 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Chronic obstructive pulmonary disease (COPD) causes significant morbidity and mortality. Cigarette smoke, the most common risk factor for COPD, induces airway and alveolar epithelial barrier permeability and initiates an innate immune response. Changes in abundance of aquaporin 5 (AQP5), a water channel, can affect epithelial permeability and immune response after cigarette smoke exposure. To determine how AQP5-derived epithelial barrier modulation affects epithelial immune response to cigarette smoke and development of emphysema, WT and AQP5−/− mice were exposed to cigarette smoke (CS). We measured alveolar cell counts and differentials, and assessed histology, mean-linear intercept (MLI), and surface-to-volume ratio (S/V) to determine severity of emphysema. We quantified epithelial-derived signaling proteins for neutrophil trafficking, and manipulated AQP5 levels in an alveolar epithelial cell line to determine specific effects on neutrophil transmigration after CS exposure. We assessed paracellular permeability and epithelial turnover in response to CS. In contrast to WT mice, AQP5−/− mice exposed to 6 months of CS did not demonstrate a significant increase in MLI or a significant decrease in S/V compared with air-exposed mice, conferring protection against emphysema. After sub-acute (4 weeks) and chronic (6 mo) CS exposure, AQP5−/− mice had fewer alveolar neutrophil but similar lung neutrophil numbers as WT mice. The presence of AQP5 in A549 cells, an alveolar epithelial cell line, was associated with increase neutrophil migration after CS exposure. Compared with CS-exposed WT mice, neutrophil ligand (CD11b) and epithelial receptor (ICAM-1) expression were reduced in CS-exposed AQP5−/− mice, as was secreted LPS-induced chemokine (LIX), an epithelial-derived neutrophil chemoattractant. CS-exposed AQP5−/− mice demonstrated decreased type I pneumocytes and increased type II pneumocytes compared with CS-exposed WT mice suggestive of enhanced epithelial repair. Absence of AQP5 protected against CS-induced emphysema with reduced epithelial permeability, neutrophil migration, and altered epithelial cell turnover which may enhance repair.
Introduction
Chronic obstructive pulmonary disease (COPD) is an irreversible disease usually occurring due to cigarette smoke exposure of variable duration and intensity. In the US, COPD accounts for more than 120,000 annual deaths,Citation1 yet treatment of COPD is primarily limited to symptom relief and removal of environmental cofactors such as cigarette smoke.Citation2 Pathologically, COPD is characterized by chronic bronchitis or emphysema. In chronic bronchitis, airway inflammation leads to increased mucus production and reduced mucociliary clearance, causing bronchoconstriction and airflow limitation. Emphysema is hallmarked by destruction of lung parenchyma.Citation3 Despite significant investigation, we do not fully understand pathologic mechanisms responsible for chronic bronchitis or emphysema.
As they serve as the interface with the outside, airway and alveolar epithelial barriers provide the first line of defense against inhaled cigarette smoke. Cigarette smoke (CS) increases epithelial barrier permeability.Citation4-Citation11 In addition, CS initiates an epithelial-derived innate immune response leading to release of pro-inflammatory mediators, recruitment of inflammatory cells, and expression of adhesion molecules.Citation12,Citation13 By modulating chemokine drive and adhesion molecule expression, epithelial cells regulate effector cell influx and thereby serve as an immunologic barrier. Both epithelial properties represent mechanisms for epithelial interactions: epithelial physical barrier is determined by how an epithelial cell interacts with neighboring epithelial cells, and epithelial immune barrier is determined by how an epithelial cell communicates with immune effector cells. How the two epithelial properties may relate to each other in response to CS has not been explored.
Communication between the epithelium and migrating inflammatory cells during smoke exposure may be crucial for subsequent development of pulmonary emphysema. Bronchial airway biopsies and lavage fluid from COPD patients with acute exacerbations and chronic smokers demonstrate elevated concentrations of IL-8, the primary neutrophil-recruiting chemokine in humans.Citation14,Citation15 Cigarette smoke extract (CSE) induced significant expression of the chemokine CXCL5 (LIX), a key murine neutrophil chemokine derived from lung epithelial cells.Citation16 Cigarette smoke exposure altered the expression of the adhesion molecule and inflammatory cell receptor ICAM-1 in mouse lungs.Citation3
Multiple studies demonstrate accumulation of neutrophils and macrophages around small and large airways in patients with COPD.Citation17,Citation18 The functional severity of COPD has been shown to correlate with an increased presence of neutrophils and macrophages in the mucosa,Citation19 although controversy exists regarding their specific contribution to smoke-induced pathology including emphysema.Citation20,Citation21 In murine models, CS induces inflammatory cell accumulation into the airways and lung parenchyma,Citation22,Citation23 and the causal role of macrophages and neutrophils in the pathogenesis of emphysema has been well-described.Citation21,Citation22 Studies investigating the role of lung epithelium on CS-induced murine emphysema have focused on cellular apoptosis,Citation24 production of inflammatory mediators, and oxidant/antioxidant or protease/anti-protease balance.Citation20 However, to date, there has been little evidence that the early changes in barrier function affect the epithelial immune responses.
Aquaporin 5 (AQP5) is an apical membrane water channel found in airway and alveolar epithelium positioned to respond to the luminal environment and participate in diverse epithelial responses. AQP5 polymorphisms can influence development and progression of COPD. A single nucleotide polymorphism of AQP5 was shown to be associated with COPD prevalence in a Chinese population.Citation25 Subsequently, Hansel et al. demonstrated that polymorphisms in AQP5 were associated with rate of lung function decline among active smokers with COPD.Citation26 Altered expression of human AQP5 in the bronchial tissue has been associated with lower lung function in the airways of subjects with COPD.Citation26,Citation27 AQP5 null mice and isolated epithelial cells deficient in AQP5 have enhanced barrier function. AQP5-mediated regulation of microtubule dynamics decreases paracellular permeability.Citation28 We hypothesized that barrier enhancement in the absence of AQP5 mitigates CS-induced emphysema by two potentially related mechanisms: one, changes in the physical barrier, and two, alterations in immune response.
By exposing mice to cigarette smoke, we provide functional data that implicate the absence of epithelial AQP5 as a protective mechanism against development of emphysema, with reduced neutrophil migration to the alveolar space and altered alveolar epithelial cell turnover. Associated differences in key signaling proteins on neutrophils and epithelial cells may be crucial to the observed response. Our findings are novel, and confer protection against CS-induced murine emphysema due to the absence of AQP5 by a reduction in epithelial barrier permeability and a blunted innate immune response.
Results
The absence of AQP5 mitigates cigarette smoke-induced pulmonary emphysema, epithelial permeability and alveolar inflammatory cell accumulation
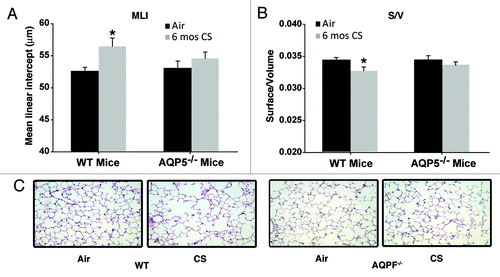
We exposed AQP5−/− mice and WT littermates to chronic (6 mo) cigarette smoke to assess for emphysema as well as the alveolar inflammatory milieu. WT mice exposed to CS for 6 mo demonstrated a significant, 8% increase in the mean linear intercept (MLI) () and a significant decrease in surface area of airspace wall per unit of lung volume (S/V) () compared with age-matched WT mice exposed to air. MLI measures interalveolar septal distance and is an index of alveolar size.Citation29,Citation30 S/V measures septal loss and is an index for alveolar destruction.Citation31,Citation32 The change in MLI and S/V were typical of previous 6 mo exposures in this facility and in other published reports.Citation24,Citation33 In contrast to WT mice, AQP5−/− mice exposed to CS for 6 mo did not have significant changes in MLI () or S/V () compared with age-matched air-exposed AQP5−/− mice. Air-exposed controls from WT or AQP5−/− had similar MLI and S/V values. The only group with a significant change in MLI and S/V were the WT CS-exposed mice, suggesting protection from emphysema in the absence of AQP5. Representative histologic lung sections stained with H&E demonstrate a qualitative increase in airspace only with CS-exposed WT mice in comparison to air exposed controls; significant airspace enlargement does not appear to occur in AQP5−/− mice irrespective of CS exposure (). Saving for some variability, 6 mo of cigarette smoke exposure has been shown to cause an 8–12% increase in MLI due to emphysema formation.Citation29
Figure 1. AQP5−/− mice are protected against cigarette-smoke induced emphysema. (A) Mean Linear Intercept (MLI) was significantly increased in WT mice exposed to 6 mo of CS compared with air-exposed WT mice and air-exposed AQP5−/− mice. n = 7–10 per group, p < 0.006. (B)Surface to volume ratio (S/V) was significantly reduced in WT mice exposed to 6 mo of CS compared with air-exposed WT mice and air-exposed AQ5−/− mice, n = 7–10 per group, *p < 0.02. (C) Representative histologic H&E lung sections (20x) demonstrate airspace size for various exposure and WT or AQP5−/− mice groups.
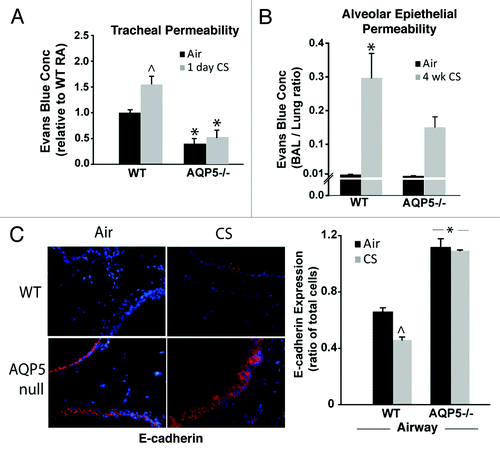
In addition to its water transport properties, our group has demonstrated that the c-terminus of AQP5 alters human airway epithelial permeability in vitro by altering microtubule assembly.Citation28,Citation34 We were interested in determining whether AQP5 also affected murine epithelial paracellular permeability. Using a previously developed assay of ex vivo tracheal permeability,Citation34 we detected less extraluminal Evans blue dye (EBD) in trachea from air-exposed AQP5−/− mice compared with trachea from air-exposed WT mice (), but suspect this difference falls within a physiologically normal range given the absence of apparent baseline functional differences in AQP5−/− mice compared with WT mice . With the addition of only 1 d of cigarette smoke exposure, trachea from WT mice had significantly more extraluminal EBD efflux compared with trachea from air-exposed WT mice. In contrast, CS-exposed AQP5−/− mice did not demonstrate a significant increase in EBD tracheal efflux when compared with air-exposed AQP5−/− mice. Next, we assessed for CS-altered permeability across airway and alveolar epithelial layers using a ratio of EBD concentration in the bronchoalveolar fluid (BAL) to EBD concentration in the lung in WT and AQP5−/− mice after 4 weeks of air or CS exposure (). Despite similar ratios in air-exposed WT and AQP5−/− mice, CS-exposed WT mice had a significant increase in the BAL to lung EBD ratio compared with air-exposed mice, consistent with increased permeability across the airway and alveolar epithelial layers; AQP5−/− mice were protected from this phenomenon. EBD lung concentration (~8–10 μg/mL), and EBD lung to EBD blood ratios (~0.05) were similar between WT and AQP5−/− mice suggesting that differences in BAL EBD efflux were not due to altered endothelial permeability (not shown). In addition to solute efflux, we assessed airway expression of an adherens junction protein, E-cadherin, in air or CS-exposed mice by lung immunofluorescence (). Air-exposed AQP5−/− mice had increased airway epithelial E-cadherin expression compared with WT mice. CS exposure significantly reduced E-cadherin airway epithelial expression only in WT mice. Similar to what was observed with EBD efflux, AQP5−/− mice resisted CS-induced changes, in this case to a key protein involved in paracellular permeability.
Figure 2. AQP5−/− mice demonstrate reduced epithelial permeability. (A) We assessed permeability in trachea by efflux of Evans’ blue dye (EBD). Trachea from AQP5−/− mice had reduced EBD efflux at baseline. Trachea from WT mice exposed to 1 d of CS had increased EBD efflux compared with trachea from WT mice exposed to air and compared with trachea from both groups of AQP5−/− mice. n = 5 per group, ^p < 0.05 compared with all other groups,*p < 0.05 compared with WT air trachea group. (B) We determined the ratio of BAL EBD to Lung EBD. EBD ratio was increased only in WT mice exposed to 4 weeks of CS exposure, in comparison to air-exposed WT mice and air-exposed AQP5−/− mice. n = 5 per group, p = 0.001. (C) We quantified E-cadherin by lung immunofluorescence. CS-exposed WT mice had a decrease in E-cadherin expression compared with air-exposed WT mice, ^p < 0.05. Both air-exposed and CS-exposed AQP5−/− mice had increased E-cadherin compared with both WT groups (*p < 0.05), and had similar E-cadherin expression to each other. n = 3.
The absence of AQP5 alters alveolar epithelial cell turnover after cigarette smoke
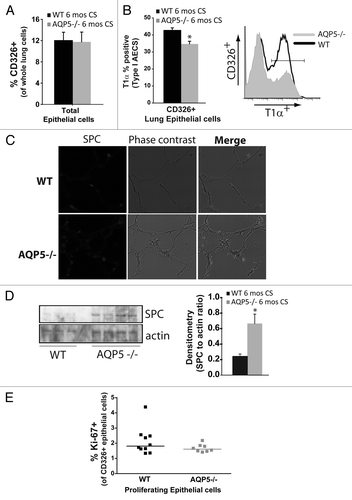
We used flow cytometry to identify and enumerate AQP5-induced differences in lung epithelial cells after chronic smoke exposure. Using total lung cells as the denominator, we found the % of CD326+ cells, a pan-epithelial cell marker, to be similar between CS-exposed strains (). When we assessed the percentage of type I alveolar epithelial cells (AECs) identified by T1α,Citation35 there were significantly fewer in CS-exposed AQP5−/− mice compared with CS-exposed WT mice; a representative flow plot is also shown (). Type II AECs are known to proliferate and transition into type I AECs. Using SPC to identify type II AECs by immunofluorescence, we observed an increase in CS-exposed AQP5−/− mice compared with CS-exposed WT mice (). We further quantified SPC using immunblots of lung homogenate (). AQP5−/− mice exposed to 6 mo of CS express higher levels of lung SPC compared with WT mice. Densitometry of SPC expressed as a ratio of SPC to actin was significantly increased in lungs from CS-exposed AQP5−/− mice. Collectively, this data suggests an increased ratio of type II to type I AEC presence in AQP5−/− mice compared with WT mice exposed to chronic cigarette smoke. We also quantified proliferation (Ki-67+) among lung epithelial cells (CD326+) after 6 mo of CS exposure in WT and AQP5−/− mice; a representative flow diagram demonstrating how CD326 positive and Ki-67 positive lung cells were selected from CS-exposed and air-exposed mice is shown (Fig. S1A). Both WT and AQP5−/− CS-exposed mice had a 5-fold increase in % of Ki-67+ cells, suggesting CS induction of epithelial cell turnover. However, % Ki-67 positivity was similar between CS-exposed WT and AQP5−/− mice among CD326+ cells (). Of note, air and CS-exposed mice had a similar number of total lung cells and a similar percentage of CD326+ cells.
Figure 3. AQP5 alters CS-induced effects on type I and II pneumocyte presence. (A) Using flow cytometry, there was a similar percentage of CD326+ cells, a pan-epithelial marker, between WT and AQP5−/− mice after 6 mo of CS exposure. n = 7–10. (B) Among CD326+ cells, there was a significant reduction in the percentage of T1α positive cells, a type I AEC marker, in AQP5−/− mice exposed to 6 mo CS compared with similarly exposed WT mice. n = 8–10, p = 0.013. A histogram representing percent positivity and mean fluorescence intensity is shown.(C) AQP5−/− mice exposed to CS had increased SPC expression by immunofluorescence (IF), a type II AEC marker, compared with CS-exposed WT mice. (D) AQP5−/− mice exposed to 6 mos CS express higher levels of SPC compared with WT mice exposed to 6 mos CS in homogenized lung; densitometry was expressed as a ratio of SPC to actin. E. Dot plot measuring the percentage of Ki-67+ cells among CD326+ cells demonstrates similar cluster and median values between WT and AQP5−/− mice after 6 mo of CS.
The absence of AQP5 blunts alveolar neutrophil accumulation after sub-acute and chronic CS exposure
In order to identify potential mechanisms by which the absence of AQP5 was protective from CS-induced damage and to provide a potential link between physical and immune barrier properties of the epithelium, we assessed whether AQP5 could influence the inflammatory cell abundance in the alveolar space with CS exposure (4 weeks vs. 6 mo) despite being expressed only on epithelial cells.Citation36 WT and AQP5−/− mice exposed to CS for 6 mo had significant but similar increases in total BAL cells compared with their respective 6 mo air-exposed controls; there was no difference between CS-exposed groups (). However, in assessing the alveolar inflammatory cellular milieu by examining BAL differentials we found that CS-exposed WT mice had significantly more BAL neutrophils compared with air-exposed WT mice or CS-exposed AQP5−/− mice (). CS-exposed WT mice had a significant increase in BAL macrophages compared with air-exposed WT mice (), but were similar compared with CS-exposed AQP5−/− mice. In contrast, CS-exposed AQP5−/− mice had a similar number of BAL macrophages compared with air-exposed AQP5−/− mice. BAL lymphocytes were increased similarly in both CS-exposed mouse strains compared with air-exposed mice ().
Figure 4. Absence of AQP5 mitigates alveolar neutrophil abundance after sub-acute and chronic cigarette smoke. (A) WT and AQP5−/− mice had an increase in total bronchoalveolar (BAL) cells after 6 mo of CS exposure, n = 7–10 per group, *p < 0.05. (B) WT mice had a ~10-fold increase in BAL neutrophils after 6 mo of cigarette smoke exposure, n = 7–10 per group, *p < 0.05 compared with all other groups. (C) WT mice and AQP5−/− mice demonstrate a significant increase in BAL macrophages and BAL lymphocytes after 6 mo of CS compared with WT air-exposed mice; macrophage increase in CS-exposed AQP5−/− mice was not significant compared with air-exposed AQP5−/− mice, n = 8–10 per group, *p < 0.05 compared with both air-exposed groups, ^p < 0.05 compared with WT air-exposed mice. (D) After sub-acute (4-week) CS exposure, WT mice have a ~5-fold increase in alveolar neutrophils compared with all other groups. n = 7–10 per group, p < 0.05. (E) AQP5−/− mice demonstrate a significant increase in BAL macrophages only when compared with air-exposed WT mice after 4-week CS exposure. n = 7–10 per group, p < 0.05. (F) Using adenovirus, we increased AQP5 expression by immunoblot (+AQP5) in A549 cells; CS exposure did not alter AQP5 expression. We measured neutrophil migration across an A549 monolayer. At baseline, there was no AQP5-mediated difference in PMN migration. After CS exposure, there was a significant increase in PMN migration across A549 cells expressing AQP5 (+AQP5) compared with A549 without AQP5 expression (control), n = 4, p < 0.05.
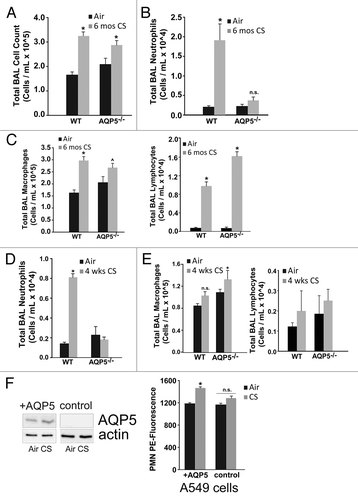
Given the altered inflammatory milieu after chronic CS exposure, we were interested in knowing whether strain differences in BAL inflammatory cell profiles were present after a sub-acute CS exposure (4 weeks) thus preceding AQP5-dependent lung structural changes occurring after chronic CS exposure. Both groups exposed to CS had significant but similar increases in total bronchoalveolar (BAL) cells compared with their respective 4-week air-exposed controls (not shown). Compared with WT air-exposed controls, WT mice exposed to cigarette smoke for 4 weeks had a significant increase in BAL neutrophils (). CS-exposed AQP5−/− mice had similar numbers of BAL neutrophils compared with air-exposed AQP5−/− mice, and significantly less than CS-exposed WT mice, a similar pattern to what we observed after 6 mo of CS. In contrast to BAL neutrophils, 4 weeks of CS exposure did not increase the number of BAL macrophages in WT mice. CS-exposed AQP5−/− mice had a significant, albeit mild, increase in BAL macrophages only when compared with WT air-exposed mice, but not AQP5−/− air-exposed mice (). Therefore, alveolar macrophage differences in WT and AQP5−/− mice were dependent on the length of CS exposure. After 4 weeks of CS exposure, the number of BAL lymphocytes was not different in any of the four groups ().
With persistent reduction in alveolar neutrophil abundance after sub-acute and chronic CS exposure as well as conferred protection from CS-induced structural changes in AQP5−/− mice, we assessed whether the presence of epithelial AQP5 directly impacts neutrophil migration using A549 cells, a human alveolar epithelial cell line (). In the presence of an adenocontrol virus, AQP5 in A549 alveolar epithelial cells is undetectable irrespective of CS exposure (control). Using an adenoviral AQP5 overexpression system, we successfully increased AQP5 expression by immunoblot in A549 cells (+AQP5), which does not change with subsequent CS exposure. There were no differences in neutrophil migration with air exposure. After CS exposure, we observed a significant increase in PMN migration across A549 cells expressing AQP5 (+AQP5) compared with A549 without AQP5 expression (control).
The absence of AQP5 decreases signaling for neutrophil migration to the alveolar space
We have shown that the presence of AQP5 in lung epithelial cells alters barrier function and neutrophil transmigration. However, it is not clear whether the changes in migration are entirely due to AQP5 effects on the paracellular space, or if in addition to this, there are changes in epithelial signaling of neutrophils. To better study this, we investigated additional aspects of epithelial recruitment of neutrophils, including expression of key adhesion molecules and signaling proteins, as well as secretion of chemokines, in response to six months of cigarette smoke exposure.
CD11b expression is critical for migration of neutrophils and macrophages to sites of inflammation especially within the lung, where it interacts with ICAM-1 and other immune modulating receptors.Citation37,Citation38 CD11b expression is sensitive to changes in inflammatory conditions and immune modulators,Citation39,Citation40 the latter of which are known to be different between the apical and basolateral surfaces of the airway/alveolar epithelium,Citation37,Citation41 necessitating measurement in each lung compartment. We utilized established macrophage (F4–80+) and neutrophil (Gr-1+) antibodies to identify each cell population in BAL and lung, and then determined cell-specific CD11b expression by flow cytometry (Fig. S1B).We observed a significant reduction in CD11b expression on BAL neutrophils from 6 mo CS-exposed AQP5−/− mice compared with BAL neutrophils from 6 mo CS-exposed WT mice and to those from AQP5−/− and WT air-exposed mice (). In contrast, CD11b expression was increased to similar levels on BAL macrophages from CS-exposed AQP5−/− and WT mice compared with those from air-exposed AQP5−/− and WT mice (), suggesting neutrophil-specific differences in signaling in AQP5−/− mice exposed to CS.
Figure 5. AQP5 alters neutrophil signaling for migration across the alveolar epithelial layer. (A) Using flow cytometry, CD11b mean fluorescence intensity (MFI) on BAL neutrophils was significantly reduced in CS-exposed AQP5−/− mice compared with CS-exposed WT mice and air-exposed AQP5−/− mice. n = 7–10 per group, *p < 0.05. CD11b MFI on BAL macrophages is similarly increased in CS-exposed WT and AQP5−/− mice. n = 7–10 per group, *p < 0.05. (B) Among total lung cells, the percentage of lung interstitial neutrophils (Gr-1+) is not increased in WT or AQP5−/− mice after 6 mo of CS exposure, but lung neutrophil expression of CD11b is significantly increased in WT and AQP5−/− mice exposed to chronic CS. n = 7–10 per group, *p < 0.05
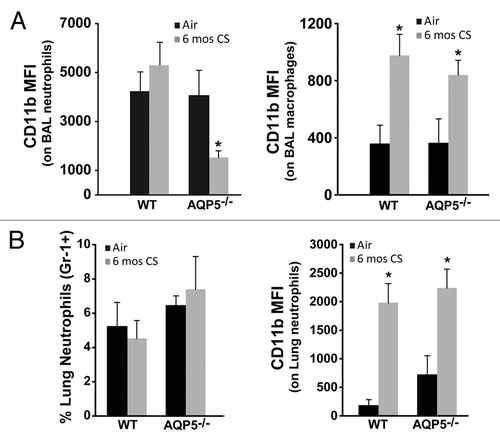
Neutrophil alveolar abundance is determined by changes in chemokine gradient, altered migration across pulmonary endothelial and epithelial layers, or altered life span and removal from the alveolar space. To assess for differences in migration across the endothelium, we measured inflammatory cells in the lung interstitial compartment.Citation42 Following bronchoalveolar lavage, we flushed the vasculature via the right ventricular outflow tract, extracted the lung, isolated cells into single cell suspension, and assessed them by flow cytometry. The total number of lung interstitial cells were similar between CS-exposed AQP5−/− and WT mice (not shown). Chronic CS-exposure did not induce differences in the percentage of lung interstitial neutrophils in either strain (). CD11b expression on lung interstitial neutrophils was significantly higher after CS exposure in both strains, but not different between CS-exposed WT and AQP5−/− mice (). This data demonstrates that significantly fewer neutrophils are present in the alveolar space of CS-exposed AQP5−/− mice despite a similar number of neutrophils with similar expression of CD11b in the lung interstitium compared with CS-exposed WT mice.
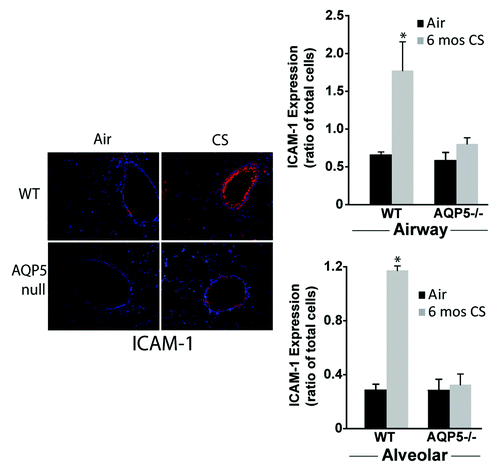
To determine if differences in neutrophil migration across the alveolar space of AQP5−/− or WT mice were influenced by differences in ICAM-1, a known receptor of CD11b, we measured it after 6 mo of CS exposure by immunofluorescence (). ICAM-1 is expressed by several different lung structural cells, so changes in abundance in the whole lung do not necessarily reflect differences in specific cell-types. WT mice exposed to CS had increased alveolar and airway epithelial cell ICAM-1 (labeled red) expression compared with WT mice exposed to air. When enumerated as a ratio of total cells (defined by labeled nuclei), ICAM-1 expression on airway and alveolar epithelial cells from WT mice was significantly increased compared with CS-exposed AQP5−/− mice, and compared with both air-exposed groups. CS-exposed AQP5−/− mice had similar ICAM-1 expression compared with air-exposed AQP5−/− mice. Collectively, this data demonstrates an AQP5-specific modulation of adhesion molecules in the context of cigarette smoke exposure that may contribute to differences in neutrophil migration.
Figure 6. AQP5 alters epithelial expression for ICAM-1 after CS. We measured ICAM-1 immunofluorescence (labeled red) in lung sections after air or chronic CS exposure, and enumerated it as a ratio of total cells (defined by labeled nuclei). WT mice exposed to CS had increased airway (top graph) and alveolar (bottom) epithelial cell ICAM-1 expression compared with air-exposed WT mice. AQP5−/− mice exposed to CS had similar ICAM-1 expression compared with air-exposed AQP5−/− mice and less than WT mice exposed to CS. n = 3, *p < 0.05.
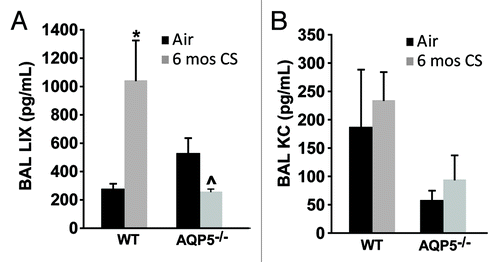
As an additional determinant of alveolar neutrophil abundance, we measured chemokine secretion into the alveolar space focusing on cytokines secreted by epithelial cells. LPS-induced chemokine (LIX) levels were altered with CS exposure (). In WT mice, there was a significant increase in LIX with CS exposure; however in AQP5−/− mice, there was a significant decrease in LIX with CS exposure, and CS-exposed AQP5−/− mice secreted significantly less LIX than their WT counterparts. Keratinocyte chemokine (KC) is secreted by epithelial cells, but also inflammatory cells including macrophages. KC did not significantly change with chronic cigarette smoke and was not different between WT and AQP5−/− at baseline for the mice sampled ().
Figure 7. The absence of AQP5 blunts epithelial-derived chemokine signal for PMN recruitment. (A) BAL LPS-induced chemokine (LIX), primarily epithelial-derived, was significantly increased in CS-exposed WT mice compared with air-exposed WT mice, and significantly reduced in CS-exposed AQP5−/− mice compared with CS-exposed WT mice and air-exposed AQP5−/− mice. n = 4–5, *p < 0.05, ^p = 0.029. (B) BAL keratinocyte chemokine (KC), both epithelial and non-epithelial cell derived, was similar between WT and AQP5−/− mice. n = 4–5.
Discussion
AQP5−/− mice are protected from cigarette smoke-induced emphysema. We demonstrate a novel, dual modulatory role for AQP5 in murine epithelial barrier permeability and innate immunity. In the absence of AQP5, the epithelial barrier is less permeable with fewer alveolar neutrophils detected after sub-acute and chronic cigarette smoke exposure. The reduction in alveolar neutrophils may have occurred due to a combined effect of decreases in paracellular space, decreases in secretion of a potent epithelial-derived neutrophil chemokine LIX, and abrogated communication between neutrophil CD11b ligand with epithelial ICAM-1 adhesion molecule. A decrease in alveolar neutrophil migration likely contributed to protection from CS-induced emphysema development in AQP5−/− mice.
AQP5 is expressed on epithelial cells and not inflammatory cells,Citation36 yet AQP5 effects on epithelial barrier function influenced communication with neutrophils during the innate immune response. There is precedence for the relationship of epithelial barrier to immune function in the gut and skin. Epithelial barrier disruption is causative in the pathogenesis of inflammatory bowel disease,Citation43,Citation44 and part of the pathogenesis of atopic dermatitis.Citation45-Citation48 Even though the lung is also at the interface with the outside world like the skin and gut, we have limited understanding of the influence of its epithelial barrier on the immune response. Historically, the alveolar epithelial barrier has been studied in the context of barrier disruption resulting from significant inflammation as in acute lung injury. In response to mild, chronic inflammation induced by smoke exposure, investigative focus has been primarily on the effects of barrier destruction on the cellular level, and the role of apoptosis in alveolar cell remodeling. Yet little attention has been paid to potential effects of early changes in barrier function on the pathogenesis of emphysema. Since we see changes in epithelial barrier function as early as after one day of cigarette smoke exposure, our data suggests that changes in epithelial barrier function predict altered immune responses to inflammatory stimuli including communication with other inflammatory cells.
Ample evidence exists to support the role of alveolar and airway neutrophils in development of emphysema in mice.Citation18,Citation21 We focused on epithelial AQP5 effects on alveolar neutrophil abundance due to several important factors. One, both macrophages and lymphocytes can proliferate locally,Citation49,Citation50 so crossing the epithelial layer is not a prerequisite for their presence in the alveolar space. Two, a significant reduction in alveolar neutrophils occurred in the context of both sub-acute and chronic CS exposure in AQP5−/− mice compared with WT mice suggesting that changes in neutrophil migration occurred early and preceded the development of emphysema. In contrast, differences in alveolar macrophages were more subtle and variable based on duration of CS exposure in either strain of mice. Furthermore, alveolar lymphocytes increased to similar levels in WT and AQP5−/− mice after chronic cigarette smoke exposure, and are therefore less likely to be dependent on the presence of AQP5. Three, there were similar numbers of neutrophils in the lung interstitium in CS-exposed AQP5−/− mice compared with CS-exposed WT mice (yet still reduced in the alveolar space of CS-exposed AQP5−/− mice), suggesting impairment of neutrophil migration specifically across AQP5-deficient alveolar and airway epithelial layers. The same relationship did not exist between lung interstitial and alveolar macrophages.
To begin to dissect this interesting finding, we assessed signaling primarily in two ways: receptor-ligand expression and chemokine gradients, while recognizing that other factors including neutrophil half-life and rates of removal can be influenced by other cell types. Expression of CD11b, the ligand primarily responsible for neutrophil migration across the alveolar epithelial layer,Citation37 was similar on lung interstitial neutrophils between WT and AQP5−/− mice after cigarette smoke exposure. However, neutrophils from CS-exposed AQP5−/− mice that successfully migrated across the basolateral surface to the apical surface of the alveolar epithelium expressed less CD11b compared with neutrophils from WT mice. On alveolar epithelial cells, ICAM-1 localizes to the apical surface and may regulate alveolar neutrophil abundance by tethering to it as has been shown in other mucosal surfaces.Citation43,Citation51 Reduced ICAM-1 expression on alveolar epithelial cells from AQP5−/− mice may also downregulate alveolar neutrophil CD11b expression, since the signal for epithelial cell – neutrophil interaction is diminished. Once the cell-cell communication is disrupted, neutrophils may be subject to enhanced apoptosis and removal as an alternative explanation for differences in alveolar neutrophil abundance after chronic CS exposure.Citation52,Citation53
While ICAM-1 is integral to communication with CD11b on the apical surface, it is not thought to be expressed on the basolateral surface of alveolar epithelial cells,Citation37,Citation54,Citation55 and therefore may not be responsible for interaction with CD11b on lung neutrophils located in the interstitium, which may explain why we did not observe differences in neutrophil CD11b expression in that compartment. We do not know how AQP5 regulates ICAM-1 expression, but one explanation may be via AQP5-directed coordination of microtubule (MT) dynamics.Citation28 AQP5-derived MT changes could rearrange cytoskeleton and alter epithelial ICAM-1 expression, which others have shown to occur especially when epithelial cells are in communication with inflammatory cells.Citation56 We also observed AQP5-mediated differences in alveolar secretion of an epithelial-derived neutrophil chemokine, LIX, after cigarette smoke exposure. Whether cross-talk between the CD11b-ICAM-1 signaling pathway and the LIX chemokine gradient exists to retard neutrophil migration in AQP5-null mice is not currently known, though it is interesting that alveolar neutrophil CD11b expression and BAL LIX secretion were lower in CS-exposed AQP5−/− mice even compared with air-exposed mice, suggesting active feedback induced by CS.
We cannot exclude the possibility that differences in macrophage sub-populations, their secreted products, or their interactions with epithelial cells were present between WT and AQP5−/− mice and may have affected emphysema development.Citation17,Citation57 Macrophages secrete proteases including MMP-2, MMP-9, MMP-12, cathepsins K, L, S, and elastaseCitation20; in COPD patients compared with normal smokers, macrophages secreted more inflammatory proteins and have greater elastolytic activity. Hautamaki demonstrated the critical role for macrophage-derived elastase, a metalloproteinase that solubilizes many extracellular matrix proteins, in emphysema development after chronic CS exposure in mice.Citation22 Microtubule stability was found to blunt elastase-derived epithelial cell detachment and IL-8 release,Citation58 an interaction which may be altered by AQP5 expression.
In addition to changes that implicate protection from lung structural damage in AQP5−/− mice, there is also evidence supporting an increase in epithelial repair. Compared with CS-exposed WT mice, CS-exposed AQP5−/− mice had an increased number of type II AECs pneumocytes and a reduced number of type I pneumocytes despite similar numbers of total and proliferating lung epithelial cells. Because the most widely accepted dogma is that type II AECs proliferate and differentiate to type 1 AECs, we postulate a decrease in type II AEC to type I AEC transition in CS-exposed AQP5−/− mice as a protective mechanism against emphysema, potentially due to type II AEC-mediated injury repair or surfactant secretion to maintain airway and alveolar stability. AQP5 expression is confined to type I AECs; we are not aware of any known role in modulating type II AEC to type I AEC transition. However, Borok et al. have shown that there is increasing AQP5 abundance during the transitionCitation59 and therefore AQP5 absence could conceivably impact on the transition to full type I AEC differentiation in models of injury and repair.
Human and murine studies have suggested that reduction in AQP5, a water channel, is associated with mucous hyperproduction and may help explain the pathology of airway obstruction.Citation27,Citation60 To our knowledge, we are the first to observe a relationship between AQP5 and development of cigarette-smoke induced structural changes consistent with emphysema. AQP5 is tightly regulated in lung epithelial cells, and dynamically responds to several pathologic stimuli including TNF-α and LPS.Citation28 Acute or chronic CS exposure may also influence alveolar epithelial AQP5 expression and subsequently impact lung structure. We have previously shown that decreased AQP5 lowers levels of assembled microtubules, leading to decreased paracellular permeability.Citation28 Published data suggests that microtubule disassembly induced by nocodazole decreases paracellular permeability in the lung epithelium due to altered membrane expression of E-cadherin.Citation61 A reduction in airway E-cadherin expression correlated with increased barrier permeability in response to CS exposure, an effect dependent on the presence of AQP5. Coordination of microtubule dynamics with resultant changes in paracellular permeability may be an additional explanation for tight AQP5 regulation in the lung epithelium, besides well-established AQP5 regulation of transmembrane water flux. Decreased microtubule assembly due to the absence of AQP5 may affect epithelial signaling pathways which modulate epithelial immune responses. Furthermore, the absence of AQP5 may have adaptive, indirect effects on prevention of permeability and emphysema. Kneidinger showed that activation of the Wnt-B-catenin pathway, recently shown to be prominent in epithelial repair,Citation63 actually protects against development of experimental emphysema. Activation of the Wnt-B-catenin pathway increases lung AQP5 levels.Citation62 One could hypothesize that with a lack of negative feedback, the absence of AQP5 may lead to constitutive upregulation of the Wnt-B-catenin pathway as an adaptive response to promote epithelial repair after cigarette smoke and protect against emphysema.
We present novel data demonstrating AQP5-induced alteration in epithelial barrier predisposing the epithelium to alter its innate immune response to cigarette smoke. Absence of AQP5 minimizes neutrophil migration to the alveolar space and protects against development of emphysema. Further understanding of AQP5-mediated mechanisms changing the epithelial barrier and subsequent innate immunity are necessary, and may provide insight into development of COPD.
Materials and Methods
Animal use and care
Male AQP5−/− mice (on a C57BL/6 background) and wild type (WT) littermates were bred and housed at the Johns Hopkins University Asthma and Allergy Center. Experiments were conducted under a protocol approved by the Johns Hopkins Animal Care and Use Committee.
Murine smoke exposure
WT (littermates) and AQP5−/− mice were exposed in the Johns Hopkins smoke exposure core.Citation24,Citation33,Citation64 Mice were exposed to cigarette smoke 5 h/day, 5 d/week for 4 weeks or 6 mo by burning 3R4F reference cigarettes (2.45 mg nicotine/cigarette; Tobacco Research Institute, University of Kentucky) using a smoking machine (Model TE-10, Teague Enterprises).Citation24,Citation64
Animal harvesting
Mice were anesthetized with intraperitoneal ketamine/acetylpromazine (150/13.5 mg/kg) prior to harvest. At specified time points after cigarette smoke or air exposure, 7–10 animals from various groups were anesthetized and killed by exsanguination from the inferior vena cava. The lungs were perfused free of blood with 1 ml of phosphate-buffered saline (PBS) unless otherwise specified.
Analysis of bronchoalveolar lavage (BAL)
BAL was obtained, total and differential cells counted, and BAL protein assessed as previously described.Citation65 Keratinocyte-derived chemokine (KC) and LPS-induced chemokine (LIX) were measured in BAL and culture medium by ELISA.
Evans blue dye (EBD) assay
In vivo, alveolar
Mice were injected with 20 mg/kg EBD (0.5% Evans blue dissolved in 4% albumin/0.9% saline (PBS, retro-orbital vein) one hour prior to harvest. At harvest, blood was collected from the aorta. The pulmonary circulation was flushed with PBS, followed by lavage and ligation of the right lung, which was then frozen (-80°C). The frozen right lung was homogenized in 1 ml PBS, and then centrifuged at 5,000 x g (7800 rpm, 30 min). The homogenate was diluted in 2 ml of formamide and incubated at 60°C for 18h.
Ex vivo, trachea
Mouse trachea were excised, and both ends of the trachea were cannulated with 18-gauge catheters. In one end, we instilled 50 μL of EBD-4% albumin into the lumen of the trachea for 20 min.Citation34 The tracheal lumen was gently flushed with PBS, homogenized, and processed as above. The supernatant (200 µl aliquots each in 96-well microtiter plates) was collected and the absorbance of tracheal, lung, or BAL supernatants, and serum (diluted 1/10) was measured by spectrophotometer at 620 nm wavelength using a standard curve prepared with serial dilutions of formamide. EBD concentration was multiplied by the dilution factor (3ml + wet weight of R lung) to account for PBS, formamide and water weight of the R lung. Data are presented as EBD µg/R lung, or as a ratio of BAL/Lung EBD.
Lung histology and morphometry
Left lungs were collected and processed for histologyCitation29,Citation66 and morphometryCitation33 by inflating to 25 cm H2O with 0.6% agarose, isolated, sectioned (5 μm), and stained with hematoxylin and formalin (H&E). Fifteen images per mouse were captured at 100X (Nikon Eclipse 50i), and mean linear intercept (MLI) and surface area of airspace wall per unit of lung volume (S/V) ratio were determined by computer-assisted morphometry using a macro designed (courtesy of Rubin Tuder) with MetaMorph software (Molecular Devices).Citation29 MLI was determined by dividing the length of a line drawn across the lung section by the total number of intercepts encountered, serving as a measure of interalveolar septal distance and as an index of alveolar size. S/V was determined by examining 10 random fields per lung using Weibel’s test grid and the formula described by Chalkley,Citation31,Citation32 serving as measure of septal loss and as an index for alveolar destruction.Citation29,Citation30
Lung immunofluorescence
Right lungs were collected, sectioned onto slides, and submerged into 4% formaldehyde/10% formalin in PBS (< 48 h, room temp), and then heated (15 min, 60°C). Slides were deparafinated and hydrated with xylene (2×, 1 min), 100% EtOH (2×, 1 min), 95% EtOH (2×, 1 min), 80% EtOH (1x, 1 min), and then water (1x, 1 min). Citrate buffer (fresh, salts) was added to the slides (20 min, 95C), followed by passive cooling to room temperature. Samples were lined with hydrophobic pen, then blocked (20% serum, 1% BSA, 0.5% Tween-20 in PBS), rinsed (0.3% Triton X-100, 1% BSA), and covered with primary antibody for E-cadherin (1:200, Cell Signaling, #3195S) or ICAM-1 (1:200, Biolegend, #116110) in PBS with 0.3% Triton X-100, 1% BSA for 18 h (4°C). We then washed (2×, 10mins, 0.3% Triton X-100, 1% BSA), and added a secondary antibody (1:400, for E-cadherin-Ax488 Donkey Anti-rabbit IgG, for ICAM-1 Ax549 Donkey anti-mouse IgG, Invitrogen) diluted in 0.3% Triton X-100, 1% BSA (2 h, 25°C) in the dark. SPC primary antibody was diluted in 4% serum/DPBS (1:100, Santa Cruz, Cat # 13979), washed (3×,10 min, DPBS), biotinylated (1:1000 Biotin goat anti-rabbit, Invitrogen, Cat # B-2770), washed again (2×, 5 min, DPBS), and then we added a secondary antibody (1:400, Alexa Fluor 488 streptavidin antibody, Invitrogen, Cat # S-11223) diluted in 4% serum/DPBS (2 h, 25°C) in the dark. After subsequent rinse (2×, 10 min in PBS, then 1×, 10 min in TBS (pH 8.6)), slides were covered with Fluoromount, sealed, and stored at 4°C. Quantification: ImageJ software by quantifying the ratio of mean fluorescence intensity between the protein of interest and the intensity of the nuclei in the same region of interest. This was done from a minimum of 3 different animals per condition.
In vitro smoke exposure and neutrophil migration
A549 cells (ATCC) were grown on collagen-coated inserts at 37°C with 5% CO2 in media (450 mL F12K, 50 mL FBS). We used a Vitrocell smoke chamber to deliver tobacco to A549 cells to mimic tobacco smoke exposure of human epithelium in smokers. The cells were cultured on a 6-well insert as described above allowing medium on the bottom of the porous insert, while the top was exposed to either air (control) or cigarette smoke. We exposed cells to 2 cigarettes (7 min/cigarette). After exposure, the inserts were inverted into a larger dish. Freshly isolated peripheral blood PMNs (courtesy of Bruce Bochner) were labeled with a commercially available fluorescent probe (carboxyfluorescein diacetate (CFDA); excitation/emission spectra similar to FITC to allow PMN tracking.Citation67 Media containing the labeled PMNs were applied to the basolateral membrane of the inverted insert. After 4 h, we collected the basolateral media; inserts were collected and lysed to determine sample fluorescence to objectively quantify PMN transmigration.
Immunoblotting
Lungs or A549 cells were harvested and lysed in RIPA buffer (50 mM TRIS-HCl pH8, 150 mM NaCl, 1% Triton X-100 + Roche Protease Inhibitor, 15 min),Citation28 followed by spin, and Bicinchoninic acid assay ((BCA) Pierce) for total protein in the whole cell lysate. Normalized proteins were loaded onto 10% gel. We then blocked with 4% BSA for 30 min, and incubated with SPC antibody (Santa Cruz Antibody, Cat # 13979), washed (3×) with TBST, then incubated with secondary in 4% BSA (Bio-Rad, Cat # 170–6515) for an hour. Followed by wash and ECL with HyGLO (Denville Scientific, Cat #E2400). Antibodies to the carboxyl-terminus of human AQP5 were generated and probed for as before.Citation28
Flow cytometry
Cells were processed for flow and analyzed as previously described,Citation68 using the following antibodies (or relevant isotypes): anti-LY6G-FITC, anti-CD11b-APCe780, anti-F4/80-APC, anti-CD86-Pacblue, anti-CD326-APC Cy7, anti-T1α-APC, and anti-Ki-67-PE. Mean fluorescence intensity (MFI) was based on the geometric mean expression for a particular marker among cells gated by positive expression.
Statistical analysis
All values are reported as mean ± SEM except , where individual values and the median for each group are shown. Multiple groups were compared using one-way ANOVA with Bonferroni t-test for multiple pairwise comparisons when data are normally distributed or with Kruskal-Wallis assessment on ranks when data are not normally distributed. Two groups were compared using the student’s t-test or Mann-Whitney rank sum test when sample size or variance was not equal. Statistical analysis was performed using Sigmaplot 11.0 (Systat Software). A p < 0.05 was used for significance.
Additional material
Download Zip (547.2 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
Research Support: FAMRI YCSA (NRA, TS, VKS), NIH R01HL089346 (LSK), and Johns Hopkins Bayview Scholars Program (LSK) NIH K08HL085763 (VKS).
Supplemental Material
Supplemental material may be found here: http://www.landesbioscience.com/journals/tissuebarriers/article/25248/
References
- Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet 2007; 370:765 - 73; http://dx.doi.org/10.1016/S0140-6736(07)61380-4; PMID: 17765526
- Podowski M, Calvi C, Metzger S, Misono K, Poonyagariyagorn H, Lopez-Mercado A, et al. Angiotensin receptor blockade attenuates cigarette smoke-induced lung injury and rescues lung architecture in mice. J Clin Invest 2012; 122:229 - 40; http://dx.doi.org/10.1172/JCI46215; PMID: 22182843
- Balamayooran G, Batra S, Cai S, Mei J, Worthen GS, Penn AL, et al. Role of CXCL5 in leukocyte recruitment to the lungs during secondhand smoke exposure. Am J Respir Cell Mol Biol 2012; 47:104 - 11; http://dx.doi.org/10.1165/rcmb.2011-0260OC; PMID: 22362385
- Burke WM, Roberts CM, Bryant DH, Cairns D, Yeates M, Morgan GW, et al. Smoking-induced changes in epithelial lining fluid volume, cell density and protein. Eur Respir J 1992; 5:780 - 4; PMID: 1499700
- Forteza RM, Casalino-Matsuda SM, Falcon NS, Valencia Gattas M, Monzon ME. Hyaluronan and layilin mediate loss of airway epithelial barrier function induced by cigarette smoke by decreasing E-cadherin. J Biol Chem 2012; 287:42288 - 98; http://dx.doi.org/10.1074/jbc.M112.387795; PMID: 23048036
- Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici-Barel Y, et al. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci 2011; 68:877 - 92; http://dx.doi.org/10.1007/s00018-010-0500-x; PMID: 20820852
- Heijink IH, Brandenburg SM, Postma DS, van Oosterhout AJM. Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. Eur Respir J 2012; 39:419 - 28; http://dx.doi.org/10.1183/09031936.00193810; PMID: 21778164
- Gangl K, Reininger R, Bernhard D, Campana R, Pree I, Reisinger J, et al. Cigarette smoke facilitates allergen penetration across respiratory epithelium. Allergy 2009; 64:398 - 405; http://dx.doi.org/10.1111/j.1398-9995.2008.01861.x; PMID: 19120070
- Van Miert E, Dumont X, Bernard A. CC16 as a marker of lung epithelial hyperpermeability in an acute model of rats exposed to mainstream cigarette smoke. Toxicol Lett 2005; 159:115 - 23; http://dx.doi.org/10.1016/j.toxlet.2005.05.007; PMID: 16165332
- Olivera DS, Boggs SE, Beenhouwer C, Aden J, Knall C. Cellular mechanisms of mainstream cigarette smoke-induced lung epithelial tight junction permeability changes in vitro. Inhal Toxicol 2007; 19:13 - 22; http://dx.doi.org/10.1080/08958370600985768; PMID: 17127639
- Rusznak C, Sapsford RJ, Devalia JL, Justin John R, Hewitt EL, Lamont AG, et al. Cigarette smoke potentiates house dust mite allergen-induced increase in the permeability of human bronchial epithelial cells in vitro. Am J Respir Cell Mol Biol 1999; 20:1238 - 50; http://dx.doi.org/10.1165/ajrcmb.20.6.3226; PMID: 10340943
- Nocker RE, Schoonbrood DF, van de Graaf EA, Hack CE, Lutter R, Jansen HM, et al. Interleukin-8 in airway inflammation in patients with asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol 1996; 109:183 - 91; http://dx.doi.org/10.1159/000237218; PMID: 8563494
- Richman-Eisenstat JB, Jorens PG, Hébert CA, Ueki I, Nadel JA. Interleukin-8: an important chemoattractant in sputum of patients with chronic inflammatory airway diseases. [T.] Am J Physiol 1993; 264:L413 - 8; PMID: 8476069
- Qiu Y, Zhu J, Bandi V, Atmar RL, Hattotuwa K, Guntupalli KK, et al. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 168:968 - 75; http://dx.doi.org/10.1164/rccm.200208-794OC; PMID: 12857718
- Morrison D, Strieter RM, Donnelly SC, Burdick MD, Kunkel SL, MacNee W. Neutrophil chemokines in bronchoalveolar lavage fluid and leukocyte-conditioned medium from nonsmokers and smokers. Eur Respir J 1998; 12:1067 - 72; http://dx.doi.org/10.1183/09031936.98.12051067; PMID: 9863998
- Jeyaseelan S, Manzer R, Young SK, Yamamoto M, Akira S, Mason RJ, et al. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am J Respir Cell Mol Biol 2005; 32:531 - 9; http://dx.doi.org/10.1165/rcmb.2005-0063OC; PMID: 15778492
- MacNee W, Wiggs B, Belzberg AS, Hogg JC. The effect of cigarette smoking on neutrophil kinetics in human lungs. N Engl J Med 1989; 321:924 - 8; http://dx.doi.org/10.1056/NEJM198910053211402; PMID: 2779614
- O’Donnell R, Breen D, Wilson S, Djukanovic R. Inflammatory cells in the airways in COPD. Thorax 2006; 61:448 - 54; http://dx.doi.org/10.1136/thx.2004.024463; PMID: 16648353
- Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 1998; 158:1277 - 85; http://dx.doi.org/10.1164/ajrccm.158.4.9802078; PMID: 9769292
- MacNee W. Pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2:258 - 66, discussion 290-1; http://dx.doi.org/10.1513/pats.200504-045SR; PMID: 16267346
- Ofulue AF, Ko M. Effects of depletion of neutrophils or macrophages on development of cigarette smoke-induced emphysema. Am J Physiol 1999; 277:L97 - 105; PMID: 10409235
- Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997; 277:2002 - 4; http://dx.doi.org/10.1126/science.277.5334.2002; PMID: 9302297
- Profita M, Sala A, Bonanno A, Riccobono L, Ferraro M, La Grutta S, et al. Chronic obstructive pulmonary disease and neutrophil infiltration: role of cigarette smoke and cyclooxygenase products. Am J Physiol Lung Cell Mol Physiol 2010; 298:L261 - 9; http://dx.doi.org/10.1152/ajplung.90593.2008; PMID: 19897740
- Le A, Zielinski R, He C, Crow MT, Biswal S, Tuder RM, et al. Pulmonary epithelial neuropilin-1 deletion enhances development of cigarette smoke-induced emphysema. Am J Respir Crit Care Med 2009; 180:396 - 406; http://dx.doi.org/10.1164/rccm.200809-1483OC; PMID: 19520907
- Ning Y, Ying B, Han S, Wang B, Wang X, Wen F. Polymorphisms of aquaporin5 gene in chronic obstructive pulmonary disease in a Chinese population. Swiss Med Wkly 2008; 138:573 - 8; PMID: 18853286
- Hansel NN, Sidhaye V, Rafaels NM, Gao L, Gao P, Williams R, et al. Aquaporin 5 polymorphisms and rate of lung function decline in chronic obstructive pulmonary disease. PLoS One 2010; 5:e14226; http://dx.doi.org/10.1371/journal.pone.0014226; PMID: 21151978
- Wang K, Feng YL, Wen FQ, Chen XR, Ou XM, Xu D, et al. Decreased expression of human aquaporin-5 correlated with mucus overproduction in airways of chronic obstructive pulmonary disease. Acta Pharmacol Sin 2007; 28:1166 - 74; http://dx.doi.org/10.1111/j.1745-7254.2007.00608.x; PMID: 17640479
- Sidhaye VK, Chau E, Srivastava V, Sirimalle S, Balabhadrapatruni C, Aggarwal NR, et al. A novel role for aquaporin-5 in enhancing microtubule organization and stability. PLoS One 2012; 7:e38717; http://dx.doi.org/10.1371/journal.pone.0038717; PMID: 22715407
- Sussan TE, Rangasamy T, Blake DJ, Malhotra D, El-Haddad H, Bedja D, et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci U S A 2009; 106:250 - 5; http://dx.doi.org/10.1073/pnas.0804333106; PMID: 19104057
- Janssens JP, Pache JC, Nicod LP. Physiological changes in respiratory function associated with ageing. Eur Respir J 1999; 13:197 - 205; http://dx.doi.org/10.1183/09031936.99.14614549; PMID: 10836348
- Chalkley HW, Cornfield J, Park H. A Method for Estimating Volume-Surface Ratios. Science 1949; 110:295 - 7; http://dx.doi.org/10.1126/science.110.2856.295; PMID: 17838566
- Choe K-H, Taraseviciene-Stewart L, Scerbavicius R, Gera L, Tuder RM, Voelkel NF. Methylprednisolone causes matrix metalloproteinase-dependent emphysema in adult rats. Am J Respir Crit Care Med 2003; 167:1516 - 21; http://dx.doi.org/10.1164/rccm.200210-1207OC; PMID: 12522028
- Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med 2011; 3:78ra32; http://dx.doi.org/10.1126/scitranslmed.3002042; PMID: 21490276
- Sidhaye VK, Schweitzer KS, Caterina MJ, Shimoda L, King LS. Shear stress regulates aquaporin-5 and airway epithelial barrier function. Proc Natl Acad Sci U S A 2008; 105:3345 - 50; http://dx.doi.org/10.1073/pnas.0712287105; PMID: 18305162
- Yamamoto K, Ferrari JD, Cao Y, Ramirez MI, Jones MR, Quinton LJ, et al. Type I alveolar epithelial cells mount innate immune responses during pneumococcal pneumonia. J Immunol 2012; 189:2450 - 9; http://dx.doi.org/10.4049/jimmunol.1200634; PMID: 22844121
- Raina S, Preston GM, Guggino WB, Agre P. Molecular cloning and characterization of an aquaporin cDNA from salivary, lacrimal, and respiratory tissues. J Biol Chem 1995; 270:1908 - 12; http://dx.doi.org/10.1074/jbc.270.4.1908; PMID: 7530250
- Zemans RL, Colgan SP, Downey GP. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am J Respir Cell Mol Biol 2009; 40:519 - 35; http://dx.doi.org/10.1165/rcmb.2008-0348TR; PMID: 18978300
- Reutershan J, Ley K. Bench-to-bedside review: acute respiratory distress syndrome - how neutrophils migrate into the lung. Crit Care 2004; 8:453 - 61; http://dx.doi.org/10.1186/cc2881; PMID: 15566616
- Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. Lung environment determines unique phenotype of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2009; 296:L936 - 46; http://dx.doi.org/10.1152/ajplung.90625.2008; PMID: 19304907
- Janssen WJ, Barthel L, Muldrow A, Oberley-Deegan RE, Kearns MT, Jakubzick C, et al. Fas determines differential fates of resident and recruited macrophages during resolution of acute lung injury. Am J Respir Crit Care Med 2011; 184:547 - 60; http://dx.doi.org/10.1164/rccm.201011-1891OC; PMID: 21471090
- Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, et al. Neutrophil transmigration triggers repair of the lung epithelium via β-catenin signaling. Proc Natl Acad Sci U S A 2011; 108:15990 - 5; http://dx.doi.org/10.1073/pnas.1110144108; PMID: 21880956
- Garibaldi BT, D’Alessio FR, Mock JR, Files DC, Chau E, Eto Y, et al. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol 2013; 48:35 - 43; http://dx.doi.org/10.1165/rcmb.2012-0198OC; PMID: 23002097
- Chin AC, Parkos CA. Neutrophil transepithelial migration and epithelial barrier function in IBD: potential targets for inhibiting neutrophil trafficking. Ann N Y Acad Sci 2006; 1072:276 - 87; http://dx.doi.org/10.1196/annals.1326.018; PMID: 17057207
- Sydora BC, Macfarlane SM, Walker JW, Dmytrash AL, Churchill TA, Doyle J, et al. Epithelial barrier disruption allows nondisease-causing bacteria to initiate and sustain IBD in the IL-10 gene-deficient mouse. Inflamm Bowel Dis 2007; 13:947 - 54; http://dx.doi.org/10.1002/ibd.20155; PMID: 17427241
- Cork MJ, Danby SG, Vasilopoulos Y, Hadgraft J, Lane ME, Moustafa M, et al. Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol 2009; 129:1892 - 908; http://dx.doi.org/10.1038/jid.2009.133; PMID: 19494826
- Jakasa I, Koster ES, Calkoen F, McLean WH, Campbell LE, Bos JD, et al. Skin barrier function in healthy subjects and patients with atopic dermatitis in relation to filaggrin loss-of-function mutations. J Invest Dermatol 2011; 131:540 - 2; http://dx.doi.org/10.1038/jid.2010.307; PMID: 20962854
- Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006; 38:441 - 6; http://dx.doi.org/10.1038/ng1767; PMID: 16550169
- Elias PM, Schmuth M. Abnormal skin barrier in the etiopathogenesis of atopic dermatitis. Curr Opin Allergy Clin Immunol 2009; 9:437 - 46; http://dx.doi.org/10.1097/ACI.0b013e32832e7d36; PMID: 19550302
- Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J Immunol 2007; 179:3488 - 94; PMID: 17785782
- D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest 2009; 119:2898 - 913; http://dx.doi.org/10.1172/JCI36498; PMID: 19770521
- Jaye DL, Parkos CA. Neutrophil migration across intestinal epithelium. Ann N Y Acad Sci 2000; 915:151 - 61; http://dx.doi.org/10.1111/j.1749-6632.2000.tb05238.x; PMID: 11193572
- Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol 2011; 2:65; http://dx.doi.org/10.3389/fimmu.2011.00065; PMID: 22566854
- Ariel A, Maridonneau-Parini I, Rovere-Querini P, Levine JS, Mühl H. Macrophages in inflammation and its resolution. Front Immunol 2012; 3:324; http://dx.doi.org/10.3389/fimmu.2012.00324; PMID: 23125842
- Kang BH, Crapo JD, Wegner CD, Letts LG, Chang LY. Intercellular adhesion molecule-1 expression on the alveolar epithelium and its modification by hyperoxia. Am J Respir Cell Mol Biol 1993; 9:350 - 5; http://dx.doi.org/10.1165/ajrcmb/9.4.350; PMID: 8104434
- Burns AR, Takei F, Doerschuk CM. Quantitation of ICAM-1 expression in mouse lung during pneumonia. J Immunol 1994; 153:3189 - 98; PMID: 7916369
- Barton WW, Wilcoxen SE, Christensen PJ, Paine R 3rd. Association of ICAM-1 with the cytoskeleton in rat alveolar epithelial cells in primary culture. Am J Physiol 1996; 271:L707 - 18; PMID: 8944713
- Di Stefano A, Caramori G, Ricciardolo FL, Capelli A, Adcock IM, Donner CF. Cellular and molecular mechanisms in chronic obstructive pulmonary disease: an overview. Clin Exp Allergy 2004; 34:1156 - 67; http://dx.doi.org/10.1111/j.1365-2222.2004.02030.x; PMID: 15298554
- Shibata Y, Nakamura H, Kato S, Tomoike H. Cellular detachment and deformation induce IL-8 gene expression in human bronchial epithelial cells. J Immunol 1996; 156:772 - 7; PMID: 8543832
- Borok Z, Lubman RL, Danto SI, Zhang XL, Zabski SM, King LS, et al. Keratinocyte growth factor modulates alveolar epithelial cell phenotype in vitro: expression of aquaporin 5. Am J Respir Cell Mol Biol 1998; 18:554 - 61; http://dx.doi.org/10.1165/ajrcmb.18.4.2838; PMID: 9533944
- Shen Y, Wang Y, Chen Z, Wang D, Wang X, Jin M, et al. Role of aquaporin 5 in antigen-induced airway inflammation and mucous hyperproduction in mice. J Cell Mol Med 2011; 15:1355 - 63; http://dx.doi.org/10.1111/j.1582-4934.2010.01103.x; PMID: 20550619
- Lorenowicz MJ, Fernandez-Borja M, van Stalborch AM, van Sterkenburg MA, Hiemstra PS, Hordijk PL. Microtubule dynamics and Rac-1 signaling independently regulate barrier function in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 2007; 293:L1321 - 31; http://dx.doi.org/10.1152/ajplung.00443.2006; PMID: 17827248
- Königshoff M, Eickelberg O. WNT signaling in lung disease: a failure or a regeneration signal?. Am J Respir Cell Mol Biol 2010; 42:21 - 31; http://dx.doi.org/10.1165/rcmb.2008-0485TR; PMID: 19329555
- Kneidinger N, Yildirim AÖ, Callegari J, Takenaka S, Stein MM, Dumitrascu R, et al. Activation of the WNT/β-catenin pathway attenuates experimental emphysema. Am J Respir Crit Care Med 2011; 183:723 - 33; http://dx.doi.org/10.1164/rccm.200910-1560OC; PMID: 20889911
- Manna SK, Rangasamy T, Wise K, Sarkar S, Shishodia S, Biswal S, et al. Long term environmental tobacco smoke activates nuclear transcription factor-kappa B, activator protein-1, and stress responsive kinases in mouse brain. Biochem Pharmacol 2006; 71:1602 - 9; http://dx.doi.org/10.1016/j.bcp.2006.02.014; PMID: 16569398
- Aggarwal NR, D’Alessio FR, Tsushima K, Files DC, Damarla M, Sidhaye VK, et al. Moderate oxygen augments lipopolysaccharide-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 2010; 298:L371 - 81; http://dx.doi.org/10.1152/ajplung.00308.2009; PMID: 20034961
- Yoshida T, Mett I, Bhunia AK, Bowman J, Perez M, Zhang L, et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat Med 2010; 16:767 - 73; http://dx.doi.org/10.1038/nm.2157; PMID: 20473305
- Davenpeck KL, Chrest FJ, Sterbinsky SA, Bickel CA, Bochner BS. Carboxyfluorescein diacetate labeling does not affect adhesion molecule expression or function in human neutrophils or eosinophils. J Immunol Methods 1995; 188:79 - 89; http://dx.doi.org/10.1016/0022-1759(95)00206-5; PMID: 8551041
- D’Alessio FR, Tsushima K, Aggarwal NR, Mock JR, Eto Y, Garibaldi BT, et al. Resolution of experimental lung injury by monocyte-derived inducible nitric oxide synthase. J Immunol 2012; 189:2234 - 45; http://dx.doi.org/10.4049/jimmunol.1102606; PMID: 22844117