
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Traditional studies of liver toxicity involve screening compounds through in vivo and in vitro tests. They need to distinguish between compounds that represent little or no health concern and those with the greatest likelihood to cause adverse effects in humans. High-throughput and toxicogenomic screening methods coupled with a plethora of circumstantial evidence provide a challenge for improved toxicity prediction and require appropriate computational methods that integrate various biological, chemical and toxicological data. We report on a data fusion approach for prediction of drug-induced liver injury potential in humans using microarray data from the Japanese Toxicogenomics Project (TGP) as provided for the contest by CAMDA 2013 Conference. Our aim was to investigate if the data from different TGP studies could be fused together to boost prediction accuracy. We were also interested if in vitro studies provided sufficient information to refrain from studies in animals. We show that our recently proposed matrix factorization-based data fusion provides an elegant computational framework for integration of the TGP and related data sets, 29 data sets in total. Fusion yields a high cross-validated accuracy (AUC of 0.819 for in vivo assays), which is above the accuracy of the established machine learning procedure of stacked classification with feature selection. Our data analysis shows that animal studies may be replaced with in vitro assays (AUC = 0.799) and that liver injury in humans can be predicted from animal data (AUC = 0.811). Our principal contribution is a demonstration that analysis of toxicogenomic data can substantially benefit from data fusion with directly and circumstantially related data sets.
Abbreviations
| CAMDA | = | Critical Assessment of Massive Data Analysis |
| DILI | = | drug-induced liver injury |
| TGP | = | Japanese Toxicogenomics Project |
| GO | = | Gene Ontology |
| AUC | = | area under the receiver operating characteristic curve |
| MF | = | matrix factorization |
| RF | = | random forests |
| LR | = | logistic regression |
| SVM | = | support vector machine |
| GBT | = | gradient boosting trees |
| PCA | = | principal component analysis |
| RSS | = | residual sum of squares |
| Evar | = | explained variance |
Introduction
Drug-induced liver injury (DILI) is the most frequent reason for drug withdrawal during early and late stages of drug development and clinical trials as well as after drugs are approved for the marketplace.Citation1 Some drugs are more likely to cause hepatic adverse events than others, and some may even lead to severe liver injuries. Development of tools for early detection of adverse effects and identification of drug's toxic potential is a major challenge within the pharmaceutical industry and clinical medicine.Citation2-4 The toxicology and drug safety evaluation communities have made great efforts in developing methodologies to assess drug toxicity risks.Citation5-7 These large-scale efforts also intend to elucidate whether animal studies can be replaced with in vitro assays and if liver injuries in humans can be predicted using toxicogenomic data from animals. Critical Assessment of Massive Data Analysis (CAMDA)Citation8 organized a challenge in 2013 to assess the performance of different analytic methods to predict the human hepatotoxic potential of drugs using the Japanese Toxicogenomics Project (TGP)Citation9 data set. The challenge aimed to foster the development of computational approaches and to promote these within the scope of tools for drug toxicity estimation.
Molecular biology abounds with data from sequencing, expression studies, function annotations, and studies of interactions between genes, proteins and drugs. These data sets are related, and analysis of one data set could benefit from the inclusion of information from others. We have recently proposed a data fusion approachCitation10 that can elegantly integrate heterogeneous data sets, representing each data set in a matrix and fusing the data sets by simultaneous matrix factorization. We here report on the fusion of 29 data sets from the TGP and related data repositories to predict DILI risk. We assess the value of combining conventional toxicogenomic data sets with circumstantial evidence for more informed prediction of adverse drug reactions and hepatotoxicity. We compare the accuracy of data fusion to that of a standard multi-classifier approach where we stack four state-of-the-art classification algorithms. We additionally investigate feature subset selection by CUR matrix decomposition applied before combining classifiers with stacking.
Results
Predictive performance of a multi-classifier approach with feature selection
Our first experiment focused on a multi-classifier approach to predict DILI risk from the preprocessed TGP microarray data. In particular, we used stacked generalizationCitation11 to combine predictions of random forests,Citation12 gradient boosting trees,Citation13 logistic regression and support vector machinesCitation14 (). We applied gene filtering to perform feature selection and to identify genes with high statistical leverage. For that we applied the CUR matrix decompositionCitation15 of the TGP microarray data sets for gene subset selection. CUR decomposition computes leverage scores for matrix columns (i.e., genes) and uses them for weighted column sampling, preferring those columns with a larger score, to assemble a low-dimensional matrix decomposition. Statistical leverage scores capture the influence of genes on the best low-rank fit of gene expression matrix. Thus, the columns selected by decomposition of the microarray data defined the reduced set of considered genes. shows the top ten genes with highest normalized statistical leverage as computed separately from animal in vitro and in vivo data.
Table 1. Predictive performance of the multi-classifier approach for DILI potential prediction with and without CUR feature subset selection (FSS)
Table 2. Genes with the highest influence on the fit of low-rank CUR decomposition of rat in vitro and rat in vivo expression data
Predictive performance of a data fusion approach
We used data fusion by matrix factorizationCitation10 to integrate various data sets. Data sets were represented as matrices, each relating objects of two types. We considered objects such as genes, gene ontology (GO) terms, drugs, and tissue samples. For instance, genes and tissue samples from rat in vivo single study were related through corresponding gene expression data matrix. Genes and drugs were related through a matrix of drug targets. All together, we considered 29 data sets that provided relations between 14 object types (). Data fusion simultaneously considers all data sets (relations) in the factorization schema and factorizes them into products of low-dimensional matrix factors, such that matrix factors are shared between relations that describe objects of the same type. Inferred latent data representation is then utilized for the prediction of DILI potential of new drugs, previously unseen by the data fusion system. Our target relation in this system was drugs’ DILI potential, which described various degrees of drug toxicity. Toxicity was provided for 101 drugs and expressed as severe, moderate or mild. In a cross-validation study, a subset of considered drugs was excluded to serve for testing of predictions of the data fusion model developed from remaining drugs and all other data sets in the factorization schema. In particular, given latent matrix factors inferred from the training data and a new drug, we estimated drug's latent profile by transforming available relations about it to inferred latent space and then used the estimated profile to predict the target relation, namely drug's DILI potential.Citation10 In that way, we avoided the unwanted information flow between the training and test sets. shows the 10-fold cross-validated accuracy for seven data fusion scenarios that considered various data sets of the complete fusion model from . The model inferred from all four TGP studies used all available data sets. Other models considered only selected toxicogenomic studies and associated non-expression data. For instance, fusion of in vivo assays omitted all data sets from in vitro studies (object types 3, 4, 7, and 8 in ).
Table 3. Predictive performance of fusing various subsets of assays for DILI potential prediction
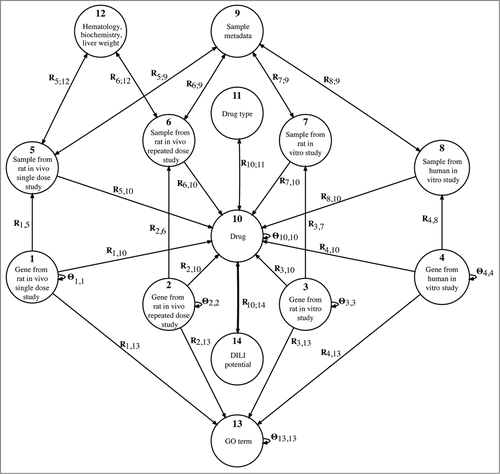
Figure 1. The fusion configuration for drug-induced liver injury prediction. Nodes represent 14 object types. Arcs denote data sets that relate objects of different types (relation matrices, Ri,j) or objects of the same type (constraints, Θi,i) for a total of 29 matrices (data sets). The bold arc (R10,14, R14,10 = RT10,14) represents relation between drugs and DILI potential that we try to augment. Fused data sets include gene annotations that are encoded in {0, 1}-matrices (R1,13, R2,13, R3,13, R4,13); expression profiles (R1,5, R2,6, R3,7, R4,8); hematology, body weight and clinical chemistry data for each rat (R5,12, R6,12, R12,5 = RT5,12, R12,6 = RT6,12); array metadata information such as dose level, dosage time and sacrifice time (R5,9, R6,9, R7,9, R8,9, R9,5 = RT5,9, R9,6 = RT6,9, R9,7 = RT7,9, R9,8 = RT8,9); drug targets (R1,10, R2,10, R3,10, R4,10); indication of medical drugs tested with samples (R5,10, R6,10, R7,10, R8,10) and structure and categorization of drugs (R10,11, R11,10 = RT10,11). Constraint matrices encode protein–protein interactions (Θ1,1, Θ2,2, Θ3,3, Θ4,4), drug interactions (Θ10,10) and the semantic structure of the Gene Ontology graph (Θ13,13).
Influence of circumstantial evidence on the quality of the fused model
We estimated the effect of circumstantial data (gene annotations, drug structural information, hematology data and sample metadata) on the quality of the fused factorized model. We observed the reconstruction quality of the target data set, which related drugs to DILI risk, through explained variance (Evar) and residual sum of squares (RSS). Better models have high Evar and low RSS. The influence of a data set was determined by observing the change in reconstruction quality of the target relation when this data set was excluded from training. Reconstruction of relation of DILI potential achieved Evar of 0.911 and RSS of 8.779 in 10-fold cross-validated study when the entire collection of data sets was considered. This quality decreased by 1.0% in Evar and 11.7% in RSS when omitting the data on hematology, biochemistry and liver weight (type 12; ) from the entire collection of data sets. In contrast, we observed a 9.6% decrease in Evar and a 12.8% increase in RSS when excluding sample metadata (type 9; ) from the collection, and a 0.7% decrease in Evar and 9.4% increase in RSS without considering related drug data (type 11; ) in the fusion. Exclusion of gene annotations (type 13; ) slightly worsened the fused model with respect to Evar (a decrease of 0.2%) but improved the RSS by 0.3%.
Discussion
From a computational perspective, the contributions of our work presented in this manuscript are 2-fold. First, we evaluated the performance of CUR matrix decomposition to select genes that exhibited high statistical leverage and employed a reduced data set using well-established classification ensemble methods. Second, we pursued a novel data fusion approach based on matrix factorization to assess the hepatotoxic risk associated with individual drugs by fusing gene expression profiles with a plethora of related and heterogeneous data sets.
In our first experiment, we considered the DILI prediction problem for each toxicogenomic study separately and pursued a multi-classifier approach (). The training data consisted of microarray profiles (independent variables) and associated drugs with given DILI potentials (dependent variable). Feature subset selection was performed independently on the training data from each fold of cross-validation and features selected by CUR matrix decomposition or constructed by PCA were then used to assess the classifier's performance on test data. Feature subset selection by CUR matrix decomposition substantially reduced the number of input features. For instance, and as averaged across folds of cross-validation, a subset of only about 300 genes was used for training the prediction models in the human in vitro study instead of the original 18,988 genes included by the FARMSCitation16 summarization. The solid performance of multi-classifier approach was not surprisingCitation17,18 as several previous studiesCitation19,20 on this data have already reported good results with single classification algorithms such as support vector machines or gradient boosting. In our case, the performance was boosted by both feature selection and classifier ensembling. Also of note is the comparable performance of data preprocessing by CUR decomposition and principal component analysis (PCA). As CUR performs feature selection rather than feature transformation, it could be a preferable procedure to identify gene biomarkers ().
Results in show that using repeated dose studies (rat, in vivo repeated dose) when forecasting the toxic potency of compounds in humans yielded more accurate models than employing single dose animal studies (rat, in vivo single dose). According to Greim et al.Citation21 and Blaauboer and Andersen,Citation22 repeated dose studies in animals represent critical data for hazard identification and risk assessment in humans. They claimed that the 28-d toxicity study, which was also used by the TGP, is the minimum requirement to evaluate the organ specific effects of compounds. Our results of the multi-classifier approach show that in the absence of such information the assessment of continuous human exposure to hazardous compounds is incomplete.
For an integrative approach that simultaneously considered all available experimental and circumstantial data, we used data fusion by matrix factorization,Citation10 an intermediate data integration approach that is able to fuse heterogeneous data sets. Intermediate integration is often the preferred integration strategyCitation23-25 as it embeds the structure of the data into a predictive model and thus often achieves higher accuracy. Data fusion surpassed the accuracy of the multi-classifier approach for predicting DILI potential in humans (). The most accurate model was inferred by fusing in vivo assays, which scored an AUC of 0.819. It is surprising that in vivo assays, which relied on an animal model, performed better than human assays, given the aim was to predict DILI potential in humans. However, Pessiot et al.Citation19 similarly observed that using in vivo animal data was more informative than using in vitro human data. Their AUC scores obtained by a linear support vector machine classifier and inferred from separate toxicogenomic studies were surpassed by those reported by our fusion-based approach.
The fusion-based model inferred from animal assays (two in vivo studies and one in vitro study) outperformed the model obtained by fusing human assays only (one human in vitro study), with the first achieving an AUC of 0.811 and the latter an AUC of 0.792. One might expect that the administration of drugs to animal models would fail to identify the risk of liver injury for drugs prescribed to humans due to differences in metabolic pathways and the current lack of suitable animal models that can reproduce human risk factors.Citation4 Our results do not confirm this hypothesis; however, differences in performance are small and further investigations seem worthwhile.
The study of influence of data sets on the reconstruction quality of target relation between drugs and DILI risk (see Results) showed that, though some data sets were small in their size, they substantially affected reconstruction of target relation. For example, sample metadata included only seven features, such as information about animal sacrifice period and dose level, yet its exclusion from data fusion resulted in a near 10% decrease in reconstruction quality of target relation. In contrast, we observed only a slight reduction in model quality when gene annotation data were omitted from the fused model despite annotation data recording associations to more than 7,000 GO terms.
Although gene expression profiling is an accepted approach for identifying drugs with potential safety problems,Citation9 our results suggest that integrating expression profiles with circumstantial data on drugs, arrays and genes can further improve predictive performance of analytic approaches and pinpoint the mechanisms that underlie drug toxicity. Our data fusion approach should be applicable to other toxicity endpoints, such as neurotoxicity, or mechanisms of action, such as regenerative hyperplasia. We anticipate that efforts in data analysis hold the promise to replace animal studies with in vitro assays and predict the outcome of liver injuries in humans using in vitro animal toxicogenomic data.
Materials and Methods
Data collections
We performed two computational experiments, one with a multi-classifier and the other with a data fusion approach. The multi-classifier approach considered gene expression data sets provided by the Japanese Toxicogenomics Project (TGP), which consisted of two in vivo studies (performed on rat cell lines) and two in vitro studies (one performed on rat and one on human cell lines). In addition to gene expression data, the data fusion approach also included data on drugs available from DrugBank (http://www.drugbank.ca), gene annotations from Gene Ontology (http://www.geneontology.org), protein–protein interactions from STRING (http://string-db.org), and hematological and clinical chemistry data for each animal and sample metadata information. Data fusion considered 14 types of objects (nodes in , e.g., genes, GO terms, or drugs) and a collection of 29 data sets, each relating a pair of object types (arcs in , e.g., gene annotations that relate genes and GO terms). We represented observations from a data source that related two distinct object types i and j in a sparse relation matrix Ri,j. For example, the matrix R1,13 encoded the annotations of genes from the rat in vivo single dose study. A data source that provided relations between objects of the same type i was represented by a constraint matrix Θi,i (e.g., Θ10,10 for DrugBank's drug interactions).
Gene expression data and sample metadata
The TGPCitation9 created a gene expression database using the Affymetrix GeneChip arrays to measure the effects of 131 chemicals, mainly medical drugs, on the liver. Approximately 20,000 samples (tissue/drug combinations) were studied both in vivo and in vitro. The in vivo study used the rat as a model organism and considered two experimental designs: a single dose study, consisting of multiple time points with multiple dose levels and a repeated dose study, consisting of multiple dose periods with multiple dose levels. The probe level intensity ratios were quantile-normalized, corrected for chemical batch effects and summarized using FARMS techniqueCitation16 to obtain expression values per genes.Citation26 Replicate measurements were collapsed to one measurement per gene, which resulted in 12,088 rat genes and 18,988 human genes. We removed samples whose corresponding chemicals were not annotated with human DILI potential and retained 4,824 samples from the rat in vivo single dose study (R1,5), 4,827 samples from the rat in vivo repeated dose study (R2,6), 2,424 samples from the rat in vitro study (R3,7) and 1,116 samples from the human in vitro study (R4,8). For each sample we considered seven metadata features (R5,9, R6,9, R7,9, R8,9), including animal sacrifice period, dose and dose level, animal age in weeks and sex type.
Histological and clinical chemistry data
Data obtained for each animal in single dose and repeated dose TGP studies included histopathology, animal weight, food consumption, hematology and blood chemistry. For each animal sample we included 41 attributes (R5,12, R6,12) describing hematology, such as the levels of monocytes and lymphocytes, biochemistry, such as the concentration of albumin (RALB), direct bilirubin (DBIL) and total bilirubin (TBIL), and body and liver weight.
Drug data
We obtained drug information data from the DrugBankCitation27 database. We related drugs to their gene targets (binary matrices R1,10, R2,10, R3,10, R4,10) and assigned structural groups (binary matrix R10,11). We considered joint adverse effects of drug pairs and DILI risk class co-membership of drugs and included them in the training set (Θ10,10). A constraint between a pair of drugs was set to (−1)ck/10−3, where k was the number of joint adverse effects of a drug pair and c indicated if the two drugs belonged to the same class of DILI risk. The DILI severity in humans was determined for 101 out of 131 drugs based on FDA-approved drug labeling.Citation2 Each drug was assigned to one of three categories resulting in 41 drugs of severe DILI concern, 51 drugs of moderate DILI concern and 8 drugs of mild or no DILI concern.
Protein–protein interaction data
We included protein–protein interactions from the STRINGCitation28 database as constraints between corresponding genes. Degrees of interaction were represented with STRING confidence scores and used to populate constraint matrices, Θ1,1, Θ2,2, Θ3,3, Θ4,4.
Gene Ontology data
We considered gene annotations from Gene Ontology (GO).Citation29 We extracted 7,056 GO terms to populate binary relation matrices R1,13, R2,13, and R3,13 with 169,816 rat gene annotations and matrix R4,13 with 288,764 human gene annotations. The hierarchical structure of GO (Θ13,13) was included by reasoning over has_part, part_of and is_a relations in the GO graph. A constraint between a pair of GO terms was set to −0.2hops, where hops was the length of the shortest path between the two GO terms.
Data fusion by matrix factorization
We applied data fusion to infer relation between drugs and DILI potential. This relation, encoded in target matrix R10,14, was observed in the context of all other data sets. Matrix
was a [0, 1]-matrix that was only partially observed. Its entries indicated the degree of membership of drugs to the three DILI severity classes. Our data fusion approach involves three main steps.Citation10 First, data are encoded in constraint and relations matrices and organized in a block-based matrix representation. In the second step, relation matrices Ri,j are simultaneously tri-factorized given constraints in Θi,i. Every relation matrix is decomposed into a product of three low-rank matrix factors, such that a relation matrix Ri,j is approximated by
Constraint matrices serve to regularize the low-rank approximations of relation matrices. The key idea of data fusion is the sharing of low-rank matrix factors between relation matrices that describe objects of common type. For instance, the latent matrix factor of drugs, G10, is shared between decompositions of all relation matrices in whose arcs point to a drug node but the matrix factor S7,10 is used only in reconstruction of the corresponding relation matrix between in vitro samples performed on rat cell lines and drugs. The resulting fused system contains factors Si,j that are specific to every relation matrix (data source) and factors Gi that are specific to every object type. Thus, low-rank matrix factors Si,j and Gi capture source- and object type-specific patterns, respectively. Finally, we use matrix factors to complete unobserved entries in relation matrices and to transform new objects to the fused latent space. We refer the reader to Žitnik and ZupanCitation10 for the derivation of factorization model and further details on computing the factorization and prediction from low-rank matrix factors. In this study, we aimed to predict the unobserved entries in R10,14. The DILI severity of d-th drug was determined as
Predictions for d-th drug in the binary classification problem of severe DILI risk against moderate or mild DILI risks were estimated by
Related matrix factorization approaches
Matrix factorization algorithms are a popular class of data analysis methods and are often used for dimensionality reduction, clustering or low-rank approximation. For example, Hochreiter et al.Citation30 proposed a factorization-inspired Bayesian approach for biclustering of transcriptomic data. In particular, nonnegative matrix factorization (NMF) algorithms, which impose the nonnegativity constraints on latent matrix factors, and their various generalizations became widely used in bioinformatics. We refer the reader to Wang and ZhangCitation31 for a comprehensive review of basic NMF algorithms and their existing modifications that can incorporate additional constraints, such as orthogonality, sparseness and preservation of local topological properties, through regularization. The approach used in this study is conceptually different from those techniques. It modifies the standard factorization formulation such that it can consider multi-relational and multi object-type data without necessitating substantial data transformation. In this way it breaks through the conventional feature-based data types and factorization of single dyadic relation. Few existing matrix factorization approaches for data integration (see Žitnik and ZupanCitation10 and references therein) can model multiple relations between the same two sets of objects (e.g., genes and drugs) or can vary object types along one dimension of data matrices. They would often require full set of pairwise relations between all pairs of object types. On the contrary, our approach can model multiple relations between multiple object types without imposing assumptions about matrix structural properties.
Multi-classifier approach and feature subset selection by CUR matrix decomposition
We employed CUR matrix decompositionCitation15 to identify a small set of information carrying genes. CUR matrix decomposition approximates target matrix A in an unsupervised manner as A ≈ CUR, where C and R are low-dimensional matrix factors that contain a subset of columns and rows from A, respectively. The advantage of CUR decomposition over some well-known low-rank matrix decompositions such as principal component analysis (PCA) or singular value decomposition (SVD) is its explicit representation in terms of a small number of actual columns and rows of target data matrix. The CUR decomposition-selected features corresponded to original gene expression profiles instead of their linear combinations as with PCA and SVD. We performed feature subset selection solely on the training set and transformed the test data to reduced feature space as defined by CUR decomposition or PCA. We then applied several state-of-the-art classifiers to predict the DILI concern in humans from the matrix factor C obtained for each toxicogenomic study separately. We used gradient tree boosting,Citation13 random forestsCitation12 and a support vector machineCitation14 with polynomial kernel to predict drug-induced toxicity. Output class probabilities generated by the classifiers were combined through stacking to compensate for classifier biases.Citation11 Stacking took as input predicted class probabilities and generalized over them with logistic regression, which increased the accuracy of the best of the individual classifiers, reduced the variance and prevented overfitting. It was shown that relatively global and simple combiners that can avoid overfitting on highly correlated input models often produce most accurate results.Citation17,32
Experimental setup
The performance of above described modeling techniques and fusion scenarios was assessed through 10-fold cross-validation and evaluated with the area under the receiver operating characteristic curve (AUC). The AUC score represents the probability that, given a pair of randomly drawn drugs from the positive and negative classes, respectively, the predictor ranks the positive drug higher than the negative drug in terms of being “positive.” The AUC is robust to class imbalance and is not biased against minority class.Citation33 In the multi-classifier approach, we considered the problem of predicting drug-induced toxicity as a binary classification of severe DILI concern against moderate or mild DILI potential. In order to compare the performance of data fusion to multi-classifier approach we casted predictions made by fusion into a binary problem as was done for the multi-classifier experiments. Feature subset selection for the multi-classifier approach was performed within cross-validation on a training data set. Parameters of the classification algorithms, such as the number of iterations and the sizes of the constituent trees in gradient boosting trees, the penalty parameter in support vector machine and the regularization term in logistic regression, were estimated through internal cross-validation on the training data. The matrix decomposition algorithm used by data fusion required a 14-tuple of factorization ranks, one value per object type, which were selected from a predefined range of values by estimating the quality of low-rank fit of target matrix
using explained variance (Evar) and residual sum of squares (RSS). Initial values of matrix factors were set uniformly at random. The algorithm terminated when the improvement in convergence of target matrix approximation between consecutive iterations measured as the Frobenius distance was below 1 × 10−5.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Slovenian Research Agency (P2-0209, J2-5480), National Institutes of Health (P01-HD39691) and European Commission (Health-F5-2010-242038).
References
- Lee WM. Drug-induced hepatotoxicity. N Engl J Med 2003; 349:474-85; PMID:12890847; http://dx.doi.org/10.1056/NEJMra021844
- Chen M, Vijay V, Shi Q, Liu Z, Fang H, Tong W. FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov Today 2011; 16:697-703; PMID:21624500; http://dx.doi.org/10.1016/j.drudis.2011.05.007
- Ju C, Reilly T. Role of immune reactions in drug-induced liver injury (DILI). Drug Metab Rev 2012; 44:107-15; PMID:22235834; http://dx.doi.org/10.3109/03602532.2011.645579
- Kaplowitz N. Avoiding idiosyncratic DILI: two is better than one. Hepatology 2013; 58:15-7; PMID:23390057; http://dx.doi.org/10.1002/hep.26295
- Dix DJ, Houck KA, Martin MT, Richard AM, Setzer RW, Kavlock RJ. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci 2007; 95:5-12; PMID:16963515; http://dx.doi.org/10.1093/toxsci/kfl103
- Yang C, Hasselgren CH, Boyer S, Arvidson K, Aveston S, Dierkes P, Benigni R, Benz RD, Contrera J, Kruhlak NL, et al. Understanding genetic toxicity through data mining: the process of building knowledge by integrating multiple genetic toxicity databases. Toxicol Mech Methods 2008; 18:277-95; PMID:20020921; http://dx.doi.org/10.1080/15376510701857502
- Shukla SJ, Huang R, Austin CP, Xia M. The future of toxicity testing: a focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov Today 2010; 15:997-1007; PMID:20708096; http://dx.doi.org/10.1016/j.drudis.2010.07.007
- Tilstone C. DNA microarrays: vital statistics. Nature 2003; 424:610-2; PMID:12904757; http://dx.doi.org/10.1038/424610a
- Uehara T, Ono A, Maruyama T, Kato I, Yamada H, Ohno Y, Urushidani T. The Japanese toxicogenomics project: application of toxicogenomics. Mol Nutr Food Res 2010; 54:218-27; PMID:20041446; http://dx.doi.org/10.1002/mnfr.200900169
- Žitnik M, Zupan B. Data fusion by matrix factorization. Submitted. Preprint available at Arxiv:1307. 0803, 2013.
- Wolpert DH. Stacked generalization. Neural Netw 1992; 5:241-59; http://dx.doi.org/10.1016/S0893-6080(05)80023-1
- Breiman L. Random forests. Mach Learn 2001; 45:5-32; http://dx.doi.org/10.1023/A:1010933404324
- Friedman JH. Stochastic gradient boosting. Comput Stat Data Anal 2002; 38:367-78; http://dx.doi.org/10.1016/S0167-9473(01)00065-2
- Cortes C, Vapnik V. Support vector machine. Mach Learn 1995; 20:273-97; http://dx.doi.org/10.1007/BF00994018
- Mahoney MW, Drineas P. CUR matrix decompositions for improved data analysis. Proc Natl Acad Sci U S A 2009; 106:697-702; PMID:19139392; http://dx.doi.org/10.1073/pnas.0803205106
- Hochreiter S, Clevert D-A, Obermayer K. A new summarization method for Affymetrix probe level data. Bioinformatics 2006; 22:943-9; PMID:16473874; http://dx.doi.org/10.1093/bioinformatics/btl033
- Džeroski S, Ženko B. Is combining classifiers with stacking better than selecting the best one? Mach Learn 2004; 54:255-73; http://dx.doi.org/10.1023/B:MACH.0000015881.36452.6e
- Pandey G, Zhang B, Chang AN, Myers CL, Zhu J, Kumar V, Schadt EE. An integrative multi-network and multi-classifier approach to predict genetic interactions. PLoS Comput Biol 2010; 6; PMID:20838583; http://dx.doi.org/10.1371/journal.pcbi.1000928
- Pessiot J-F, Wong PS, Maruyama T, Morioka R, Aburatani S, Tanaka M, Fujibuchi W. The impact of collapsing data on microarray analysis and DILI prediction. Syst Biomed 2013; 1:1-7; http://dx.doi.org/10.4161/sysb.24255
- Bowles M, Shigeta R. Statistical models for predicting liver toxicity from genomic data. Syst Biomed 2013; 1:1-6; http://dx.doi.org/10.4161/sysb.24254
- Greim H, Arand M, Autrup H, Bolt HM, Bridges J, Dybing E, Glomot R, Foa V, Schulte-Hermann R. Toxicological comments to the discussion about REACH. Arch Toxicol 2006; 80:121-4; PMID:16411136; http://dx.doi.org/10.1007/s00204-005-0039-z
- Blaauboer BJ, Andersen ME. The need for a new toxicity testing and risk analysis paradigm to implement REACH or any other large scale testing initiative. Arch Toxicol 2007; 81:385-7; PMID:17262219; http://dx.doi.org/10.1007/s00204-006-0175-0
- van Vliet MH, Horlings HM, van de Vijver MJ, Reinders MJT, Wessels LFA. Integration of clinical and gene expression data has a synergetic effect on predicting breast cancer outcome. PLoS One 2012; 7:e40358; PMID:22808140; http://dx.doi.org/10.1371/journal.pone.0040358
- Gevaert O, De Smet F, Timmerman D, Moreau Y, De Moor B. Predicting the prognosis of breast cancer by integrating clinical and microarray data with Bayesian networks. Bioinformatics 2006; 22:e184-90; PMID:16873470; http://dx.doi.org/10.1093/bioinfor-matics/btl230
- Lanckriet GRG, De Bie T, Cristianini N, Jordan MI, Noble WS. A statistical framework for genomic data fusion. Bioinformatics 2004; 20:2626-35; PMID:15130933; http://dx.doi.org/10.1093/bioinfor-matics/bth294
- Clevert D-A, Heusel M, Mitterecker A, Talloen W, G¨ohlmann H, Wegner J, Mayr A, Klambauer G, Hochreiter S. Exploiting the Japanese toxicogenomics project for predictive modelling of drug toxicity. CAMDA 2012; 2012:26-9
- Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res 2011; 39:D1035-41; PMID:21059682; http://dx.doi.org/10.1093/nar/gkq1126
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013; 41:D808-15; PMID:23203871; http://dx.doi.org/10.1093/nar/gks1094
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al.; The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat Genet 2000; 25:25-9; PMID:10802651; http://dx.doi.org/10.1038/75556
- Hochreiter S, Bodenhofer U, Heusel M, Mayr A, Mitterecker A, Kasim A, Khamiakova T, Van Sanden S, Lin D, Talloen W, et al. FABIA: factor analysis for bicluster acquisition. Bioinformatics 2010; 26:1520-7; PMID:20418340; http://dx.doi.org/10.1093/bioinformatics/btq227
- Wang Y, Zhang Y. Nonnegative matrix factorization: A comprehensive review. IEEE Trans Knowl Data Eng 2013; 25:1336-53; http://dx.doi.org/10.1109/TKDE.2012.51
- Reid S, Grudic G. “Regularized linear models in stacked generalization,” In Multiple Classifier Systems, vol. 5519, pp. 112-121. Springer Berlin Heidelberg, 2009
- Guo X, Yin Y, Dong C, Yang G, Zhou G. “On the class imbalance problem,” In Fourth International Conference on Natural Computation, ICNC’08, vol. 4, pp. 192-201, 2008