Abstract
Technological advances have enabled researchers to probe gene regulatory pathways on an unprecedented scale. Here, we summarize our recent work that exploits a systematic screening approach in the budding yeast to discover regulators of a promoter of interest. We discuss future applications of our approach based on emerging themes in the literature.
Introduction
Decades of research have produced a general view of the mechanisms of genome programming—that is, how cell identity, morphology and biochemistry are determined by regulating gene expression. The general idea that has emerged is that combinations of proteins, including chromatin proteins, transcription factors (TFs) and other regulators, must conspire to confer specific recognition of genes and their regulatory sequences.Citation1,Citation2 As the roster of regulators involved in controlling gene expression continues to grow, the challenge becomes how to predict functional gene elements and the targets of regulatory proteins from genome sequence. The enormity of this challenge has been emphasized by recent genome-scale analyses which have provided some intriguing insights: (1) comparisons among mammalian genomes shows that most of the conserved and presumably functional sequence lies outside protein-coding regions and likely contains binding sites for regulatory proteins;Citation3,Citation4 (2) metazoans tend to possess a larger number of TFs and more conserved non-coding sequence than simpler organisms, while the number of genes does not increase as greatly, suggesting that complexity in biology must be dictated to a large degree by gene regulation;Citation3,Citation5,Citation6 (3) contextual cues must play a significant role in determining the sequence specificity of TFs since individual TFs typically do not contain enough independent sequence specificity to enable accurate prediction of their in vivo targets.Citation7 Our approach to deciphering the “code” that dictates TF function and specificity is to develop and exploit new technologies and approaches that allow systematic exploration of TF pathways in an experimentally tractable model, the budding yeast Saccharomyces cerevisiae. Below, we briefly describe an approach we developed called Reporter-Synthetic Genetic Array (R-SGA) analysis that allows us to probe the non-essential yeast deletion collection for specific regulators of a promoter-reporter gene of interest.
R-SGA Screening
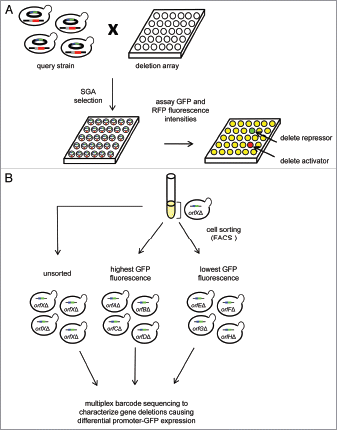
Genetic screens using reporter genes are a powerful tool for discovery of transacting regulatory proteins and upstream signals that confer promoter regulation, although limitations to conventional approaches exist.Citation8 Our major effort has been to develop a sensitive genomicsbased assay for systematic analysis of transcriptional regulatory pathways in yeast. Our approach, called R-SGA analysis, allows examination of the consequences of genetic perturbations (e.g., gene deletion) on any promoter of interest.Citation9 R-SGA involves construction of an otherwise wild type “query” yeast strain that harbors a control promoter (e.g., ACT1 or RPL39) fused to RFP as well as any test promoter of interest fused to GFP. We then use a functional genomics approach, the synthetic genetic array (SGA) methodCitation10–Citation12, to screen the yeast deletion collection for regulators of promoters of interest. SGA allows introduction of a query gene of interest into any appropriately marked array of yeast mutants through a series of robotic replica-pinning steps.Citation10–Citation12 In R-SGA, both the RFP control and GFP test reporter genes are introduced into the set of ∼5,000 haploid deletion mutants. After appropriate selection, an output array is produced where both reporter genes are combined with each deletion mutant. GFP and RFP intensities from the strains on the output array could be rapidly and simply assessed by scanning fluorescence intensities directly from these colonies arrayed on agar plates using a scanning fluorimager.Citation9 After fluorescence quantification, the GFP:RFP ratio is computed. A decreased GFP:RFP ratio identifies potential activators of the test promoter while an increased GFP:RFP ratio may reflect deletion of a repressor. Thus R-SGA allows rapid, saturating surveys of the yeast genome for genes that control transcription of specific promoters.
In previous work, we applied the R-SGA approach to study transcriptional control of histone genes,Citation13 which show peak expression during S phase of the cell cycle and are rapidly repressed as cells transit through subsequent phases. Cell cycle-dependent expression of histone genes is a universal feature of eukaryotic cell cycles and reflects the biological importance of ensuring that adequate histones are available to assemble nucleosomes de novo during S-phase and to prevent the catastrophic effect of inappropriate expression of histone genes at other times during the cell cycle.Citation14–Citation16 Budding yeast contains two copies of each core histone gene, each of which is arranged in opposite orientation to a gene encoding its dimer partner within the nucleosome: HHT1-HHF1 and HHT2-HHF2, the two gene pairs that encode H3/H4, and HTA1-HTB1 and HTA2-HTB2, the two pairs that encode H2A/H2B. The four-component HIR complex (Hir1, Hir2, Hir3 and Hpc2) associates with another histone chaperone Asf1 to repress transcription of three of the four histone gene pairs outside of S-phase.Citation17–Citation20 Despite the identification of the HIR genes and their role in histone gene expression more than 20 years ago, the precise mechanisms controlling histone gene expression have remained unclear. Because of this gap in our knowledge, we decided to explore histone gene expression as a test-case for our R-SGA method. We fused the promoter of the HTA1 gene to GFP and carried out an R-SGA screen to identify new regulators of histone gene transcription. We examined our R-SGA data for potential new regulators of histone gene expression.Citation13 We noticed that deletion of a histone chaperone, Rtt106, caused a similar derepression of histone gene transcription to that seen in HIR mutants. We used transcript profiling by quantitative PCR and ChIP experiments to discover that Rtt106 is a previously unappreciated member of the HIR regulatory pathway, which acts through a defined cis-regulatory site (NEG) in the promoters of most histone genes to create a repressive chromatin structure.
We scanned our R-SGA results for factors that relieve Rtt106-HIR-mediated repression and noticed that deletion of the bromodomain-containing protein Yta7 is required for proper activation of histone H2A (HTA1) transcription. The bromodomain is found in chromatin-associated proteins and histone acetyltransferases and functions as a protein module known to bind acetyl-lysine motifs.Citation21 We then used chromatin-immunoprecipitation (ChIP) experiments to show that Yta7 acts as a chromatin boundary protein that is required to restrict Rtt106 to the NEG site within the promoter of histone genes.Citation13 Deletion of YTA7 causes lateral spreading of Rtt106 from its position on the promoter into the ORF of histone genes where it is normally not present. Our experiments suggest that HIR/Rtt106-mediated repressive chromatin domains are the predominant means of cell cycle regulation of histone promoters and that Yta7 acts to prevent the spreading of repressive chromatin.
Since R-SGA screens identify input signals that control individual promoters, we next asked if the HIR/Rtt106 pathway operates at other promoters throughout the genome by performing a genome-wide nucleosome occupancy experiment in hir1Δ and rtt106Δ mutants. In brief, mononucleosomal samples were prepared from wild-type or mutant yeast cultures and samples of nucleosomal and total genomic DNA were hybridized to an Affymetrix tiling array with 4 bp resolution.Citation22 We found depletion of nucleosomes at a number of promoters throughout the genome including those of histone genes, as expected from our primary R-SGA screen.Citation13 The nucleosome profiles suggested that important aspects of the HIRRtt106 regulatory pathway remain to be discovered. We identified a subset of promoters whose nucleosome occupancy profile mirrored that of the histone genes, showing dependence on both HIR and Rtt106. Other promoters were sensitive to only Rtt106 or Hir1, but not both. These results suggest that the HIR-Rtt106-Yta7 pathway may include other components and likely represents a chromatin regulatory mechanism broadly applied across the genome, an idea we are examining in more detail by carrying out ChIP-seq experiments on TAP-tagged versions of Rtt106, Hir1 and Yta7.
Expansion of the R-SGA toolkit.
So far, we have used R-SGA to query the yeast deletion mutant collection, which has arguably revolutionized the functional characterization of yeast genes.Citation23,Citation24 However, deletions are only one type of genetic reagent, and in some cases other types of alleles are more useful. In an extreme case, ∼1,000 yeast genes (∼18%) are essential for haploid viabilityCitation25 and the biological attributes of this important gene set cannot be analyzed with deletion alleles. However, collections of yeast strains containing conditional alleles of essential genes are available in a format compatible with R-SGA. These collections include TET-repressible promoter replacement allelesCitation26,Citation27 as well as temperature sensitive alleles of many essential genes.Citation28 The effect of essential gene perturbation on transcription factor pathways could be explored using R-SGA with only minor changes to the protocol.
In other work, we have expanded the general SGA platform to include the capacity to systematically assay the effects of gene overexpression, which also allows assessment of essential genes. This effort involved construction of an “overexpression array” of 5,280 yeast strains, each containing an inducible copy of a yeast gene, covering 80% of the yeast genome.Citation29,Citation30 The first overexpression array we constructed expresses a different yeast ORF tagged at its amino terminus with GST from the inducible GAL1/10 promoter on a multicopy plasmid.Citation29,Citation31 This array can be manipulated using the SGA method to combine any marked gene of interest, including GFP and RFP reporter genes, with overexpression of each ORF when grown in the presence of galactose. In this case, we expect overexpressing an activator of the promoter of interest to result in increased GFP:RFP fluorescence while overexpressing a repressor will cause decreased GFP:RFP fluorescence. This array has been used in systematic overexpression genetic studies to identify kinase targetsCitation29,Citation32,Citation33 and to explore TF pathways. The exploration of the effects of gene overexpression on promoters using R-SGA is likely to be quite fruitful since deletion of many transcription factors results in little effect on target genes examined by gene expression microarrays, likely because the transcription factor is inactive under the conditions tested.Citation34 However, overexpression often results in a gene expression pattern above noise indicating that artificially activating expression of these proteins bypasses the need for specific activating conditions and can be used to identify target genes of transcription factors.Citation34
Other Applications of the R-SGA Approach
Several studies have reported variations in gene expression among individuals,Citation35–Citation40 but the contribution that cis-regulatory elements play in mediating this variation has only recently been explored. Two recent studies examined variation in transcription factor binding in both yeastCitation41 and humans.Citation42 In one study, binding of the transcription factor Ste12 was monitored using ChIP-seq in segregants of a cross between two highly diverged yeast strains relative to both parental strains.Citation41 Most binding variation appeared to be the result of polymorphisms [usually single nucleotide polymorphisms (SNPs) or inserts or deletions (indels)] in Ste12 binding sites or other sites that might bind cofactors of Ste12. Analysis of gene expression by microarrays confirmed that binding variation often correlated with transcription, indicating that variation in TF binding due to binding site variation is in fact biologically relevant. In a related study, NFκB and RNA polymerase II binding were monitored in ten lymphoblastoid cell lines using ChIP-seq, again revealing binding differences.Citation42 These differences were often attributed to SNPs and genomic structural variants. Similar to what was observed in yeast, variation in binding affected gene expression, indicating binding variation is functional.
The results of these and other studies suggest differences in transcriptional control play a role in phenotypic differences among individuals and that polymorphisms in cis-regulatory sequence may contribute significantly to these differences. One way to explore the functional consequences of polymorphisms on cis-regulatory sequences in more detail is to carry out studies to identify trans-acting regulators and upstream pathways of these regulators that act through transcription factor binding sites and ultimately control gene expression. Above, we described our R-SGA approach which could be used to screen promoter-GFP reporter genes harboring polymorphisms identified in cisregulatory sequences like those described for Ste12 and NFκB. This type of reporter screen would likely yield new regulatory pathways that control gene expression in individuals and could pinpoint the underlying molecular mechanisms occurring as a consequence of transcription factor binding variation. In future work we plan to further develop the R-SGA strategy to allow pooled reporter screens by combining bar-coded gene disruption libraries (e.g., yeast deletion library or RNAi gene knockdown libraries), fluorescent reporter genes and fluorescence activated cell sorting (FACS) (see and ref. Citation8). This methodology would allow systematic reporter screens in mammalian cells enabling, for example, a panel of promoter-GFP constructs with variable NFκB binding sites to be screened for novel regulators.
Abbreviations
| TFs | = | transcription factors |
| R-SGA | = | reporter-synthetic genetic array |
| SGA | = | synthetic genetic array |
| NEG | = | negative regulatory site |
| SNPs | = | single nucleotide polymorphisms |
| indels | = | inserts or deletions |
| RNAi | = | RNA interference |
| FACS | = | fluorescence activated cell sorting |
Figures and Tables
Figure 1 Quantitative promoter-reporter screening to identify regulators of gene expression. (A) The Reporter-Synthetic Genetic Array (R-SGA) approach (reviewed in ref. Citation9). (B) Combing FACS with pooled cultures of deletion mutants to identify transcriptional regulators of a promoter-GFP reporter gene. The reporter gene can be combined with each deletion mutant using the SGA methodology (see part A) and the resulting array of output strains can be pooled into a single culture. The cells can be sorted physically using FACS into different populations depending on the level of GFP expression in each strain. Cells are expected to be sorted into the brightest population if the deleted gene is a repressor of the promoter driving GFP expression while cells which have a deletion in a gene required for activation of the promoter driving GFP expression are expected to be sorted into the dimmest population of cells. Since each deletion mutant is barcoded,Citation25 the various deletion strains in each population can be identified by sequencing each barcode.Citation43
References
- Barrera LO, Ren B. The transcriptional regulatory code of eukaryotic cells—insights from genome-wide analysis of chromatin organization and transcription factor binding. Curr Opin Cell Biol 2006; 18:291 - 298
- Veitia RA. One thousand and one ways of making functionally similar transcriptional enhancers. Bioessays 2008; 30:1052 - 1057
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm and yeast genomes. Genome Res 2005; 15:1034 - 1050
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002; 420:520 - 562
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J. Initial sequencing and analysis of the human genome. Nature 2001; 409:860 - 921
- Larroux C, Luke GN, Koopman P, Rokhsar DS, Shimeld SM, Degnan BM. Genesis and expansion of metazoan transcription factor gene classes. Mol Biol Evol 2008; 25:980 - 996
- Megraw M, Pereira F, Jensen ST, Ohler U, Hatzigeorgiou AG. A transcription factor affinity-based code for mammalian transcription initiation. Genome Res 2009; 19:644 - 656
- Kainth P, Andrews B. Quantitative cell array screening to identify regulators of gene expression. Brief Funct Genomics 2010; 9:13 - 23
- Kainth P, Sassi HE, Peña-Castillo L, Chua G, Hughes TR, Andrews B. Comprehensive genetic analysis of transcription factor pathways using a dual reporter gene system in budding yeast. Methods 2009; 48:258 - 264
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Pagé N, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 2001; 294:2364 - 2368
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, et al. Global mapping of the yeast genetic interaction network. Science 2004; 303:808 - 813
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, et al. The genetic landscape of a cell. Science 2010; 327:425 - 431
- Fillingham J, Kainth P, Lambert JP, van Bakel H, Tsui K, Peña-Castillo L, et al. Two-color cell array screen reveals interdependent roles for histone chaperones and a chromatin boundary regulator in histone gene repression. Mol Cell 2009; 35:340 - 351
- Gunjan A, Verreault A. A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 2003; 115:537 - 549
- Gunjan A, Paik J, Verreault A. Regulation of histone synthesis and nucleosome assembly. Biochimie 2005; 87:625 - 635
- Hereford LM, Osley MA, Ludwig TR 2nd, McLaughlin CS. Cell cycle regulation of yeast histone mRNA. Cell 1981; 24:367 - 375
- Osley MA, Lycan D. Trans-acting regulatory mutations that alter transcription of Saccharomyces cerevisiae histone genes. Mol Cell Biol 1987; 7:4204 - 4210
- Green EM, Antczak AJ, Bailey AO, Franco AA, Wu KJ, Yates JR 3rd, et al. Replication-independent histone deposition by the HIR complex and Asf1. Curr Biol 2005; 15:2044 - 2049
- Prochasson P, Florens L, Swanson SK, Washburn MP, Workman JL. The HIR corepressor complex binds to nucleosomes generating a distinct protein/DNA complex resistant to remodeling by SWI/SNF. Genes Dev 2005; 19:2534 - 2539
- De Koning L, Corpet A, Haber JE, Almouzni G. Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol 2007; 14:997 - 1007
- Mujtaba S, Zeng L, Zhou MM. Structure and acetyllysine recognition of the bromodomain. Oncogene 2007; 26:5521 - 5527
- Lee W, Tillo D, Bray N, Morse RH, Davis RW, Hughes TR, et al. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet 2007; 39:1235 - 1244
- Fisk DG, Ball CA, Dolinski K, Engel SR, Hong EL, Issel-Tarver L, et al. Saccharomyces cerevisiae S288C genome annotation: a working hypothesis. Yeast 2006; 23:857 - 865
- Scherens B, Goffeau A. The uses of genome-wide yeast mutant collections. Genome Biol 2004; 5:229
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002; 418:387 - 391
- Mnaimneh S, Davierwala AP, Haynes J, Moffat J, Peng WT, Zhang W, et al. Exploration of essential gene functions via titratable promoter alleles. Cell 2004; 118:31 - 44
- Davierwala AP, Haynes J, Li Z, Brost RL, Robinson MD, Yu L, et al. The synthetic genetic interaction spectrum of essential genes. Nat Genet 2005; 37:1147 - 1152
- Ben-Aroya S, Coombes C, Kwok T, O'Donnell KA, Boeke JD, Hieter P. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol Cell 2008; 30:248 - 258
- Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell 2006; 21:319 - 330
- Zhu H, Klemic JF, Chang S, Bertone P, Casamayor A, Klemic KG, et al. Analysis of yeast protein kinases using protein chips. Nat Genet 2000; 26:283 - 289
- Sopko R, Papp B, Oliver SG, Andrews BJ. Phenotypic activation to discover biological pathways and kinase substrates. Cell Cycle 2006; 5:1397 - 1402
- Huang D, Kaluarachchi S, van Dyk D, Friesen H, Sopko R, Ye W, et al. Dual regulation by pairs of cyclin-dependent protein kinases and histone deacetylases controls G1 transcription in budding yeast. PLoS Bio 2009; 7:1000188
- Sopko R, Huang D, Smith JC, Figeys D, Andrews BJ. Activation of the Cdc42p GTPase by cyclindependent protein kinases in budding yeast. EMBO J 2007; 26:4487 - 4500
- Chua G, Morris QD, Sopko R, Robinson MD, Ryan O, Chan ET, et al. Identifying transcription factor functions and targets by phenotypic activation. Proc Natl Acad Sci USA 2006; 103:12045 - 12050
- Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, et al. Genetic analysis of genome-wide variation in human gene expression. Nature 2004; 430:743 - 747
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature 2003; 422:297 - 302
- Brem RB, Yvert G, Clinton R, Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science 2002; 296:752 - 755
- Yvert G, Brem RB, Whittle J, Akey JM, Foss E, Smith EN, et al. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat Genet 2003; 35:57 - 64
- Ronald J, Brem RB, Whittle J, Kruglyak L. Local regulatory variation in Saccharomyces cerevisiae. PLoS Genet 2005; 1:25
- Li Y, Alvarez OA, Gutteling EW, Tijsterman M, Fu J, Riksen JA, et al. Mapping determinants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet 2006; 2:222
- Zheng W, Zhao H, Mancera E, Steinmetz LM, Snyder M. Genetic analysis of variation in transcription factor binding in yeast. Nature 2010; 464:1187 - 1191
- Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, et al. Variation in transcription factor binding among humans. Science 2010; 328:232 - 235
- Smith AM, Heisler LE, Mellor J, Kaper F, Thompson MJ, Chee M, et al. Quantitative phenotyping via deep barcode sequencing. Genome Res 2009; 19:1836 - 1842