metabolism, complete loss of SMN is embryonic lethal.Citation19
SMA is a model genetic disease in which correction of aberrant splicing of a single exon (i.e., SMN2 exon 7) holds the promise for cure of more than 90% of the affected population.Citation20 Consistently, SMN2 exon 7 splicing has been extensively studied and remains one of the highly contested topics among genetic diseases linked to defective splicing. Many excellent reviews published recently highlight approaches of SMN2 exon 7 splicing correction in SMA.Citation20–Citation24 Here we focus on emerging novel aspects of splicing regulation of SMN2 exon 7 that happens to be the last coding as well as the last internal exon. The 54-nt long human SMN exon 7 is flanked by a 5.76 kb intron 6 and a 444 nt intron 7 ().Citation25 Of note, human gene is referred to as SMN, whereas mouse gene is referred to as Smn. Despite the same number of exons, there are substantial differences in gene organization between SMN and Smn. For instance, the 51-nt long Smn exon 7 is flanked by a 3.22 kb intron 6 and a 1.64 kb intron 7. Unlike the alternatively spliced SMN exon 7, Smn exon 7 is constitutively spliced. The 3-nucleotide difference within exon 7 and other variations in the downstream intron 7 between SMN and Smn account for the differential splicing of human and mouse exon 7.Citation25–Citation28 Given the multifunctional role of SMN in cellular metabolism, it is plausible that gene duplication may have triggered a differential splicing mechanism or vice versa to tightly regulate SMN levels in humans.
According to conventional point of view, the exon definition model is applicable to the splicing of small internal exons that are flanked by large intronic sequences.Citation29–Citation31 The exon definition model requires simultaneous recognition of both splice sites through precise crosstalk involving both ends of an exon. As per new hypothesis supporting the exon definition model, intronic sequences immediately upstream and downstream of an exon may enforce exon definition through additional cross-communication.Citation32 However, such intronic communications may also serve the opposite purpose as has been documented in case of hnRNP A1-mediated exon skipping event through a looping out mechanism.Citation33 The exon definition model requires a highly regulated transition process from splice site recognition across short exons to assembly of the spliceosome across long introns.Citation34 However, regulation of this transition process is poorly understood.
Considering both splice sites are simultaneously selected in the exon definition model, it is rather unlikely that a disproportionate intron retention product will accumulate during splicing via exon definition mechanism. Thus, exon definition model provides a basis for challenge if disproportionate intron retention products are easily detectable in a reliable RT-PCR reaction. Also, the effect of mutations within deep intronic sequences on splicing could not be explained by the simplistic view of crosstalk between two ends of an exon (; described below). The intron definition model is often used to describe mechanism of splicing of large internal exons flanked by at least one small intron.Citation30,Citation35–Citation38 Based on the wealth of information accumulated to date, we propose that splicing of SMN2 exon 7 is driven by the intron definition mechanism. Of note, much of the results described here pertain to the widely studied SMN intron 7, which is only 444-nt long. However, this size exceeds the length of a typical intron (∼250 nts) defined by an intron definition model.Citation30 Our argument supporting the intron definition model may be relevant to other short exons flanked by large intronic sequences.
Exon Skipping is Linked to a Critical Exonic Position
The initial finding that C6U mutation alone is sufficient to cause SMN2 exon 7 skipping uncovered the significance of a single position within exon 7.Citation5,Citation6 Close proximity of C6U mutation to the 3′ ss led to an early hypothesis that a weak 3′ ss of SMN2 exon 7 is the probable cause of exon 7 skipping.Citation39 Subsequently, C6U mutation was linked to either loss of an SF2/ASF-associated enhancer or gain of an hnRNP A1-associated silencer ().Citation40,Citation41 Based on an in vitro study, SF2/ASF and hnRNP A1-associated hypotheses may not be mutually exclusive.Citation42 Depletion of hnRNP A1 has been shown to restore SMN2 exon 7 inclusion in an engineered cell line lacking SF2/ASF. These findings support C6U-associated gain of hnRNP A1 motif and clearly demonstrate the prominent role of hnRNP A1 in SMN2 exon 7 splicing in vivo. Recent studies revealed interactions of hnRNP A1 with additional sites located within SMN intron 7 ( and described below). Supporting the C6U-associated gain of an inhibitory element coupled with the loss of a stimulatory element, bifunctional antisense oligonucleotides (ASOs) that blocked C6U region and presumably recruited stimulatory factors promoted SMN2 exon 7 inclusion.Citation43,Citation44 Recently, Sam68 has been proposed to be an additional C6U-interacting factor responsible for SMN2 exon 7 skipping.Citation45 These findings are in agreement with the role of an extended inhibitory context (Exinct) that spans from 3rd to 15th positions of exon 7.Citation46 Interestingly, hnRNP Q was shown to stimulate SMN2 exon 7 inclusion by binding to C6U region that falls within Exinct.Citation47 It is possible that low levels of exon 7 inclusion in SMN2 mRNA is maintained by factors such as hnRNP Q through interaction with a hotspot like Exinct.
In vivo selection of entire SMN1 exon 7 further supported the presence of Exinct and revealed two additional regulatory regions within exon 7.Citation26 These are: conserved tract (a positive cis-element) in the middle of exon 7 and 3′-cluster (a negative cis-element) located toward the end of the exon (). Presence of Exinct, conserved tract and 3′-cluster was independently corroborated by a complementary approach of an antisense micro-walk covering the entire exon 7.Citation48 ASOs that blocked Exinct and 3′-cluster promoted exon 7 inclusion, whereas ASOs that blocked conserved tract promoted exon 7 skipping. Conserved tract fully overlaps with the binding site of splicing factor Tra2-β1 ().Citation26 Overexpression of Tra2-β1 and its interacting factors SRp30c, hnRNP G and TDP-43 has been shown to promote SMN2 exon 7 inclusion.Citation49–Citation52 However, a recent report employing an engineered murine embryonic fibroblast cell line showed no effect of Tra2-β1 loss on SMN2 exon 7 splicing.Citation53 This surprising result underscores the redundancy in exon 7-interacting stimulatory factors, some of which, for example Tra2-β1, are totally dispensable for exon 7 inclusion.
Intron Retention Supports Intron Definition Model
Minigenes containing shortened intron 6 of SMN1 and SMN2 recapitulate the splicing pattern of endogenous genes and are useful tools for detection of intron-retained intermediates.Citation26,Citation46 In addition to predominant exon 7-included products, SMN1 minigene produces detectable level of intron 7-retained intermediates ().Citation26 These observations suggest that the 5′ ss of exon 7 is weak and inclusion of exon 7 from SMN1 is primarily (if not exclusively) driven by the removal of intron 6, supporting the intron definition model. Thus, it is possible that the loss of intron 6 definition due to C6U mutation (that creates a weak 3′ ss) in SMN2 coupled with an inherently weak 5′ ss causes exon 7 skipping.
If the intron definition mechanism were to hold true, strengthening of the 5′ ss alone would guarantee SMN2 exon 7 inclusion even in the presence of a very weak 3′ ss. Indeed, results of in vivo selection of entire exon 7 affirmed intron definition model as a majority of selected sequences carried an A-to-G substitution (A54G) at the last position on exon 7.Citation26 Further, A54G mutation in SMN2 promoted exon 7 inclusion despite the loss of additional positive cis-elements at the 3′ ss.Citation26 The likely cause of the stimulatory effect of A54G mutation was the strengthened 5′ ss that allowed an efficient U1 snRNP recruitment through a strong RNA:RNA duplex formed between U1 snRNA and the 5′ ss (abbreviated as “U1:5′ ss duplex”). Of note, A54G mutation increases the size of U1:5′ ss duplex by two base pairs: G:C and G:U. In a counter experiment, a mutant U1 snRNA that increased U1:5′ ss duplex by five base pairs also restored SMN2 exon 7 inclusion.Citation54 Similar to A54G mutation, mutant U1 snRNA promoted SMN2 exon 7 inclusion despite the simultaneous loss of additional positive cis-elements responsible for strengthening of the 3′ ss. Notably, mutant U1 snRNA that effectively defined intron 7 produced a disproportionate increase in intron 6-retained intermediate due to the existing weak 3′ ss.Citation54 These results are perfectly in line with the intron definition model, according to which removal of intron 7 eliminates the 3′ ss (of exon 8) that otherwise would be used for exon 7 skipping. On the other hand, based on the results of U1 snRNP recruitment, one may argue that intron and exon definition models are not mutually exclusive. For instance, it is likely that an effective recruitment of U1 snRNP at the strengthened 5′ ss of SMN2 exon 7 also promotes exon definition process through cross-exon talk. However, exon definition in this case appears to be dependent upon and secondary to the intron definition process.
Distributed Signals of Intron Definition
A number of reported cis-elements are likely to prevent intron 7 definition by weakening the 5′ ss of exon 7. These include an inhibitory terminal stem-loop structure (TSL2), intronic splicing silencer N1 (ISS-N1) and a GC-rich sequence ( and ).Citation27,Citation54,Citation55 TSL2 sequesters the 5′ ss of exon 7 and mutations that abrogate TSL2 promote SMN2 exon 7 inclusion. The 15-nt long ISS-N1 spans from 10th to 24th positions of intron 7 and serves as a major inhibitory cis-element responsible for SMN2 exon 7 skipping ().Citation27 Deletion of ISS-N1 fully restores SMN2 exon 7 inclusion even in the absence of positive exonic cis-elements at the 3′ ss of exon 7. Further, ASO-mediated blocking of ISS-N1 fully restores SMN2 exon 7 inclusion in SMA patient cells.Citation27 ISS-N1 contains two putative hnRNP A1 binding sites ().Citation56 It is likely that hnRNP A1 in complex with ISS-N1 renders the 5′ ss of exon 7 inaccessible. It is also possible that ISS-N1 is a complex regulatory element with multiple overlapping motifs. For instance, ISS-N1 partially overlaps with an 8-nt long inhibitory GC rich sequence that spans from 7th to 14th positions of intron 7.Citation55 An 8-mer ASO targeting GC-rich sequence fully restores SMN2 exon 7 inclusion in SMA patient cells.Citation55 Although mechanism by which GC-rich sequence exerts its negative impact is yet to be determined, the stimulatory effect due to blocking of GC-rich sequence supports the intron definition model. Of note, SMN exon 7 lacks specific intronic motif pairs that have been recently implicated in exon definition.Citation32
Long-distance interactions (LDIs) involving deep intronic sequences could play pivotal role in pre-mRNA splicing. Depending upon the nature, such interactions could enhance or slow intron definition process. First LDI within SMN2 intron 7 was discovered through an ultra-refined antisense microwalk in which two 14-mer ASOs (F14 and L14) annealing to targets differing by a single nucleotide produced antagonistic effects on SMN2 exon 7 splicing.Citation57 F14 promoted SMN2 exon 7 inclusion by sequestering a 14-nt long sequence that starts with a cytosine residue at the 10th intronic position (10C), whereas L14 that did not block 10C promoted SMN2 exon 7 skipping. Further experiments confirmed that 10C makes a negative LDI with downstream intronic sequences.Citation57 Deletion of those downstream sequences reversed the negative effect of L14. Interestingly, 10C falls within the GC-rich sequence and occupies the first ISS-N1 position that is not a part of hnRNP A1 motif (). In any case, 10C-associated LDI suggests a tightly regulated intron definition process in which motifs across intron 7 show mutual dependency.
One may envision several other scenarios of LDIs involving sequences of intron 7, intron 6 and exon 7. For example, a SMN2 specific A-to-G mutation at the 100th position (A100G) of intron 7 creates an hnRNP A1 motif.Citation58 Of note, A100G is a deep intronic mutation that does not create a cryptic splice site and yet imparts a significant impact on SMN2 exon 7 splicing. It is likely that hnRNP A1 bound to this motif (at the 100th position of intron 7) communicates with another hnRNP A1 molecule interacting with ISS-N1 and/or C6U location. Further, intron 6 has been reported to contain inhibitory Element 1 that encompasses binding site for PTB.Citation59,Citation60 It is probable that PTB bound to Element 1 communicates with another PTB molecule interacting with intron 7. The intron definition model would necessitate abrogation of these and other negative LDIs through stimulatory functions of intron-interacting factors. We have recently reported TIA1, a glutamine-rich domain containing RNA-binding protein, as the first splicing factor to activate SMN2 exon 7 inclusion by binding to intron 7.Citation25 The stimulatory effect of TIA1 was able to counteract the inhibitory effect of PTB. Interestingly, site of TIA1 interaction within intron 7 is distinct from the traditional TIA1 binding sites located immediately downstream of 5′ ss. TIA1 interacts with U-rich clusters (URC1 and URC2) downstream of ISS-N1 ().Citation25 Such interaction is not consistent with the exon definition model that generally requires a host of stimulatory cis-elements adjacent to splice sites. Of note, there is substantial sequence conservation within URC1 and URC2 between human and mouse genes.Citation61 However, non-conserved residues seem to enforce TIA1 specificity in SMN intron 7.Citation25 Consistently, a TIA1 knockout mouse cell line showed no effect on splicing of Smn exon 7.Citation25 Binding site of TIA1 is situated adjacent to Element 2, an earlier reported stimulatory element within intron 7.Citation62 It is likely that splicing activators interacting with Element 2 as well as with other yet undefined motifs within intron 7 communicate with TIA1. Once again, such communications would be consistent with intron definition model.
Concluding Remarks
The exon definition model has been considered as the conventional mechanism of splicing of small exons flanked by large intronic sequences.Citation29–Citation32 The simultaneous recognition of both splice sites of an internal exon is the minimum prerequisite for this model. Consistently, a mutation within an exon leading to its skipping provides a likely example of the exon definition model. However, the competing intron definition model could be applicable in situations where intronic sequences appear to play a significant role in splice site selection. Employing SMN exon 7 splicing as one of the best-studied systems, we propose that a small exon flanked by large intronic sequences could be spliced by the intron definition mechanism. The strongest support for the intron definition model came from intronic motifs, whose blocking promoted SMN2 exon 7 inclusion.Citation27,Citation55 The independently validated stimulatory impact of the ASO-mediated intron definition is so enormous that the approach has a potential for the treatment of SMA.Citation27,Citation55,Citation63–Citation65 Additional support of the intron definition model came from the observation that TIA1 restored SMN2 exon 7 inclusion by binding to intronic sequences away from the 5′ ss. The intron definition model is further supported by the presence of the disproportionately trapped intron-retained intermediates that are uncharacteristic of the exon definition model. Based on the evidence of a complex regulatory network and LDIs within SMN2 intron 7, one may envision intronic motifs as the powerhouse of splicing regulation.
Splicing is coupled with transcription, capping, micro-RNA processing, polyadenylation and nuclear export.Citation66–Citation71 Large introns may encompass critical information for several layers of regulation that links splicing to the above mentioned processes. Many unrelated small compounds have been reported to modulate SMN exon 7 splicing.Citation23 Mechanisms of action of these compounds remain unknown. Not limited by the constraints of a size, intron definition provides an attractive model to accommodate the role of a large number of factors, small compounds and structural rearrangements in pre-mRNA splicing. It remains to be seen if compelling evidence supporting the intron definition model for a small exon in SMN is part of a larger trend of pre-mRNA splicing regulation in higher eukaryotes. Overall, the intron and exon definition models may not be mutually exclusive. It is likely that an intron definition process in SMN leads to an exon definition process. However, there are several missing links and we are rather at the very early stages of understanding the mechanism of interconnectivity between these two very attractive and simplistic models.
Figures and Tables
Figure 1 Organization of human (SMN) and mouse (Smn) genes covering the last 3 exons. (A) Comparison of exon 7 and introns 6 and 7 of SMN and Smn genes. Exons are shown as colored boxes, whereas introns are shown as broken lines. Sizes of introns and exon 7 are given. (B) Results of RT-PCR for transcripts derived from SMN1 and SMN2 minigenes.Citation26 Note the presence of intron 7 retained intermediate due to a weak 5′ ss in SMN1. As per intron definition model, splicing of SMN1 exon 7 was mainly driven by intron 6 definition due to a strong 3′ ss. C6U mutation causes a loss on intron 6 definition in SMN2 leading to skipping of exon 7. (C) Partial view of factors responsible for intron definition. In vivo selection of entire SMN exon 7 revealed three major regulatory regions: Exinct, conserved tract and 3′-cluster.Citation26 SF2/ASF, hnRNP A1, Sam68 and hnRNP Q interact within the region corresponding to Exinct.Citation40,Citation41,Citation45,Citation47 Binding site of tra2-β1 and associated factors hnRNP G, SRp30c and TDP-43 fall within conserved tract.Citation49–Citation52 The 3′-cluster falls within the terminal stem loop 2 (TSL2) structure that also sequesters the 5′ ss of exon 7.Citation54 ISS-N1 is a major intronic inhibitory elements that harbors 2 hnRNP A1 motifs.Citation27,Citation56 ISS-N1 also overlaps with a GC-rich sequences that provides the shortest antisense target for correction of SMN2 exon 7 splicing in SMA.Citation55 The cytosine residue at the 10th intronic position (10C) has been implicated in a long-distance interaction (LDI) with deep intronic sequences.Citation57 TIA1 binds to an intronic region downstream of ISS-N1.Citation25 Cis-elements and factors that are likely to define either intron 6 or intron 7 have been grouped. (+) and (−) indicates that a given cis-element or splicing factor promotes and suppresses exon 7 inclusion, respectively. Exon 7 sequences are shown in capital letters, whereas intronic sequences are shown in small case letters.
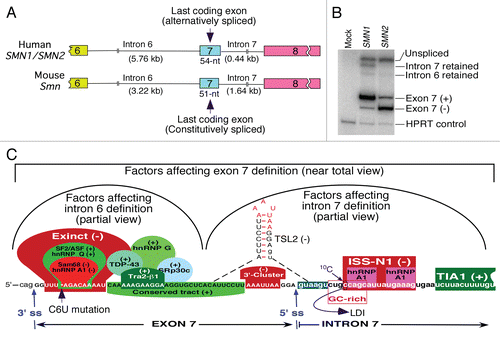
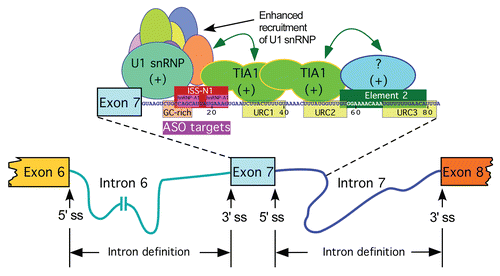
Figure 2 Intron definition model for splicing of SMN exon 7. As per intron definition model, both splice sites of an intron could be defined without participation of a cross-exon interaction. Shown are splicing cis-elements and transacting factors that affect intron 7 definition. These include ISS-N1, GC-rich, Element 2 and hnRNP A1 and TIA1 motifs.Citation25,Citation27,Citation55,Citation56,Citation62 (+) indicates that a given cis-element or splicing factor promotes exon 7 inclusion. Supporting intron definition model, binding of TIA1 to URC1/URC2 within intron 7 brings a change in the context leading to recruitment of U1 snRNP at the 5′ ss of exon 7. Similar stimulatory changes and a forced intron 7 definition could be brought about by ASOs targeting ISS-N1 or GC-rich sequence.Citation27,Citation55
Acknowledgments
Work in Singh group is supported by a grant from United States National Institutes of Health (R01NS055925). R.N.S. acknowledges support of Salsbury Endowment at Iowa State University, Ames, IA.
References
- Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick?. Nat Rev Neurosci 2009; 10:597 - 609; http://dx.doi.org/10.1038/nrn2670
- Park GH, Kariya S, Monani UR. Spinal muscular atrophy: new and emerging insights from model mice. Curr Neurol Neurosci Rep 2010; 10:108 - 117; http://dx.doi.org/10.1007/s11910-010-0095-5
- Wee CD, Kong L, Sumner CJ. The genetics of spinal muscular atrophies. Curr Opin Neurol 2010; 23:450 - 458; http://dx.doi.org/10.1097/WCO.0b013e32833e1765
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy determining gene. Cell 1995; 80:155 - 165
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 1999; 96:6307 - 6311; http://dx.doi.org/10.1073/pnas.96.11.6307
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999; 8:1177 - 1183; http://dx.doi.org/10.1093/hmg/8.7.1177
- Lorson CL, Rindt H, Shababi M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet 2010; 19:111 - 118; http://dx.doi.org/10.1093/hmg/ddq147
- Cho S, Dreyfuss G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev 2010; 24:438 - 442; http://dx.doi.org/10.1101/gad.1884910
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet 2000; 9:333 - 339; http://dx.doi.org/10.1093/hmg/9.3.333
- Battle DJ, Kasim M, Yong J, Lotti F, Lau CK, Mouaikel J, et al. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol 2006; 71:313 - 320; http://dx.doi.org/10.1101/sqb.2006.71.001
- Winkler C, Eggert C, Gradl D, Meister G, Giegerich M, Wedlich D, et al. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev 2005; 19:2320 - 2330; http://dx.doi.org/10.1101/gad.342005
- Strasswimmer J, Lorson CL, Breiding DE, Chen JJ, Le T, Burghes AH, et al. Identification of survival motor neuron as a transcriptional activator-binding protein. Hum Mol Genet 1999; 8:1219 - 1226; http://dx.doi.org/10.1093/hmg/8.7.1219
- Rossoll W, Kröning AK, Ohndorf UM, Steegborn C, Jablonka S, Sendtner M. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons?. Hum Mol Genet 2002; 11:93 - 105; http://dx.doi.org/10.1093/hmg/11.1.93
- Jodelka FM, Ebert AD, Duelli DM, Hastings ML. A feedback loop regulates splicing of the spinal muscular atrophy-modifying gene, SMN2. Hum Mol Genet 2010; 19:4906 - 4917; http://dx.doi.org/10.1093/hmg/ddq425
- Bowerman M, Shafey D, Kothary R. Smn depletion alters profilin II expression and leads to upregulation of the RhoA/ROCK pathway and defects in neuronal integrity. J Mol Neurosci 2007; 32:120 - 131; http://dx.doi.org/10.1007/s12031-007-0024-5
- Peter CJ, Evans M, Thayanithy V, Taniguchi-Ishigaki N, Bach I, Kolpak A, et al. The COPI vesicle complex binds and moves with survival motor neuron within axons. Hum Mol Genet 2011; 20:1701 - 1711
- Fallini C, Zhang H, Su Y, Silani V, Singer RH, Rossoll W, et al. The survival of motor neuron (SMN) protein interacts with the mRNA-Binding protein HuD and regulates localization of Poly(A) mRNA in primary motor neuron axons. J Neurosci 2011; 31:3914 - 3925; http://dx.doi.org/10.1523/JNEUROSCI.3631-10.2011
- Zou T, Yang X, Pan D, Huang J, Sahin M, Zhou J. SMN deficiency reduces cellular ability to form stress granules, sensitizing cells to stress. Cell Mol Neurobiol 2011; 31:541 - 550
- Schrank B, Gotz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA 1997; 94:9920 - 9925; http://dx.doi.org/10.1073/pnas.94.18.9920
- Shababi M, Mattis VB, Lorson CL. Therapeutics that directly increase SMN expression to treat spinal muscular atrophy. Drug News Perspect 2010; 23:475 - 482
- Bäumer D, Ansorge O, Almeida M, Talbot K. The role of RNA processing in the pathogenesis of motor neuron degeneration. Expert Rev Mol Med 2010; 12:21; http://dx.doi.org/10.1017/S1462399410001523
- NlendNlend R, Meyer K, Schümperli D. Repair of pre-mRNA splicing: prospects for a therapy for spinal muscular atrophy. RNA Biol 2010; 7:430 - 440; http://dx.doi.org/10.4161/rna.7.4.12206
- Bebee TW, Gladman JT, Chandler DS. Splicing regulation of the survival motor neuron genes and implications for treatment of spinal muscular atrophy. Front Biosci 2010; 15:1191 - 1204; http://dx.doi.org/10.2741/3670
- Burghes AH, McGovern VL. Antisense oligonucleotides and spinal muscular atrophy: skipping along. Genes Dev 2010; 24:1574 - 1579; http://dx.doi.org/10.1101/gad.1961710
- Singh NN, Seo J, Ottesen EW, Shishimorova M, Bhattacharya D, Singh RN. TIA1 prevents skipping of a critical exon associated with spinal muscular atrophy. Mol Cell Biol 2011; 31:935 - 954; http://dx.doi.org/10.1128/MCB.00945-10
- Singh NN, Androphy EJ, Singh RN. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA 2004; 10:1291 - 1305; http://dx.doi.org/10.1261/rna.7580704
- Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol 2006; 26:1333 - 1346; http://dx.doi.org/10.1128/MCB.26.4.133346.2006
- Singh RN. Evolving concepts on human SMN pre-mRNA splicing. RNA Biol 2007; 4:7 - 10; http://dx.doi.org/10.4161/rna.4.1.4535
- Berget SM. Exon recognition in vertebrate splicing. J Biol Chem 1995; 270:2411 - 2414
- Fox-Walsh KL, Dou Y, Lam BJ, Hung SP, Baldi PF, Hertel KJ. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc Natl Acad Sci USA 2005; 102:16176 - 16181; http://dx.doi.org/10.1073/spnas.0508489102
- Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 2010; 11:345 - 355; http://dx.doi.org/10.1038/nrg2776
- Ke S, Chasin LA. Intronic motif pairs cooperate across exons to promote pre-mRNA splicing. Genome Biol 2010; 11:84; http://dx.doi.org/10.1186/gb-201011-8-r84
- Martinez-Contreras R, Fisette JF, Nasim FU, Madden R, Cordeau M, Chabot B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol 2006; 4:21; http://dx.doi.org/10.1371/journal.pbio.0040021
- Schellenberg MJ, Ritchie DB, MacMillan AM. PremRNA splicing: a complex picture in higher definition. Trends Biochem Sci 2008; 33:243 - 246; http://dx.doi.org/10.1016/j.tibs.2008.04.004
- Xiao X, Wang Z, Jang M, Burge CB. Coevolutionary networks of splicing cis-regulatory elements. Proc Natl Acad Sci USA 2007; 104:18583 - 18588; http://dx.doi.org/10.1073/pnas.0707349104
- Královicová J, Vorechovsky I. Global control of aberrant splice-site activation by auxiliary splicing sequences: evidence for a gradient in exon and intron definition. Nucleic Acids Res 2007; 35:6399 - 6413; http://dx.doi.org/10.1093/nar/gkm680
- Sharma S, Kohlstaedt LA, Damianov A, Rio DC, Black DL. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat Struct Mol Biol 2008; 15:183 - 191; http://dx.doi.org/10.1038/nsmb.1375
- Dhir A, Buratti E, van Santen MA, Lührmann R, Baralle FE. The intronic splicing code: multiple factors involved in ATM pseudoexon definition. EMBO J 2010; 29:749 - 760; http://dx.doi.org/10.1038/emboj.2009.397
- Lim SR, Hertel KJ. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J Biol Chem 2001; 276:45476 - 45483; http://dx.doi.org/10.1074/jbc.M107632200
- Cartegni L, Krainer AR. Disruption of an SF2/ASFdependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 2002; 30:377 - 384; http://dx.doi.org/10.1038/ng854
- Kashima T, Manley JL. A negative element in SMN2 exon inhibits splicing in spinal muscular atrophy. Nat Genet 2003; 34:460 - 463; http://dx.doi.org/10.1038/ng1207
- Martins de Araújo M, Bonnal S, Hastings ML, Krainer AR, Valcárcel J. Differential 3′ splice site recognition of SMN1 and SMN2 transcripts by U2AF and U2 snRNP. RNA 2009; 15:515 - 523; http://dx.doi.org/10.1261/rna.1273209
- Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci USA 2003; 100:4114 - 4119; http://dx.doi.org/10.1073/pnas.0633863100
- Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol 2003; 10:120 - 125; http://dx.doi.org/10.1038/nsb887
- Pedrotti S, Bielli P, Paronetto MP, Ciccosanti F, Fimia GM, Stamm S, et al. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J 2010; 29:1235 - 1247; http://dx.doi.org/10.1038/emboj.2010.19
- Singh NN, Androphy EJ, Singh RN. An extended inhibitory context causes skipping of exon 7 of SMN2 in spinal muscular atrophy. Biochem Biophys Res Commun 2004; 315:381 - 388; http://dx.doi.org/10.1016/j.bbrc.2004.01.067
- Chen HH, Chang JG, Lu RM, Peng TY, Tarn WY. The RNA binding protein hnRNP Q modulates the utilization of exon 7 in the survival motor neuron 2 (SMN2) gene. Mol Cell Biol 2008; 28:6929 - 6938; http://dx.doi.org/10.1128/MCB.01332-08
- Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol 2007; 5:73; http://dx.doi.org/10.1371/journal.pbio.0050073
- Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-β1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc Natl Acad Sci USA 2000; 97:9618 - 9623; http://dx.doi.org/10.1073/pnas.160181697
- Hofmann Y, Wirth B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-β1. Hum Mol Genet 2002; 11:2037 - 2049; http://dx.doi.org/10.1093/hmg/11.17.2037
- Young PJ, DiDonato CJ, Hu D, Kothary R, Androphy EJ, Lorson CL. SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2b1. Hum Mol Genet 2002; 11:577 - 587; http://dx.doi.org/10.1093/hmg/11.5.577
- Bose JK, Wang IF, Hung L, Tarn WY, Shen CK. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem 2008; 283:28852 - 28859; http://dx.doi.org/10.1074/jbc.M805376200
- Mende Y, Jakubik M, Riessland M, Schoenen F, Rossbach K, Kleinridders A, et al. Deficiency of the splicing factor Sfrs10 results in early embryonic lethality in mice and has no impact on full-length SMN/Smn splicing. Hum Mol Genet 2010; 19:2154 - 2167; http://dx.doi.org/10.1093/hmg/ddq094
- Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res 2007; 35:371 - 389; http://dx.doi.org/10.1093/nar/gkl1050
- Singh NN, Shishimorova M, Cao LC, Gangwani L, Singh RN. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol 2009; 6:341 - 350; http://dx.doi.org/10.4161/rna.6.3.8723
- Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer correctsSMN2 splicing in transgenic mice. Am J Hum Genet 2008; 82:834 - 848; http://dx.doi.org/10.1016/j.ajhg.2008.01.014
- Singh NN, Hollinger K, Bhattacharya D, Singh RN. An antisense microwalk reveals critical role of an intronic position linked to a unique long-distance interaction in pre-mRNA splicing. RNA 2010; 16:1167 - 1181; http://dx.doi.org/10.1261/rna.2154310
- Kashima T, Rao N, Manley JL. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci USA 2007; 104:3426 - 3431; http://dx.doi.org/10.1073/pnas.0700343104
- Miyajima H, Miyaso H, Okumura M, Kurisu J, Imaizumi K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J Biol Chem 2002; 277:23271 - 23277; http://dx.doi.org/10.1074/jbc.M200851200
- Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet 2009; 18:1600 - 1611; http://dx.doi.org/10.1093/hmg/ddp076
- Gladman JT, Chandler DS. Intron 7 conserved sequence elements regulate the splicing of the SMN genes. Hum Genet 2009; 126:833 - 841; http://dx.doi.org/10.1007/s00439-009-0733-7
- Miyaso H, Okumura M, Kondo S, Higashide S, Miyajima H, Imaizumi K. An intronic splicing enhancer element in Survival Motor Neuron (SMN) pre-mRNA. J Biol Chem 2003; 278:15825 - 15831; http://dx.doi.org/10.1074/jbc.M209271200
- Williams JH, Schray RC, Patterson CA, Ayitey SO, Tallent MK, Lutz GJ. Oligonucleotide-mediated survival of motor neuronprotein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J Neurosci 2009; 29:7633 - 7638; http://dx.doi.org/10.1523/JNEUROSCI.0950-09.2009
- Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 2010; 24:1634 - 1644; http://dx.doi.org/10.1101/gad.1941310
- Passini MA, Bu J, Richards AM, Kinnecom C, Sardi SP, Stanek LM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 2011; 3:72
- Rigo F, Martinson HG. Polyadenylation releases mRNA from RNA polymerase II in a process that is licensed by splicing. RNA 2009; 15:823 - 836; http://dx.doi.org/10.1261/rna.1409209
- Ip JY, Schmidt D, Pan Q, Ramani AK, Fraser AG, Odom DT, et al. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res 2011; 21:390 - 401; http://dx.doi.org/10.1101/gr.111070.110
- Mapendano CK, Lykke-Andersen S, Kjems J, Bertrand E, Jensen TH. Crosstalk between mRNA 3′ end processing and transcription initiation. Mol Cell 2010; 40:410 - 422; http://dx.doi.org/10.1016/j.molcel.2010.10.012
- Muñoz MJ, de la Mata M, Kornblihtt AR. The carboxy terminal domain of RNA polymerase II and alternative splicing. Trends Biochem Sci 2010; 35:497 - 504; http://dx.doi.org/10.1016/j.tibs.2010.03.010
- Niwa M, Berget SM. Mutation of the AAUAAA polyadenylation signal depresses in vitro splicing of proximal but not distal introns. Genes Dev 1991; 5:2086 - 2095; http://dx.doi.org/10.1101/gad.5.11.2086
- Pawlicki JM, Steitz JA. Nuclear networking fashions pre-messenger RNA and primary microRNA transcripts for function. Trends Cell Biol 2010; 20:52 - 61; http://dx.doi.org/10.1016/j.tcb.2009.10.004