Abstract
Claudins are tight junction membrane proteins that regulate paracellular permeability to ions and solutes in many physiological systems. The electric property of claudin is the most interesting and pertains to two important organ functions: the renal and sensorineural functions. The kidney comprises of three major segments of epithelial tubules with different paracellular permeabilities: the proximal tubule (PT), the thick acending limb of Henle’s loop (TALH) and the collecting duct (CD). Claudins act as ion channels allowing selective permeation of Na+ in the PT, Ca2+ and Mg2+ in the TALH and Cl− in the CD. The inner ear, on the other hand, expresses claudins as a barrier to block K+ permeation between endolymph and perilymph. The permeability properties of claudins in different organs can be attributed to claudin interaction within the cell membrane and between neighboring cells. The first extracellular loop of claudins contains determinants of paracellular ionic permeability. While analogous to transmembrane ion channels in many ways, the biophysical and biochemical properties of claudin based paracellular channels remain to be fully characterized.
Introduction
A fundamental question in cell biology is how cells transport ions in highly complex tissue structures such as the kidney tubule, the gastrointestinal tract, the lung alveolus and the organ of Corti of the inner ear. The cells lining these ductal structures coordinate with each other to acquire the apicobasal polarity through the formation of a key cellular organelle—the tight junction (TJ) that separates the extracellular space into different compartments. The tight junction is responsible for the barrier to movement of ions and molecules between these compartments.Citation1 The TJ in thin section electron microscopy is composed of a series of direct membrane contacts.Citation2 Freeze-fracture electron microscopy reveals the membrane protein interactions at the tight junction as a branching and anastomosing reticulum of “fibrils” or “strands” on the P fracture face.Citation3 These fibrils have been demonstrated to be partly composed of integral membrane proteins directly involved in cell-cell interaction. Deep-etch microscopy and subcellular fractionation expose a matrix of densely packed globular proteins on the cytoplasmic surfaces of the junctional membranes that are detergent resistant and sandwiched between the membrane surface and the actin cytoskeleton.Citation4
The known integral membrane proteins of the tight junction include occludin (a 65 kDa membrane protein bearing four transmembrane domains and two uncharged extracellular loops),Citation5 the Junctional Adhesion Molecules (JAMs), a four-member group of glycosylated proteinsCitation6 and the claudins. A remarkably complex submembrane scaffold protein complex such as ZO-1 interacts with cytoplasmic portions of the integral proteins.Citation7
Claudins are 21–28-kDa proteins consisting of a large family of at least 26 members in humans that were first identified by Furuse et al. in 1998.Citation8 Topological analyses revealed a common structure in claudins made of four transmembrane domains, two extracellular loops, amino- and C-terminal cytoplasmic domains and a short cytoplasmic turn.Citation9 The first extracellular loop (ECL1) of claudin consists of ~50 amino acids with charged residues that contribute to paracellular ion selectivity.Citation10 The second extracellular loop (ECL2) consists of ~25 amino acids with a predicted helix-turn-helix motifCitation11 that mediate trans claudin interactions and claudin interactions with the Clostridium perfringens enterotoxin (CPE).Citation12 The C-terminal domain of claudin contains a PDZ (postsynaptic density 95/discs large/zonula occludens-1)-binding motif (YV) that is critical for interaction with the submembrane scaffold protein ZO-1 and intracellular trafficking.Citation13
Claudin mutations have serious consequences, consistent with its primary role in ion homeostasis. Claudin-1 deficient mice die within one day of birth and show a loss of the water barrier of skin.Citation14 Claudin-2 knockout mice lose salt through the kidney, accompanied by hypercalciuria and polyuria.Citation15 Targeted deletion of claudin-5, which is predominantly expressed in vascular endothelia, results in a selective increase in the blood-brain barrier to small molecules.Citation16 Knockout of claudin-11 results in male infertility and severe demyelination in the central nervous system, consistent with its function to maintain proper ion balance in Sertoli tight junctions and at the Nodes of Ranvier.Citation17 Mutations in claudin-14 cause nonsyndromic recessive deafness DFNB29, ostensibly due to a failure in ion balance in the organ of Corti.Citation18 Mutations in claudin-16 have been associated with human FHHNC syndrome (familial hypomagnesemia with hypercalciuria and nephrocalcinosis), a severe renal disease due to uncontrolled loss of serum Mg2+ and Ca2+.Citation19
Electric Properties of Claudin
The electric properties of claudin are defined by its ability to alter the ion permeability and selectivity of the tight junction. Measurement of paracellular permeability using cell membrane impermeable tracers indicates that there are 7−8 Å diameter size-selective pores in the tight junction that allow passage of small charged or uncharged solutes.Citation20-Citation22 Most inorganic ions are permeable through the tight junction, including major extracellular ions – Na+, K+, Cl−, Ca2+ and Mg2+. In a non-selective epithelium, the paracellular conductance represents the overall permeability of tight junction to all ions present in the extracellular space. The conductance of tight junction (GTJ) is the reciprocal of its resistance (RTJ) that can be determined using a direct current (DC) circuit according to Ohm’s law (). A more accurate measurement takes cell membrane capacitance into account by using an alternating current (AC) circuit (). An alternating current (I) with an angular frequency (ω) generates an oscillating potential (E) across the tight junction with the same frequency but different phase. The impedance (ZTJ), deriving from E/I and its reciprocal (1/ZTJ) reflect tight junction conductance when ω approaches zero (). Numerous recordings have led to an important conclusion: the permeability of an ion across the tight junction is significantly different from its free-water mobility. The paracellular transport is not a simple diffusion but requires interaction and facilitation from proteins in the tight junction. Claudin is the primary factor underlying the conductance process. The best example is claudin-2. Amasheh et al. showed that ectopic expression of claudin-2 in high-resistance MDCK I cells increased paracellular conductance by over 20-fold.Citation23 The tight junction also demonstrates selectivity allowing permeation of only a small number of ions. The paracellular ion selectivity clearly depends on claudins. For example, overexpression of claudin-16 in anion selective LLC-PK1 cells reversed the tight junction selectivity to cation.Citation24 The structural basis for paracellular ion selectivity is encoded in the ECL1 of claudins. Through a series of chimera studies, Colegio et al. showed that claudin-4 adopted the ion selectivity of claudin-2 when the ECL1 domains of claudin-2 and -4 were swapped.Citation25 Yu et al. proposed a single-pore model to explain the observed ion selectivity of claudin-2, in which the pore is a cylinder with conical vestibules and charged side chains from ECL1 positioned facing into the lumen and electrostatically interacting with permeating ions.Citation26 While Yu’s model well explained the cation over anion selectivity of claudin-2, emphasizing a role of the effective charge in its extracellular domain, the paracellular conductance appeared not solely dependent upon the extracellular charge. In addition to ion permeability and selectivity, claudins demonstrate pH dependence, thermodynamics and anomalous mole fraction effects similar to conventional ion channels,Citation20 which led to a new concept of claudins forming the paracellular channel, a novel class of channels oriented perpendicular to the membrane plane and joining two extracellular compartments.
Figure 1. Equivalent electrical circuits of transepithelial recording methods. (A) A DC circuit records transepithelial resistance RTJ and Rcell arranged in parallel. When Rcell > > RTJ, transepithelial conductance is dictated by the tight junction. (B) An AC circuit takes cell membrane capacitance into account. An alternating current (I) with an angular frequency (ω) generates an oscillating potential (E) across the epithelia. The impedance (ZTJ), deriving from E/I, reflects transepithelial resistance under different frequencies. (C) Nyquist diagram (plot of the real and the imaginary axis of the impedance, Zre, Zim). At low frequencies (ω -> 0), Zre approaches RTJ. The capacitance C can be calculated from the frequency at which |Zim| reaches a maximum value: C = 1 / (ω|Zim|max•RTJ). (D) Schematic of relative position of an SICM nanopipette probe to a membrane pore. When the nanopipette is positioned over the membrane pore, the pipette and pore resistances are divided by the access resistance to result in a measured change in potential on the pipette that depends on the conductance of the pore in the membrane. PE: pipette electrode, RE: reference electrode and WE: working electrode.
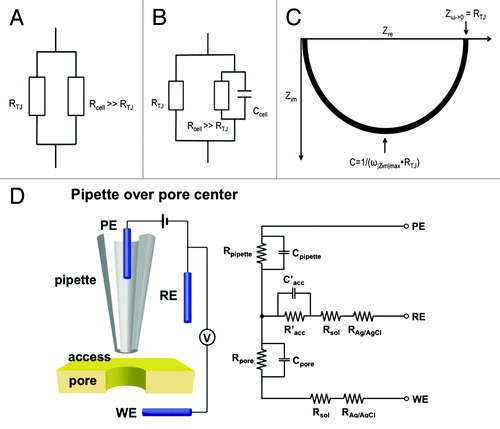
The existence of paracellular channels in the tight junction has long been debated, largely due to the lack of recording technology that allows definitive separation paracellular from transcellular conductance. Conductance scanning, first introduced by Frömter in 1972, is based on the analysis of local differences in current density above the surface of an epithelial monolayer, allowing quantitative determination of the size and distribution of local conductivity.Citation27 The limiting factor is spatial resolution. Earlier efforts had to rely upon optic lens to position the recording probe that neither provided sufficient lateral resolution nor allowed precise control of probe-surface distance.Citation28,Citation29 A breakthrough was made by Chen et al. in 2013 by using scanning ion conductance microscopy (SICM) to record the paracellular conductance in MDCK cells.Citation30 SICM makes use of a “virtual seal” to monitor the access resistance of the gap between the probe and surface (Racc) ().Citation31 Racc in the PE-RE circuit can be recorded to draw a topographic image of the surface and position the probe in a fixed distance above surface. When the pipette resistance (Rpipette), access resistance (Racc) and resistance of a conducting pore (Rpore) in a membrane are of comparable scale (order of magnitude), the conductance of the pore (Gpore = 1/Rpore) can be separated through a circuit PE-WE. In MDCK cell monolayers, Chen et al. recorded local conductance over an area within nominal radii of 265nm at discrete locations: cell junction (for paracellular pathway) and cell center (for transcellular pathway).Citation30 The recorded paracellular conductance differed significantly from the transcellular conductance in size and distribution. Deletion of claudin-2 gene abolished the paracellular conductance without affecting the transcellular conductance. Further improvements to increase the spatial resolution to below 10 nm will allow capture of single paracellular channel conductance, open probability, ion selectivity and most importantly the structure-function relationship.
Claudin Interaction Plays More Than an Adhesion Role
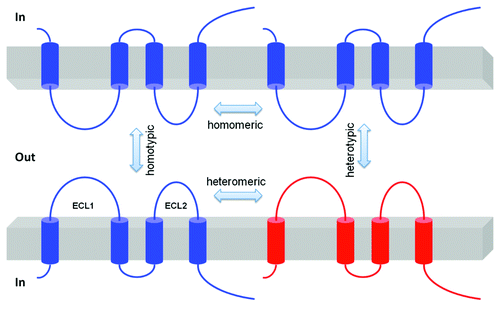
Claudins associate by cis interactions within the plasma membrane of the same cell into dimers or higher oligomeric statesCitation32,Citation33 and by trans interactions between claudins in adjacent cells. Additional cis interactions likely assemble claudin oligomers into tight junction strands. The cis interaction can involve a single type of claudin (homomeric interaction) or different types of claudins (heteromeric interaction); the trans interaction can occur in homotypic or heterotypic mode ().Citation34 Earlier anatomical studies revealed the membrane protein interactions at the tight junction as a branching and anastomosing reticulum of “fibrils” or “strands” on the freeze-fracture face.Citation3 These fibrils had been hypothesized to be partly composed of integral membrane proteins directly involved in cell-cell adhesion. Using the fibroblast cells that normally have no tight junction, Furuse et al. have demonstrated that ectopically introduced claudins were able to form well-elaborated strands at cell-cell junctions that resemble the tight junction strands.Citation34 It was clear at that time that claudin trans interaction played a crucial adhesion role. Co-culture experiments revealed that trans interactions of claudin-1 and -3 were permitted, but that interactions between claudin-1 and -2 were not observed, demonstrating considerable selectivity in heterotypic claudin-claudin interactions.Citation34 The ECL2 domain is required for trans claudin interaction. Whereas claudin-5 heterotypically interacts with claudin-3, but not with claudin-4, chimeras exchanging the ECL2 of claudin-4 with that of claudin-3 confer the ability to bind to claudin-5.Citation35 Mutagenesis has identified a locus of amino acids in ECL2 (F147, Y148, Y158) critical for homotypic claudin-5 interaction.Citation11 The role of cis claudin interaction in tight junction strand formation was not evident until the discovery made by Hou et al. in 2009 demonstrating that genetic ablation of either claudin-16 or claudin-19 prevents the assembly of both claudins into tight junction strands.Citation36 Although no data are available to demonstrate the critical loci for cis interaction within claudin-16 or -19 molecules, the extracellular loops (ECL1 and ECL2) appear not to be involved.Citation33
Figure 2. Schematic presentation of interactions between claudin molecules. The cis interaction includes homomeric or heteromeric interaction; the trans interaction includes homotypic or heterotypic interaction.
Beyond the structural role played in tight junction formation, claudin interactions directly influence paracellular ion conductance. When expressed alone in the LLC-PK1 epithelial cells, claudin-16 functions as a Na+ channel whereas claudin-19 functions as a Cl− blocker.Citation37 Co-expression of claudin-16 and claudin-19 generates a cation channel with its ion selectivity higher than that of each claudin combined.Citation37 Disruption of claudin-16 and -19 cis interaction abolishes this synergistic effect, rendering channel ion selectivity similar to that of claudin-16 or -19 alone.Citation37 A third claudin – claudin-14 was later found to interact with claudin-16 and negatively regulate its cation selectivity.Citation38 In triply transfected LLC-PK1 cells, claudin-14 integrates into the claudin-16/19 channel to form a higher oligomeric complex with novel physiological signature.Citation38 Preliminary biochemical studies of claudin-4 in insect Sf9 cellsCitation32 and of claudin-5 in human fibroblast NIH/3T3 cellsCitation39 show that claudins preferentially form hexamers via cis interaction. It is not known by what stoichiometry claudin-14, -16 and -19 oligomerize in the tight junction. Another hypothesis of claudin cis interaction is the regulation of trafficking. In a number of epithelial cell models, claudin-16 was found to co-localize with claudin-19 subcellularly in the endoplasmic reticulum (ER), the endosome or lysosomes and the plasma membrane.Citation37 Knockout of claudin-16 rendered claudin-19 mis-localization to intracellular vesicles in adult kidney tubules.Citation36 The dependence of TJ localization on claudin cis interaction is even more evident in the case of claudin-4 and -8.Citation40 Depletion of claudin-8 in mouse collecting duct cells blocked claudin-4 trafficking to the tight junction. Claudin-4 was confined to the ER and the Golgi apparatus.Citation40 By contrast, the loss of trans interaction between claudin-5 in neighboring cells reduced tight junction strand density, even though cis interaction remained intact, compatible with a primary role of trans interaction in cell junctional adhesion.Citation11
Claudins are known to interact with the TJ peripheral proteins such as ZO-1, ZO-2, ZO-3 and MUPP1 through the PDZ (postsynaptic density 95/discs large/zonula occludens-1)-binding motif (YV) located at the carboxyl end.Citation13 The interaction appears to be important for claudin localization to the tight junction. For example, Müller et al. have identified a mutation in the PDZ binding motif (TRV mutated to RRV) of claudin-16 from patients of the FHHNC syndrome, which caused claudin-16 mis-trafficking to the lysosome due to a selective loss of its binding affinity with ZO-1.Citation41 In a more general study, Umeda et al. have knocked out both ZO-1 and ZO-2 in mammary epithelia and demonstrated their functional requirement for de novo TJ formation and claudin localization.Citation42 The model of ZO-1 facilitated claudin localization was soon contested by Hou et al. using double claudin knockout animals.Citation36 Despite normal distribution of ZO-1 and occludin in claudin-16 or -19 KO animal, loss of either claudin gene rendered mis-localization of both claudin proteins on the level of tight junction, suggesting a ZO-1 independent mechanism for claudin trafficking.Citation36
The Channel Theory of Claudin in the Kidney
The Kidney functions by initially excreting many salts and small molecules found in the blood, then selectively taking back those that need to be conserved while allowing others to be excreted in the urine.
The traditional view of the renal reabsorption process is that of a tandem array of ion channels and transporters located in the plasma membrane of tubular epithelial cell conducting ion transport in a coordinated manner at the expense of energy. While many mutations have been found in these transmembrane ion channels or transporters causing human diseases of electrolyte imbalances, there are growing numbers of cases caused by mutations in the claudin genes, pointing to a new mechanism by which the kidney utilizes the paracellular channel to conduct ion transport. It should also be emphasized that in addition to an independent transport route, the paracellular pathway provides a feedback mechanism to coordinate with the transcellular pathway and form a complete electric circuit.
There are segment-specific claudin expression profiles along the length of the nephron. Several studies using probes specific for claudin-1–19 mRNAs and proteins reveal that claudin-6, -9 and -13 are not detectable, and claudin-5 and -15 only in endothelial cells; the rest are specifically expressed in different segments of the nephron.Citation36,Citation38,Citation43,Citation44 While claudin-3, -10, -11, -14, -16 and -19 were observed in the thick ascending limb of Henle’s loop (TALH) and claudin-3, -4, -7 and -8 in the distal tubules and the collecting duct, claudin-2, -10 and -17 is highly expressed in the “leaky” proximal tubule consistent with its high cation permselectivity when expressed in MDCK cells.Citation23 Claudin-4 and -8 are expressed primarily along the entire “tight” distal nephron and in inner medullary segments of the thin descending limbs of juxtamedullary nephrons. This pattern of expression is consistent with their role as paracellular cation barrier.Citation45,Citation46 Immunofluorescence analysis has shown that claudin-7 is expressed in the TALH and the collecting ducts of porcine and rat kidneys,Citation47 although another study described claudin-7 in the distal nephron as located primarily on the basolateral membrane.Citation48 Claudin-14, -16 and -19 are expressed exclusively in the TALH where the tight junction has low resistance but high cation selectivity.Citation36,Citation38 In summary, while there are still some conflicting published data, claudin-2, -10 and -17 are expressed in proximal tubules, while claudin-3, -4, -7, -8, -10, -14, -16 and -19 are found in the distal nephron including the TALH, the distal tubule and the collecting duct. It is also clear that the patterns of claudin expression along the nephron changes with development, with claudin-7 and -8 upregulated postnatally.Citation49
A single-pore model in the proximal tubule
The proximal tubule of the kidney is a leaky epithelium that reabsorbs up to 60% of the filtered Na+ and Cl− load as well as 60–65% of filtered Ca2+ and 25–30% of filtered Mg2+. Almost half of the NaCl is reabsorbed through the paracellular pathway.Citation50 The driving force for paracellular reabsorption is not clear. Unlike the transcellular pathway that utilizes ion transporters in the membrane at the expense of energy, the paracellular pathway is passive and only depends upon the electrochemical gradient across the epithelium. A prevailing hypothesis is that paracellular reabsorption of NaCl is driven by the Cl− electrochemical gradient that builds up owing to preferential reabsorption of HCO3− over Cl−.Citation51 The early proximal tubule reabsorbs Na+ via the Na+/H+ exchanger (NHE3) and HCO3− via the Na+/HCO3− co-transporter (NBC). This makes the luminal fluid delivered to the mid to late proximal tubule low in HCO3− concentration but higher in Cl− concentration compared with the levels in the plasma. Here, the lumen to basolateral Cl− concentration gradient drives paracellular Cl− reabsorption and in turn generates a lumen-positive potential.Citation52 This electrical gradient then drives paracellular reabsorption of Na+ ().
Figure 3. Na+ transport mechanism in the proximal tubule. (A) In the early portion of the proximal tubule, both Na+ and HCO3− are reabsorbed via the Na+/H+ exchanger NHE3 on the apical membrane and a Na+-HCO3− cotransporter (NBC) on the basolateral membrane. This process increases the Cl− concentration in late proximal luminal fluid, driving passive paracellular Cl− reabsorption down the chemical gradient of Cl−. Paracellular Cl− reabsorption creates a lumen-positive voltage, which then drives paracellular reabsorption of Na+ through claudin-2. (B) A single pore model of claudin-2 channel that is made of two negatively charged carboxyl groups on the residue aspartate-65 of ECL1 from two apposing claudin molecules in adjacent cells.
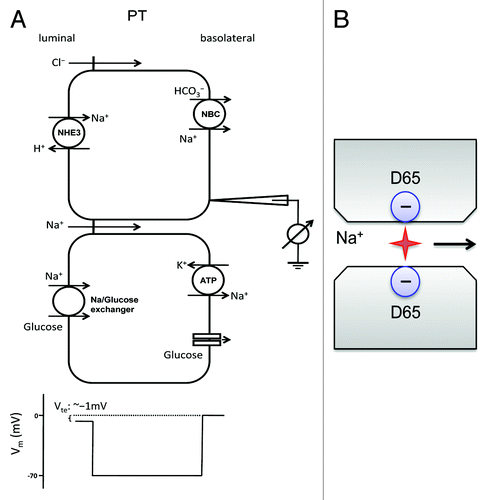
Claudin-2, which acts as a paracellular cation pore, is highly expressed in the proximal tubule. Knockout of claudin-2 decreases the paracellular cation selectivity of the proximal tubule, causing urinary loss of both Na+ and Cl− when fed with high-salt diet.Citation15 The molecular insight of claudin-2 channel permeability is provided by Yu et al.Citation26 Using Brownian dynamics, Yu’s single-pore model suggests that the channel pore of claudin-2 is made of two protruding carboxyl groups on the residue aspartate-65 of ECL1 from two apposing claudin molecules in adjacent cells (). The electrostatic field generated by the apposing carboxyl groups is predicted to be cylindrical and symmetrical. Ablating the charge on aspartate-65 by mutation to serine indeed abolished the cation selectivity of claudin-2 and reduced its permeability to Na+ (PNa). Yu’s model also predicts a single-pore conductance of 70 pS at physiological Na+ concentrations. Ca2+ permeability, on the other hand, is predicted to be low owing to its high activation energy barrier for dehydration. Paradoxically in claudin-2 knockout animals, urinary loss of Ca2+ is much more pronounced than Na+, suggesting claudin-2 channel is prone to divalent cation.Citation15 In fact, extracellular Ca2+ competes with Na+ and inhibits Na+ permeability in transfected claudin-2 channels.Citation53 It appears that aspartate 65 has higher than predicted charge density, thus favoring binding of Ca2+ over that of Na+. This hypothesis was soon corroborated by van Itallie et al. in a biochemical study demonstrating that claudin-2 forms a homodimer through cis-interaction of the second transmembrane domain.Citation33
The molecule underlying paracellular Cl− transport remains elusive. In addition to claudin-2, the proximal tubule also expresses claudins 10a,Citation54 12,Citation55 and 17.Citation44 Claudin-10a and -17 can function as anion-selective pores in vitro and may be responsible for paracellular Cl− reabsorption in the proximal tubule.Citation44,Citation54 In intestinal epithelium, claudin-12 interacts with claudin-2 and functions as a Ca2+-selective pore.Citation56 This claudin interaction may play a similar role in paracellular Ca2+ reabsorption in the proximal tubule.
An anti-porter model in the loop of Henle
The thick ascending limb of the loop of Henle (TALH) reabsorbs 25–40% of filtered Na+, 50–60% of filtered Mg2+ and 30–35% of filtered Ca2+. The TALH actively transports Na+ and Cl− via the transcellular route and provides a paracellular pathway for reabsorption of Mg2+ and Ca2+.Citation57,Citation58 Na+, K+ and Cl− enter the cell through the Na+-K+-2Cl− cotransporter (NKCC2) in the luminal membrane. Na+ exits the cell through the Na+/K+-ATPase in exchange for K+ entry. K+ is secreted into the lumen through the renal outer medullary potassium channel (ROMK). Cl− leaves the cell through a basolateral Cl− channel (ClC-kb and barttin). Mg2+ and Ca2+ are passively reabsorbed through the paracellular channel, driven by a lumen-positive transepithelial voltage (Vte) (). The generation of this lumen-positive Vte can be attributed to two mechanisms: (a) the active-transport Vte due to apical K+ secretion and basolateral Cl− exit and (b) the diffusion Vte generated because of the transepithelial NaCl concentration gradient and the cation selectivity of the paracellular channel in the TALH.Citation59 Because the TALH expresses no aquaporin, continuous NaCl reabsorption generates a diluted luminal fluid that facilitates the generation of a NaCl concentration gradient from basolateral down to lumen. The backflux of NaCl through a cation selective paracellular pathway generates a lumen-positive diffusion potential according to the Goldman-Hodgkin-Katz equation. This diffusion Vte is the major driving force of Mg2+ and Ca2+ reabsorption.
Figure 4. Ca2+ and Mg2+ transport mechanism in the thick ascending limb of Henle’s loop (TALH). (A) NaCl is reabsorbed through a concerted action of the Na+-K+-2Cl− cotransporter (NKCC2), the renal outer medullary potassium channel (ROMK) and basolateral Cl− channel (ClC-kb and barttin). Because water is not reabsorbed in the TALH, continuous NaCl reabsorption generates a diluted luminal fluid that facilitates the generation of a NaCl concentration gradient from basolateral down to lumen. The backflux of Na+ through a cation selective paracellular channel generates a lumen-positive diffusion potential that drives Mg2+ and Ca2+ reabsorption. (B) An anti-porter model of claudin-16 channel that is made of two apposing hemi-channers each with five negatively charged residues in ECL1 protruding into the junctional space.
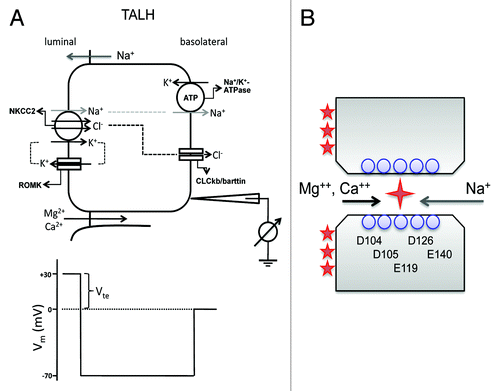
The molecular basis for TALH divalent cation reabsorption remained unknown until the discovery of mutations in claudin-16 and -19 as the cause of the rare autosomal recessive renal disorder FHHNC without ocular involvement (OMIM#248250)Citation19 and with ocular involvement OMIM#248190),Citation60 respectively. FHHNC is characterized by uncontrolled loss of serum Mg2+ and Ca2+ through the kidney. Renal immunohistological studies reveal that claudin-16 and claudin-19 are exclusively localized in the TALH.Citation36 It was initially hypothesized that claudin-16 formed a selective paracellular Mg2+ and Ca2+ channel. Several studies tested this hypothesis in vitro but only came to ambiguous conclusion. The mild effect and peculiar unidirectional rectification of transfected claudin-16 in MDCK cells precluded definitive conclusion.Citation61,Citation62 Hou et al. transfected a model cell line, LLC-PK1, that has low endogenous paracellular cation permeability and found that claudin-16 profoundly increased Na+ permeability (PNa) accompanied by only moderately enhanced Mg2+ permeability (PMg).Citation24 PNa was greatly reduced or completely disappeared in all FHHNC disease mutants of claudin-16, suggesting that changes in PNa, rather than changes in PCa or PMg, are pathogenic for this disease.Citation24 Claudin-16 deficient KD mice showed significantly reduced plasma Mg2+ levels, increased urinary excretion of Mg2+ and Ca2+ and nephrocalcinosis.Citation63 This phenotype recapitulated that of FHHNC. Consistent with the hypothesis that claudin-16 acts primarily as a PNa pathway, when TALH segments were isolated and perfused ex vivo, the paracellular ion selectivity (PNa/PCl) of the TALH was significantly reduced in claudin-16 deficient mice.Citation63 The structural insight of claudin-16 function was provided by Hou et al.Citation24 Unlike claudin-2 that is dependent upon one charged amino acid in ECL1 to permeate Na+, claudin-16 appears to require a string of negatively charged residues interspersed along ECL1 (D104S, D105S, E119T, D126S and E140T). Mutation of each of the five functionally important residues had a modest effect (11–33% reduction in PNa), and combining the mutations generated additive effects.Citation24 Because in the TALH reabsorption of Mg2+ and Ca2+ depends upon backflux of Na+ in order to generate the diffusion potential, claudin-16 must be able to permeate Mg2+ or Ca2+ with Na+ at the same time but in opposite direction (). This anti-porter mode requires that Mg2+ or Ca2+ does not inhibit Na+ permeation by blocking the channel pore as in the case of claudin-2. Because of the size selectivity of claudin channel, it is less likely that the claudin-16 channel pore allows simultaneous occupation of two cations. Because Mg2+ and Ca2+ concentrations are far less than Na+ on both sides of the epithelia, an alternation mechanism allowing absorption of divalent cations will not affect the overall level of Na+ backflux or diffusion potential.
The other major cause of FHHNC is mutations in claudin-19. Hou et al. found that claudin-19 profoundly decreased Cl− permeability (PCl) and functioned as a Cl− barrier when expressed in LLC-PK1 cells.Citation37 The FHHNC mutations from human patients either partially or completely abolished the claudin-19 effects on PCl. Coexpression of claudin-16 and claudin-19 in LLC-PK1 cells resulted in the simultaneous upregulation of PNa and downregulation of PCl and hence in a large increase in PNa/PCl, generating a highly cation-selective paracellular pathway.Citation37 Mutations in claudin-19 allows backflux of Cl−, which neutralizes the positive potential of the tubular lumen and eliminates the driving force for Mg2+ and Ca2+ reabsorption. In addition to demonstrating cooperativity between two different claudins, these data also suggest that, whereas some claudins form pores that allow preferential passage of specific ions, other claudins may contribute to barriers that prevent passage of specific ions. Claudin-19 KD animals phenocopy claudin-16 KD animals and develop the FHHNC manifestations of reduced plasma Mg2+ levels and excessive renal wasting of Mg2+ and Ca2+.Citation36 The phenotypic similarities of claudin-19 KD with claudin-16 KD can be explained by the cis heteromeric interaction between the two claudins.Citation37 Several FHHNC mutations in claudin-16 and claudin-19 disrupted this interaction and abolished their cation selectivity when coexpressed in LLC-PK1 cells, suggesting a role for claudin interaction in the development of FHHNC.Citation37
A recent genome-wide association study (GWAS) identified claudin-14 as a major risk gene for hypercalciuric nephrolithiasis and bone mineral loss.Citation64 Gong et al. revealed predominant gene expression of claudin-14 in the TALH of the kidney.Citation38 Using several biochemical and genetic criteria, Gong et al. show that the claudin-14 protein interacts with claudin-16 but not with claudin-19. In transfected LLC-PK1 cells, claudin-14 diminishes the cation permeability of the claudin-16 channel.Citation38 Thus, claudin-14 physiologically binds to the claudin-16/claudin-19 complex and acts as a negative regulator of divalent cation reabsorption in the TALH. Consistent with this, claudin-14 knockout mice developed hypermagnesemia, hypomagnesiuria and hypocalciuria on a high Ca2+ diet, exactly the opposite phenotype of that of claudin-16 KD mice.Citation38
Claudin-10b, which is also expressed in the TALH, functions as a cation channel in cultured epithelial cells.Citation54 Though it does not interact with claudin-16,Citation36 claudin-10b appears to play a negative regulatory role in paracellular Ca2+ and Mg2+ reabsorption in the TALH. Knockout of claudin-10b in the kidney generated a phenotype of hypermagnesemia, hypocalciuria and nephrocalcinosis.Citation65 In ex vivo perfused TALH tubules, loss of claudin-10b decreased PNa, and to lesser extent decreased PCa and PMg,Citation65 consistent with Hou et al.’s hypothesis that the paracellular channel in the TALH is Na+ selective rather than Mg2+ or Ca2+ selective.Citation24 The disproportionately smaller effect on divalent cation suggests that claudin-10b does not contribute to the conducting pore of the paracellular channel.Citation26 The furosemide-inhibitable Vte increased dramatically in claudin-10b KO TALH, pointing to an unknown mechanism that coupled transcellular to paracellular pathways.Citation65 Nevertheless, a definitive conclusion of claudin-10b function in vivo would have to come from a comprehensive study of claudin-10b in claudin-14 KO background, because loss of claudin-10b induced an over 20-fold increase in claudin-14 gene expression.Citation65
A chloride shunt in aldosterone sensitive distal nephron
The aldosterone-sensitive distal nephron (ASDN) encompasses the distal convoluted tubule (DCT), the connecting tubule (CNT) and the collecting duct (CD). It is collectively responsible for the reabsorption of approximately 5% of filtered NaCl and plays a vital role in the regulation of extracellular fluid volume (ECFV) and blood pressure.Citation66 The Na+ reabsorption in the CNT and the CD is driven by apical entry through the epithelial Na+ channel (ENaC) and by basolateral efflux through the Na+/K+-ATPase and in the principal cell.Citation67 The unidirectional reabsorption of Na+ generates a lumen-negative transepithelial potential (Vte) of −25mV. The Cl− transport in the CNT and the CD depends on anion channels or transporters located in both principal cells and intercalated cells, dictated by two independent cellular mechanisms. The transcellular mechanism utilizes the Na+-driven Cl– bicarbonate exchanger (NDCBE; encoded by the Slc4a8 gene) in the apical membrane of β-intercalated cells to reabsorbs Na+ and HCO3− in exchange of Cl− secretionCitation68 and the pendrin exchanger (encoded by the Slc26a4 gene) in the apical membrane of β-intercalated cells to takes back Cl− in exchange of HCO3− secretion.Citation69 The net result is an equal quantity of Cl− and Na+ reabsorbed with no effect on Vte. The paracellular mechanism depends upon the lumen-negative Vte and a Cl− conductance in the tight junction (“chloride shunt”) (). In principle, the level of Cl− reabsorption must match that of Na+ reabsorption so as to maintain luminal fluid electroneutrality, because the NDCBE–pendrin-mediated transcellular route carries no net charge. Conversely, defects in Cl– transport will increase the magnitude of Vte, depolarize the luminal membrane and subsequently inhibit ENaC.
Figure 5. Cl− transport mechanism in the collecting duct. (A) Na+ is reabsorbed through two parallel transcellular pathways: the amiloride sensitive ENaC pathway and the thiazide sensitive NDCBE–pendrin pathway. The unidirectional reabsorption of Na+ generates a lumen-negative transepithelial potential (Vte) of −25mV that drives paracellular Cl− reabsorption. (B) An anion pore model of claudin-4 channel that is made of two positively charged amine groups on the residue lysine-65 of protruding ECL1, a locus homologous to aspartate-65 in claudin-2.
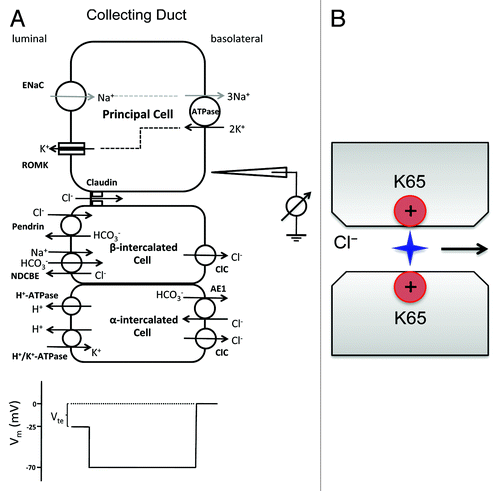
The molecular mechanism of chloride shunt has been revealed by a systematic study of claudin function in the collecting duct. Several claudins, including claudin-3, -4, -7 and -8, are expressed in the collecting duct.Citation43,Citation48 Using RNA interference, Hou et al. found that loss of claudin-4 significantly decreased paracellular anion permeabilities, including PCl, in cultured collecting duct cells (M-1 and mIMCD3).Citation40 Mutagenesis has identified a key amino acid – lysine-65 in ECL1 of claudin-4 required for Cl− permeability ().Citation40 Lysine-65 in claudin-4 is homologous to aspartate-65 in claudin-2. This positively charged residue confers anion selectivity to claudin channel pore in a similar Brownian model. A direct support for claudin-4’s role in chloride shunt comes from its KO study. Constitutive knockout of claudin-4 resulted in higher urinary excretion of Cl− as well as Ca2+.Citation70 Increased urine volume, accompanied by urothelial hyperplasia, caused urinary tract obstruction and hydronephrosis as animals aged. Claudin-4 was found in the TJs of the ureter in addition to the DCT, the CNT and the CD.Citation70 Despite the electrolyte imbalances, the morphology of TJ in urothelia and renal epithelia appeared to be normal.Citation70 Claudin-8 also plays a potentially important role in collecting duct PCl. In both M-1 and mIMCD3 cells, claudin-8 KD decreased PCl to a level similar to that in claudin-4 KD.Citation40 Claudin-8 does not appear to make an individual channel to permeate Cl−. Instead, it interacts with claudin-4 and recruits claudin-4 to the tight junction.Citation40 Loss of claudin-4 in KO animals reduced the TJ localization level of claudin-8, providing in vivo support for their interaction.Citation70
To determine the role of claudin-3 and -7, Hou et al. knocked down their expression in collecting duct M-1 and mIMCD3 cells.Citation40 Whereas claudin-3 KD showed no significant effect on PNa or PCl, claudin-7 KD resulted in a 30% decrease in TER, with no significant change in ion selectivity. Consistent with this, claudin-7 knockout mice exhibited severe renal wasting of Na+, K+, Cl− and water, accompanied by increased aldosterone synthesis, and died within 12 d after birth.Citation71 The renal loss of Na+ and Cl− suggests that claudin-7 acts as a nonselective paracellular ion barrier to prevent backleak of interstitial NaCl through the collecting duct. The tight junction localization of claudin-4 is not affected in claudin-7 knockout mice, consistent with the biochemical results that claudin-3 and -7 do not interact with claudin-4.Citation71
The pathophysiological role of chloride shunt in the distal nephron has been the focal point of extensive investigation over the past decades. Abnormal increases in chloride shunt conductance will cause salt retention and ECFV expansion, while decreasing K+ and H+ secretion. Phenotypic manifestation of chloride shunt derangement includes hyperkalemia, hyperchloremia, hypertension and metabolic acidosis, clinically known as the chloride shunt syndrome or pseudohypoaldosteronism type II (PHAII).Citation72 Recent genetic studies have identified the causative genes for PHAII – WNK kinases.Citation73 One of the WNKs, WNK4, is present in the tight junction of the collecting duct.Citation73 The PHAII-related mutations in WNK4 increase paracellular Cl− conductance in transfected MDCK cells, compatible with the original hypothesis that a gain-of-function in chloride shunt conductance causes PHAII.Citation74 Nevertheless, transgenic animals expressing a gain-of-function mutation in WNK4 failed to show any increase in transepithelial Cl− permeation in the collecting duct.Citation75 In ex vivo perfused cortical collecting ducts, the tight junction was only slightly anion-selective (PCl/PNa: 1.64 in presence of amiloride),Citation75 not substantially different from the ratio of their free-water mobilities (1.54). WNK4 stimulation increased amiloride sensitive Na+ current but not the paracellular anion permselectivity,Citation75 contrasting with the in vitro data.
The Barrier Theory of Claudin in Sensory Systems
In contrast with the kidney, the functions of sensory organs rely upon highly specialized signal transduction mechanisms employed by neurons and receptor cells, which require an independent environment well insulated from general circulation. Tight junctions fulfill such a role. The claudins made by the sensory system therefore must play a very different role from those in the kidney. Interestingly, some claudins play a conserved role from renal to sensory systems while others are unique to each organ. On the basis of whether their expression increases or decreases TER in epithelial recordings, claudins are classified as channel or barrier claudins. This loose categorization helps to separate the renal from neural claudins.
A K+ barrier in the organ of Corti
The cochlea of the inner ear has two compartments with different ionic composition (). The perilymph of the scala vestibule and scala tympani is low in K+ concentration but high in Na+ concentration, similar to the cerebrospinal fluid.Citation76 The endolymph of the scala media is unusually rich in K+ (~150mM) and is held at a positive potential of +80 to +100 mV relative to the perilymph.Citation77 The endocochlear K+ and potential levels are critical for the depolarization of sensory hair cells in the organ of Corti. Hearing depends upon two types of mechanosensitive hair cells – inner hair cell (IHC) and outer hair cell (OHC). Both types of hair cells respond to movements of their apical hair bundles by modulation of cation influx through mechanosensitive channels. The depolarizing current across the apical membrane of hair cells is carried by K+, instead of Na+.Citation77,Citation78 K+ is then taken up by the apposing supporting cells, secreted into the perilymph of scala tympani and recycled back to the scala media across the stria vascularis. The enrichment of endocochlear K+ and the development of endocochlear potential are owed to the transport activity of the multilayered epithelium of the stria vascularis (). The stria vascularis comprises of four epithelial layers: the fibrocyte, the basal cell, the intermediate cell and the marginal cell.Citation77 K+ is actively taken up from the intrastrial space into the marginal cell through the Na+/K+-ATPase and the Na+/K+/2Cl− cotransporter (NKCC1) and are then secreted into the endolymph through apical KCNQ1/KCNE1 K+ channels. This uptake creates a low K+ concentration in the intrastrial space,Citation79,Citation80 creating a large K+ diffusion potential across the apical membrane of the intermediate cell that prominently expresses the K+ channel Kir4.1.Citation80,Citation81 There are two diffusion barriers formed by tight junctions in the stria vascularis, one between the apical membrane of the marginal cell and the other between the basal cell (). Tight junctions are also found between sensory hair cells and neighboring supporting cells or between supporting cells ().
Figure 6. Structure of the cochlea and its tight junction. (A) Schematic presentation of the cochlea and extracellular fluid compartments. (B) Cellular model of the stria vascularis that generates the endocochlear potential (EP) through K+ secretion from perilymph to endolymph. The stria vascularis is a multilayered epithelium in which tight junctions between the marginal cells, which face directly the endolymph, as well as tight junctions between the basal cells isolate an intrastrial space that has a low K+ concentration. (C) Tight junctions are also found in the organ of Corti between hair cells and supporting cells and between supporting cells.
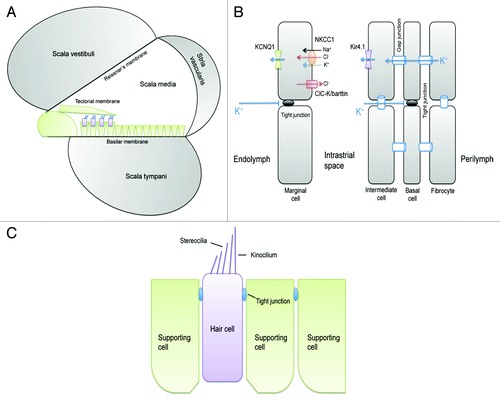
Claudin-11 is only found in the tight junction between basal cells of the stria vascularis.Citation82 Two independent lines of claudin-11 KO animals revealed the same phenotypes.Citation82,Citation83 The tight junction completely disappeared from the basal cells; the endocochlear potential was less than 30mV; and the hearing thresholds were elevated 50 dB across the entire frequency spectrum, despite a normal K+ concentration in the endolymph.Citation82,Citation83 There was no morphological abnormality in sensory hair cells or degeneration of hair cells. Normal K+ accumulation in endolymph suggests that (1) tight junctions in the basal cells are not required for barrier to K+ backflux; or (2) the uptake of K+ by the margional cell is very efficient and not affected in absence of basal cell tight junction. The major source of endocochlear potential is the K+ diffusion potential across the apical membrane of intermediate cell through Kir4.1 (). Loss of claudin-11 presumably creates a shunt between basal cells that allows current passage and rapid dissipation of the apical potential. It is not known which ion carries the current. Recording of claudin-11 permeability has not been performed either in vitro in epithelial cell cultures or in vivo in the stria vascularis.
The first clinical evidence of claudin mutations in deafness comes from a linkage study of claudin-14 in nonsyndromic recessive deafness DFNB29.Citation18 In two Pakistani consanguineous cohorts, two causal mutations (398delT and T254A) were found in the claudin-14 gene.Citation18 The 398delT mutation causes a frameshift within the codon of Met133, which substitutes 23 incorrect amino acids and prematurely terminates translation 69 nucleotides later. The T254A mutation substitutes aspartic acid for valine (V85D). Valine 85 is a conserved site within the second transmembrane domain (TM2) of claudin-14 protein. Aspartic acid at position 85 is predicted to affect hydrophobicity and disrupt the secondary structure of TM2. Therefore, both 398delT and T254A are loss-of-function mutations. Claudin-14 gene expression in the inner ear was developmentally regulated.Citation18 At P4, claudin-14 was found primarily in the IHC and OHC and to a lesser extent, in the supporting cells. By P8, claudin-14 expression was observed in IHC, OHC and supporting cells throughout the organ of Corti. In contrast, no expression of claudin-14 was found in the stria vascularis or the Reissner’s membrane. Claudin-14 KO mice have a normal endocochlear potential but are deaf due to degeneration of OHC.Citation84 The tight junction between supporting cells and between supporting cells and hair cells are normal in absence of claudin-14, suggesting these TJs contain other redundant claudins. The K+ level in endolymph was not recorded for claudin-14 KO. In vitro, claudin-14 functions as a cation barrier, reducing PK and PNa.Citation84 One hypothesis of OHC degeneration is the diffusion of K+ via OHC tight junctions from the endolymph to the space of Nuel, which is normally filled with low K+ cortilymph.Citation85 Long-term exposure of the basolateral membrane of OHC to high K+ levels is toxic and causes OHC depolarization and degeneration.Citation86
A recent large-scale mutagenesis has identified a novel claudin – claudin-9, mutation of which causes deafness (nmf329) in mice.Citation87 Positional cloning revealed a missense mutation (F35L) in claudin-9 from the nmf329 strain. In vitro, heterologous expression of wildtype claudin-9 reduced PK and PNa in cultured MDCK cells while the F35L mutant abolished this cation barrier function without affecting TJ localization.Citation87 Claudin-9 is predominantly expressed by the IHC, OHC and supporting cells, the same location as of claudin-14. Consistent with a barrier defect, the perilymphatic K+ concentration was elevated in the nmf329 mouse line, despite a normal endolymphatic K+ level and endocochlear potential.Citation87 Similar to claudin-14 KO mice, the claudin-9 mutant mice are deaf owing to the degeneration of OHC. The mutant phenotype of hair cell loss was rescued when explanted organ of Corti was cultured in a low K+ milieu or when endolymphatic K+ level was reduced.Citation87
Claudins and Human Diseases
Claudins can play direct causative roles in various human diseases. The best example is the claudin trio – claudin-14, -16 and -19. The loss-of-function mutations in claudin-14 are found in children with inherited deafness,Citation18 owing to a K+ barrier defect in the organ of Corti.Citation84 Its genetic locus is also linked to a common disease – kidney stone by a recent GWAS study.Citation64 Using biochemical and biophysical analyses, Gong et al. have elegantly shown that claudin-14 interacts with and inhibits the claudin-16 and claudin-19 channel permeability,Citation38 both genes found in a fatal inherited disease due to uncontrolled loss of serum Ca2+ and Mg2+.Citation19,Citation66 Claudins may be involved in the signaling pathways influenced by many disease conditions. For example, CaSR, the Ca2+ sensing receptor, has been recognized with pivotal importance in extracellular Ca2+ metabolism and causing familial hypocalciuric hypercalcemia (FHH) and neonatal severe hyperparathyroidism (NSHPT), two inherited conditions characterized by altered calcium homeostasis.Citation88 The loss of CaSR in the kidney significantly reduces the gene expression level of claudin-14, hence upregulating claudin-16 and -19 permeability for urinary Ca2+ reabsorption.Citation89 WNK4, the causative gene for pseudohypoaldosteronism type II,Citation73 has been found to hyperphosphorylate claudin-4 and concomitantly increase paracellular Cl− permeability in the collecting duct of the kidney.Citation74
Future Directions
The single-channel conductance of a paracellular channel has never been recorded. With recent advances in SICM technology, paracellular conductance can be revealed at sub-micron scales. Further refinement in spatial resolution will allow capture of “discrete” paracellular channels along the tight junction strand.
The oligomeric nature of paracellular channel is a major mystery. It is now well known that claudin function is dictated by its cellular background and combination with other claudin molecules. What is the basic functional unit of claudin along the tight junction strand? How is the tight junction strand organized from these claudin units? Recent advances in super-resolution microscopy may provide the much-needed resolution in claudin molecule localization and interaction.
How does claudin switch between channel and barrier function? Because all claudins possess a similar extracellular loop (ECL1) that presumably dictates channel or barrier function, how is the ECL1 structurally arranged to achieve such a diverse function? The single-pore theory lacks the complexity to be extended to organ systems beyond the renal proximal tubule.
How is claudin function regulated? Current knowledge of claudin regulation is limited to its intracellular C-terminus where numerous phosphorylation and interaction sites have been characterized. These modifications seem only to affect claudin protein trafficking. A bigger challenge would be to identify regulatory mechanisms that alter claudin function extracellularly or through structural changes.
The clinical evidence for claudin dysfunction in human diseases is still very limited. Despite recent progresses in human genomic studies, causative mutation or SNP has yet been found in claudin genes from common diseases such as kidney stone or hypertension. Physiological analyses of transgenic animals for claudin dysfunction will provide pathogenic candidate genes that can be examined under higher genomic resolution and within more focused population.
No drugs that modulate claudin function have been developed. Despite several attempts to target the ECL2 domain of claudin with Clostridium perfringens enterotoxin in cancer therapy, our knowledge of drugs that can target the larger ECL1 domain is limited. Screening for paracellular channel inhibitors will be challenging but rewarding. Such inhibitors will not only provide new treatments to related diseases but also allow structural analyses of paracellular channel permeation.
Finally and most importantly, no structural information is available for claudin. Since the discovery of the first claudin in 1998, no lab has ever been able to crystalize a claudin molecule. The technical difficulties will eventually be overcome with growing knowledge of claudin protein biochemistry both in Petri dish and under native environments.
Acknowledgments
This work was supported by the National Institutes of Health Grants RO1DK084059 and P30 DK079333 and American Heart Association Grant 0930050N.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol 1963; 17:375 - 412; http://dx.doi.org/10.1083/jcb.17.2.375; PMID: 13944428
- Miller F. Hemoglobin absorption by the cells of the proximal convoluted tubule in mouse kidney. J Biophys Biochem Cytol 1960; 8:689 - 718; http://dx.doi.org/10.1083/jcb.8.3.689; PMID: 13770760
- Goodenough DA, Revel JP. A fine structural analysis of intercellular junctions in the mouse liver. J Cell Biol 1970; 45:272 - 90; http://dx.doi.org/10.1083/jcb.45.2.272; PMID: 4105112
- Stevenson BR, Goodenough DA. Zonulae occludentes in junctional complex-enriched fractions from mouse liver: preliminary morphological and biochemical characterization. J Cell Biol 1984; 98:1209 - 21; http://dx.doi.org/10.1083/jcb.98.4.1209; PMID: 6425301
- Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 1993; 123:1777 - 88; http://dx.doi.org/10.1083/jcb.123.6.1777; PMID: 8276896
- Ebnet K, Suzuki A, Ohno S, Vestweber D. Junctional adhesion molecules (JAMs): more molecules with dual functions?. J Cell Sci 2004; 117:19 - 29; http://dx.doi.org/10.1242/jcs.00930; PMID: 14657270
- Stevenson BR, Anderson JM, Goodenough DA, Mooseker MS. Tight junction structure and ZO-1 content are identical in two strains of Madin-Darby canine kidney cells which differ in transepithelial resistance. J Cell Biol 1988; 107:2401 - 8; http://dx.doi.org/10.1083/jcb.107.6.2401; PMID: 3058723
- Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 1998; 141:1539 - 50; http://dx.doi.org/10.1083/jcb.141.7.1539; PMID: 9647647
- Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta 2008; 1778:631 - 45; http://dx.doi.org/10.1016/j.bbamem.2007.10.018; PMID: 18036336
- Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol 2006; 68:403 - 29; http://dx.doi.org/10.1146/annurev.physiol.68.040104.131404; PMID: 16460278
- Piontek J, Winkler L, Wolburg H, Müller SL, Zuleger N, Piehl C, et al. Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J 2008; 22:146 - 58; http://dx.doi.org/10.1096/fj.07-8319com; PMID: 17761522
- Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tsukita S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett 2000; 476:258 - 61; http://dx.doi.org/10.1016/S0014-5793(00)01744-0; PMID: 10913624
- Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 1999; 147:1351 - 63; http://dx.doi.org/10.1083/jcb.147.6.1351; PMID: 10601346
- Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol 2002; 156:1099 - 111; http://dx.doi.org/10.1083/jcb.200110122; PMID: 11889141
- Muto S, Hata M, Taniguchi J, Tsuruoka S, Moriwaki K, Saitou M, et al. Claudin-2-deficient mice are defective in the leaky and cation-selective paracellular permeability properties of renal proximal tubules. Proc Natl Acad Sci U S A 2010; 107:8011 - 6; http://dx.doi.org/10.1073/pnas.0912901107; PMID: 20385797
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 2003; 161:653 - 60; http://dx.doi.org/10.1083/jcb.200302070; PMID: 12743111
- Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, et al. CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell 1999; 99:649 - 59; http://dx.doi.org/10.1016/S0092-8674(00)81553-6; PMID: 10612400
- Wilcox ER, Burton QL, Naz S, Riazuddin S, Smith TN, Ploplis B, et al. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell 2001; 104:165 - 72; http://dx.doi.org/10.1016/S0092-8674(01)00200-8; PMID: 11163249
- Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 1999; 285:103 - 6; http://dx.doi.org/10.1126/science.285.5424.103; PMID: 10390358
- Tang VW, Goodenough DA. Paracellular ion channel at the tight junction. Biophys J 2003; 84:1660 - 73; http://dx.doi.org/10.1016/S0006-3495(03)74975-3; PMID: 12609869
- Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, et al. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci 2008; 121:298 - 305; http://dx.doi.org/10.1242/jcs.021485; PMID: 18198187
- Watson CJ, Rowland M, Warhurst G. Functional modeling of tight junctions in intestinal cell monolayers using polyethylene glycol oligomers. Am J Physiol Cell Physiol 2001; 281:C388 - 97; PMID: 11443038
- Amasheh S, Meiri N, Gitter AH, Schöneberg T, Mankertz J, Schulzke JD, et al. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J Cell Sci 2002; 115:4969 - 76; http://dx.doi.org/10.1242/jcs.00165; PMID: 12432083
- Hou J, Paul DL, Goodenough DA. Paracellin-1 and the modulation of ion selectivity of tight junctions. J Cell Sci 2005; 118:5109 - 18; http://dx.doi.org/10.1242/jcs.02631; PMID: 16234325
- Colegio OR, Van Itallie C, Rahner C, Anderson JM. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am J Physiol Cell Physiol 2003; 284:C1346 - 54; PMID: 12700140
- Yu AS, Cheng MH, Angelow S, Günzel D, Kanzawa SA, Schneeberger EE, et al. Molecular basis for cation selectivity in claudin-2-based paracellular pores: identification of an electrostatic interaction site. J Gen Physiol 2009; 133:111 - 27; http://dx.doi.org/10.1085/jgp.200810154; PMID: 19114638
- Frömter E. The route of passive ion movement through the epithelium of Necturus gallbladder. J Membr Biol 1972; 8:259 - 301; http://dx.doi.org/10.1007/BF01868106; PMID: 5084117
- Cereijido M, Stefani E, Palomo AM. Occluding junctions in a cultured transporting epithelium: structural and functional heterogeneity. J Membr Biol 1980; 53:19 - 32; http://dx.doi.org/10.1007/BF01871169; PMID: 7373646
- Gitter AH, Bertog M, Schulzke J, Fromm M. Measurement of paracellular epithelial conductivity by conductance scanning. Pflugers Arch 1997; 434:830 - 40; http://dx.doi.org/10.1007/s004240050472; PMID: 9306019
- Chen CC, Zhou Y, Morris CA, Hou J, Baker LA. Scanning ion conductance microscopy measurement of paracellular channel conductance in tight junctions. [Epub ahead of print] Anal Chem 2013; 85:3621 - 8; http://dx.doi.org/10.1021/ac303441n; PMID: 23421780
- Hansma PK, Drake B, Marti O, Gould SA, Prater CB. The scanning ion-conductance microscope. Science 1989; 243:641 - 3; http://dx.doi.org/10.1126/science.2464851; PMID: 2464851
- Mitic LL, Unger VM, Anderson JM. Expression, solubilization, and biochemical characterization of the tight junction transmembrane protein claudin-4. Protein Sci 2003; 12:218 - 27; http://dx.doi.org/10.1110/ps.0233903; PMID: 12538885
- Van Itallie CM, Mitic LL, Anderson JM. Claudin-2 forms homodimers and is a component of a high molecular weight protein complex. J Biol Chem 2011; 286:3442 - 50; http://dx.doi.org/10.1074/jbc.M110.195578; PMID: 21098027
- Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol 1999; 147:891 - 903; http://dx.doi.org/10.1083/jcb.147.4.891; PMID: 10562289
- Daugherty BL, Ward C, Smith T, Ritzenthaler JD, Koval M. Regulation of heterotypic claudin compatibility. J Biol Chem 2007; 282:30005 - 13; http://dx.doi.org/10.1074/jbc.M703547200; PMID: 17699514
- Hou J, Renigunta A, Gomes AS, Hou M, Paul DL, Waldegger S, et al. Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc Natl Acad Sci U S A 2009; 106:15350 - 5; http://dx.doi.org/10.1073/pnas.0907724106; PMID: 19706394
- Hou J, Renigunta A, Konrad M, Gomes AS, Schneeberger EE, Paul DL, et al. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest 2008; 118:619 - 28; PMID: 18188451
- Gong Y, Renigunta V, Himmerkus N, Zhang J, Renigunta A, Bleich M, et al. Claudin-14 regulates renal Ca⁺⁺ transport in response to CaSR signalling via a novel microRNA pathway. EMBO J 2012; 31:1999 - 2012; http://dx.doi.org/10.1038/emboj.2012.49; PMID: 22373575
- Coyne CB, Gambling TM, Boucher RC, Carson JL, Johnson LG. Role of claudin interactions in airway tight junctional permeability. Am J Physiol Lung Cell Mol Physiol 2003; 285:L1166 - 78; PMID: 12909588
- Hou J, Renigunta A, Yang J, Waldegger S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc Natl Acad Sci U S A 2010; 107:18010 - 5; http://dx.doi.org/10.1073/pnas.1009399107; PMID: 20921420
- Müller D, Kausalya PJ, Claverie-Martin F, Meij IC, Eggert P, Garcia-Nieto V, et al. A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Am J Hum Genet 2003; 73:1293 - 301; http://dx.doi.org/10.1086/380418; PMID: 14628289
- Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, et al. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 2006; 126:741 - 54; http://dx.doi.org/10.1016/j.cell.2006.06.043; PMID: 16923393
- Kiuchi-Saishin Y, Gotoh S, Furuse M, Takasuga A, Tano Y, Tsukita S. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J Am Soc Nephrol 2002; 13:875 - 86; PMID: 11912246
- Krug SM, Günzel D, Conrad MP, Rosenthal R, Fromm A, Amasheh S, et al. Claudin-17 forms tight junction channels with distinct anion selectivity. Cell Mol Life Sci 2012; 69:2765 - 78; http://dx.doi.org/10.1007/s00018-012-0949-x; PMID: 22402829
- Van Itallie C, Rahner C, Anderson JM. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J Clin Invest 2001; 107:1319 - 27; http://dx.doi.org/10.1172/JCI12464; PMID: 11375422
- Yu AS, Enck AH, Lencer WI, Schneeberger EE. Claudin-8 expression in Madin-Darby canine kidney cells augments the paracellular barrier to cation permeation. J Biol Chem 2003; 278:17350 - 9; http://dx.doi.org/10.1074/jbc.M213286200; PMID: 12615928
- Alexandre MD, Lu Q, Chen YH. Overexpression of claudin-7 decreases the paracellular Cl- conductance and increases the paracellular Na+ conductance in LLC-PK1 cells. J Cell Sci 2005; 118:2683 - 93; http://dx.doi.org/10.1242/jcs.02406; PMID: 15928046
- Li WY, Huey CL, Yu AS. Expression of claudin-7 and -8 along the mouse nephron. Am J Physiol Renal Physiol 2004; 286:F1063 - 71; http://dx.doi.org/10.1152/ajprenal.00384.2003; PMID: 14722018
- Reyes JL, Lamas M, Martin D, del Carmen Namorado M, Islas S, Luna J, et al. The renal segmental distribution of claudins changes with development. Kidney Int 2002; 62:476 - 87; http://dx.doi.org/10.1046/j.1523-1755.2002.00479.x; PMID: 12110008
- Alpern RJ, Howlin KJ, Preisig PA. Active and passive components of chloride transport in the rat proximal convoluted tubule. J Clin Invest 1985; 76:1360 - 6; http://dx.doi.org/10.1172/JCI112111; PMID: 4056034
- Liu FY, Cogan MG. Axial heterogeneity in the rat proximal convoluted tubule. I. Bicarbonate, chloride, and water transport. Am J Physiol 1984; 247:F816 - 21; PMID: 6496747
- Green R, Giebisch G. Reflection coefficients and water permeability in rat proximal tubule. Am J Physiol 1989; 257:F658 - 68; PMID: 2801963
- Yu AS, Cheng MH, Coalson RD. Calcium inhibits paracellular sodium conductance through claudin-2 by competitive binding. J Biol Chem 2010; 285:37060 - 9; http://dx.doi.org/10.1074/jbc.M110.146621; PMID: 20807759
- Van Itallie CM, Rogan S, Yu A, Vidal LS, Holmes J, Anderson JM. Two splice variants of claudin-10 in the kidney create paracellular pores with different ion selectivities. Am J Physiol Renal Physiol 2006; 291:F1288 - 99; http://dx.doi.org/10.1152/ajprenal.00138.2006; PMID: 16804102
- Abuazza G, Becker A, Williams SS, Chakravarty S, Truong HT, Lin F, et al. Claudins 6, 9, and 13 are developmentally expressed renal tight junction proteins. Am J Physiol Renal Physiol 2006; 291:F1132 - 41; http://dx.doi.org/10.1152/ajprenal.00063.2006; PMID: 16774906
- Fujita H, Sugimoto K, Inatomi S, Maeda T, Osanai M, Uchiyama Y, et al. Tight junction proteins claudin-2 and -12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes. Mol Biol Cell 2008; 19:1912 - 21; http://dx.doi.org/10.1091/mbc.E07-09-0973; PMID: 18287530
- Hebert SC, Culpepper RM, Andreoli TE. NaCl transport in mouse medullary thick ascending limbs. I. Functional nephron heterogeneity and ADH-stimulated NaCl cotransport. Am J Physiol 1981; a 241:F412 - 31; PMID: 7315965
- Hebert SC, Culpepper RM, Andreoli TE. NaCl transport in mouse medullary thick ascending limbs. II. ADH enhancement of transcellular NaCl cotransport; origin of transepithelial voltage. Am J Physiol 1981; b 241:F432 - 42; PMID: 7315966
- Greger R. Ion transport mechanisms in thick ascending limb of Henle’s loop of mammalian nephron. Physiol Rev 1985; 65:760 - 97; PMID: 2409564
- Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 2006; 79:949 - 57; http://dx.doi.org/10.1086/508617; PMID: 17033971
- Ikari A, Matsumoto S, Harada H, Takagi K, Hayashi H, Suzuki Y, et al. Phosphorylation of paracellin-1 at Ser217 by protein kinase A is essential for localization in tight junctions. J Cell Sci 2006; 119:1781 - 9; http://dx.doi.org/10.1242/jcs.02901; PMID: 16608877
- Kausalya PJ, Amasheh S, Günzel D, Wurps H, Müller D, Fromm M, et al. Disease-associated mutations affect intracellular traffic and paracellular Mg2+ transport function of Claudin-16. J Clin Invest 2006; 116:878 - 91; http://dx.doi.org/10.1172/JCI26323; PMID: 16528408
- Hou J, Shan Q, Wang T, Gomes AS, Yan Q, Paul DL, et al. Transgenic RNAi depletion of claudin-16 and the renal handling of magnesium. J Biol Chem 2007; 282:17114 - 22; http://dx.doi.org/10.1074/jbc.M700632200; PMID: 17442678
- Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet 2009; 41:926 - 30; http://dx.doi.org/10.1038/ng.404; PMID: 19561606
- Breiderhoff T, Himmerkus N, Stuiver M, Mutig K, Will C, Meij IC, et al. Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc Natl Acad Sci U S A 2012; 109:14241 - 6; http://dx.doi.org/10.1073/pnas.1203834109; PMID: 22891322
- Guyton AC. Blood pressure control--special role of the kidneys and body fluids. Science 1991; 252:1813 - 6; http://dx.doi.org/10.1126/science.2063193; PMID: 2063193
- Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994; 367:463 - 7; http://dx.doi.org/10.1038/367463a0; PMID: 8107805
- Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, et al. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 2010; 120:1627 - 35; http://dx.doi.org/10.1172/JCI40145; PMID: 20389022
- Hadchouel J, Büsst C, Procino G, Valenti G, Chambrey R, Eladari D. Regulation of extracellular fluid volume and blood pressure by pendrin. Cell Physiol Biochem 2011; 28:505 - 12; http://dx.doi.org/10.1159/000335116; PMID: 22116364
- Fujita H, Hamazaki Y, Noda Y, Oshima M, Minato N. Claudin-4 deficiency results in urothelial hyperplasia and lethal hydronephrosis. PLoS One 2012; 7:e52272; http://dx.doi.org/10.1371/journal.pone.0052272; PMID: 23284964
- Tatum R, Zhang Y, Salleng K, Lu Z, Lin JJ, Lu Q, et al. Renal salt wasting and chronic dehydration in claudin-7-deficient mice. Am J Physiol Renal Physiol 2010; 298:F24 - 34; http://dx.doi.org/10.1152/ajprenal.00450.2009; PMID: 19759267
- Gordon RD, Klemm SA, Tunny TJ, Stowasser M. In: Laragh JH, Brenner BM, editors. Hypertension: pathophysiology, diagnosis, and management, 2nd ed. New York: Raven; 1995. pp. 2111–2123.
- Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, et al. Human hypertension caused by mutations in WNK kinases. Science 2001; 293:1107 - 12; http://dx.doi.org/10.1126/science.1062844; PMID: 11498583
- Yamauchi K, Rai T, Kobayashi K, Sohara E, Suzuki T, Itoh T, et al. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc Natl Acad Sci U S A 2004; 101:4690 - 4; http://dx.doi.org/10.1073/pnas.0306924101; PMID: 15070779
- Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, et al. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 2007; 5:331 - 44; http://dx.doi.org/10.1016/j.cmet.2007.03.009; PMID: 17488636
- Ryan AF, Wickham MG, Bone RC. Element content of intracochlear fluids, outer hair cells, and stria vascularis as determined by energy-dispersive roentgen ray analysis. Otolaryngol Head Neck Surg (1979) 1979; 87:659 - 65; PMID: 503532
- Hibino H, Kurachi Y. Molecular and physiological bases of the K+ circulation in the mammalian inner ear. Physiology (Bethesda) 2006; 21:336 - 45; http://dx.doi.org/10.1152/physiol.00023.2006; PMID: 16990454
- Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol 2006; 576:11 - 21; http://dx.doi.org/10.1113/jphysiol.2006.112888; PMID: 16857713
- Ikeda K, Morizono T. Electrochemical profiles for monovalent ions in the stria vascularis: cellular model of ion transport mechanisms. Hear Res 1989; 39:279 - 86; http://dx.doi.org/10.1016/0378-5955(89)90047-6; PMID: 2753832
- Nin F, Hibino H, Doi K, Suzuki T, Hisa Y, Kurachi Y. The endocochlear potential depends on two K+ diffusion potentials and an electrical barrier in the stria vascularis of the inner ear. Proc Natl Acad Sci U S A 2008; 105:1751 - 6; http://dx.doi.org/10.1073/pnas.0711463105; PMID: 18218777
- Marcus DC, Wu T, Wangemann P, Kofuji P. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol 2002; 282:C403 - 7; PMID: 11788352
- Gow A, Davies C, Southwood CM, Frolenkov G, Chrustowski M, Ng L, et al. Deafness in Claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J Neurosci 2004; 24:7051 - 62; http://dx.doi.org/10.1523/JNEUROSCI.1640-04.2004; PMID: 15306639
- Kitajiri S, Miyamoto T, Mineharu A, Sonoda N, Furuse K, Hata M, et al. Compartmentalization established by claudin-11-based tight junctions in stria vascularis is required for hearing through generation of endocochlear potential. J Cell Sci 2004; 117:5087 - 96; http://dx.doi.org/10.1242/jcs.01393; PMID: 15456848
- Ben-Yosef T, Belyantseva IA, Saunders TL, Hughes ED, Kawamoto K, Van Itallie CM, et al. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum Mol Genet 2003; 12:2049 - 61; http://dx.doi.org/10.1093/hmg/ddg210; PMID: 12913076
- Johnstone BM, Patuzzi R, Syka J, Syková E. Stimulus-related potassium changes in the organ of Corti of guinea-pig. J Physiol 1989; 408:77 - 92; PMID: 2778743
- Zenner HP, Reuter G, Zimmermann U, Gitter AH, Fermin C, LePage EL. Transitory endolymph leakage induced hearing loss and tinnitus: depolarization, biphasic shortening and loss of electromotility of outer hair cells. Eur Arch Otorhinolaryngol 1994; 251:143 - 53; http://dx.doi.org/10.1007/BF00181826; PMID: 8080633
- Nakano Y, Kim SH, Kim HM, Sanneman JD, Zhang Y, Smith RJ, et al. A claudin-9-based ion permeability barrier is essential for hearing. PLoS Genet 2009; 5:e1000610; http://dx.doi.org/10.1371/journal.pgen.1000610; PMID: 19696885
- Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, et al. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 1993; 75:1297 - 303; http://dx.doi.org/10.1016/0092-8674(93)90617-Y; PMID: 7916660
- Toka HR, Al-Romaih K, Koshy JM, DiBartolo S 3rd, Kos CH, Quinn SJ, et al. Deficiency of the calcium-sensing receptor in the kidney causes parathyroid hormone-independent hypocalciuria. J Am Soc Nephrol 2012; 23:1879 - 90; http://dx.doi.org/10.1681/ASN.2012030323; PMID: 22997254