Transcription-Associated Mutagenesis at UV-Photolesions
Transcription is quintessential for life. Although at least 50% of the mammalian genome is transcribed, frequently into regulatory RNA molecules,Citation1 only a minor fraction of the genome consists of protein-encoding genes that can be transcribed by a multiprotein complex that includes RNA polymerase II. In the last decades, evidence has accumulated that there is also a downside to transcription. Research conducted mainly in bacteria and lower eukaryotes has demonstrated that a high level of transcription at undamaged templates induces genome instability, including nucleotide substitution mutations, deletions and chromosome rearrangements.Citation2,Citation3 In mammals these classes of mutations are associated with the oncogenic derailment of stem cells. Also transcription at damaged templates is associated with genomic instability. Ultraviolet (UV) light is a prevalent environmental mutagen and carcinogen. We have recently found in mouse embryonic stem (ES) cells that UV light-induced lesions at the template strand for transcription in a reporter gene become much more mutagenic when the gene is transcribed.Citation4 Two novel classes of UV-induced transcription-associated mutations (UV-TAM) were found. The first class was comprised of nucleotide substitutions, by a pathway dubbed UV-induced transcription-associated point mutagenesis (UV-TAP). Specifically, frequencies of CC to TT and C to T transition mutations, derived from photolesions residing at the template strand for transcription, were increased when the gene was transcribed during and after UV exposure of the cells.Citation5 We have found that dicytosines, embedded within a cyclobutane pyrimidine dimer (CPD) photolesion, are prone to deamination to UU when the template is transcribed.Citation5 During replication, these deoxyuridine residues within a CPD photolesion direct the incorporation of deoxyadenosine, leading to the observed CC to TT transition mutations. Although these were not separately investigated, transcription-associated single C to T transitions are most probably generated by the same mechanism. Based on these and previousCitation6 studies we infer that it is the prolonged presence of cytosine-containing CPDs in single-stranded DNA at the stalled transcription complex that predisposes them to spontaneous deamination. Since particularly cytosines that reside in photolesions are prone to deamination,Citation6 this class of TAM is probably restricted to UV-induced DNA damage. Supporting this hypothesis, Benzo(a)pyrene diolepoxyde (BPDE) induces transcription-associated transversion mutations originating from lesions at the non-transcribed strand for transcription (Hendriks et al., in preparation),Citation4 in sharp contrast to UV that induces transcription-associated transitions at the transcribed strand.
In addition to inducing nucleotide transitions, UV exposure at an active reporter gene induced transcriptionassociated deletions (UV-TAD) that are most likely caused by error-prone end joining of double stranded DNA breaks (DSBs). These DSBs may mechanistically be related to those that underlie transcription-associated recombination (TAR) in mammalian cells, in the absence of template damageCitation7 or at UV-damaged templates.Citation8 At undamaged templates, DNA-RNA hybrids (R-loops) that persist behind the transcribing RNA polymerase II, are involved in the induction of DSBs.Citation9 The dependence of TAR on replication indicates that a replication fork that encounters an R-loop may result in the collapse of the fork and the generation of a DSB.Citation7 We hypothesize that, when RNA polymerase II remains stalled at photolesions within the template.Citation10 R loops persist, which increases their chance of collision with replication forks. Also BPDE induces TAD in ES cells, suggesting that TAD may be triggered by any lesion that arrests transcription (manuscript in preparation).
Transcription-Coupled Nucleotide Excision Repair Provides Specific Protection Against UV-TAM
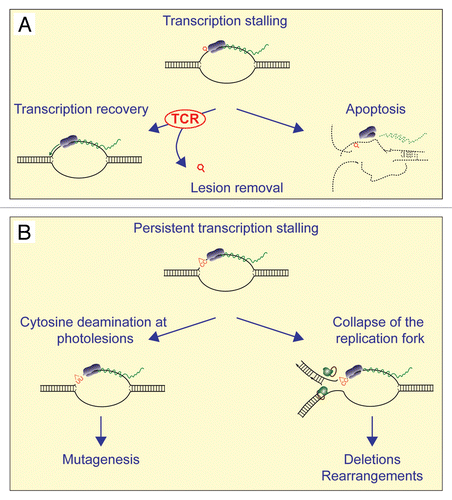
DNA lesions that arrest transcription can be removed from the template DNA by the transcription-coupled sub-pathway of nucleotide excision repair (TCR).Citation11 TCR is initiated by stalling of the transcription machinery at a DNA lesion, resulting in the recruitment of nucleotide excision repair (NER) factors that remove the lesion from the DNA. In this fashion, the recovery of transcription after exposure to DNA-damaging agents is ensured, and this generally is considered to be the primary function of TCR ().Citation11 However, the specific characteristics of UV-TAM provide an additional raison d'être for TCR (). Most DNA-damaging agents induce nucleotide substitution mutagenesis via error-prone translesion synthesis opposite the template lesion during DNA replication. In contrast, UV-TAM is determined by deamination during transcription and by subsequent error-free (but mutagenic) translesion synthesis, presumably by DNA polymerase η.Citation12 Since they are extremely mutagenic, cytosinecontaining CPD photolesions at a stalled transcription complex or their deaminated derivatives, should efficiently be repaired. CPDs are a relatively poor substrate for global-genome NER.Citation13,Citation14 This puts the burden of preventing UV-induced TAP on TCR, which efficiently removes CPDs from the transcriptional template. In conclusion, TCR forms the major barrier against the deamination-dependent mutagenicity of cytosine-containing CPDs, in addition to its roles in transcription recovery and in preventing UV-TAR and presumably TAD.Citation4,Citation5,Citation8
Our finding of UV-TAP and its prevention by TCR explains previous data from others. For instance, most UV-induced C to T transitions at the Hprt locus in TCR-deficient hamster and human cells are derived from photolesions at the transcribed strand.Citation15 Cells that are deficient for Cockayne Syndrome B (CSB) protein, a key factor in TCR, show a strong bias in mutations induced by photolesions at the template strand for transcription and an excessive apoptotic response when exposed to UV light.Citation16
Transcription Arrest, Apoptosis and TAM
TCR-deficient epidermal cells in situ display high levels of apoptosis upon the infliction of DNA damage, indicating that the inability to remove lesions at the template strand for transcription efficiently initiates DNA damage responses.Citation19 Indeed, persistent stalling of the elongating transcription complex induces strong p53 activation.Citation20 It was hypothesized that the transcription arrest-induced apoptosis serves as a general DNA damage sensor, resulting in the elimination of cells that contain high levels of DNA damage (). This would provide additional protection against the possible mutagenic and carcinogenic consequences of unrepaired genomic DNA damage in mammals.Citation16,Citation21,Citation22 Although attractive, such a hypothesis contains a paradoxical element. As argued above, mutations usually are generated during error-prone replication of damaged DNA, rather than during transcription. This suggests that only replicating cells are at risk for mutagenesis upon the infliction of DNA damage and need to initiate apoptosis when the damage load entails a too high risk for losing genome integrity. In agreement, when replication is arrested at lesions, DNA damage responses are efficiently induced.Citation20,Citation23 What would be the added benefit of the induction of DNA damage responses by arrested transcription complexes in non-replicating cells, when genome stability is threatened only during replication? The finding that UV-TAP is determined at the level of (arrested) transcription, rather than during replication, may provide an answer to this conundrum. If the transcriptionarresting UV lesions are not repaired efficiently by TCR, deamination of cytosine-containing CPDs may occur and this will inevitably result in the induction of transition mutations when the cell would subsequently enter S phase. Similarly, the persistence of R-loops at transcription complexes, arrested at lesions within the template, entails a very high risk of replication fork collapse, genomic rearrangements and deletions. To avoid such detrimental TAM, transcription arrests sensitize the cells to apoptosis, so that if the cells enter S phase, the apoptotic pathway is effectuated. In support, the efficient induction of apoptosis by arrested transcription complexes requires replication.Citation21 Summarizing, the induction of apoptosis by transcription-arresting lesions depends on the integration of DNA damage responses that are induced by both transcription arrests and replication.
Our and others' dataCitation4,Citation5,Citation8 indicate that TCR provides a rather moderate level of protection against UV-TAM. UV doses that cause similar frequencies of UV-TAM in NER-proficient and NER-deficient ES cells, reflecting similar numbers of persisting lesions, induce very similar cell death.Citation4,Citation5 This finding indicates the presence of a threshold level of lesions at the template strands for transcription above which transcriptional stalling will induce apoptosis, when cells enter S phase. We hypothesize that this strong apoptotic response will eliminate cells with an excessive risk of UV-TAM and, by implication, prevents associated (skin) carcinogenesis.
Despite their TCR deficiency, CSBdeficient humans develop no UV-induced skin cancer.Citation17 These patients are proficient for the global genome NER that might remove a fraction of the mutagenic CPD lesions from transcribed templates in the absence of TCR, thereby suppressing carcinogenesis, as was recently found for CSB-deficient mice.Citation18 Nevertheless, the remarkable lack of skin cancer supports the idea that the efficient induction of apoptosis by arrested transcription complexes, despite the presence of TAP, provides protection against oncogenic transformation of the cell.
Conclusions and Perspective
The discovery of two novel classes of UV-TAM may provide a new rationale for the interacting roles of TCR and of transcription arrest-induced apoptosis in responses to UV light-induced DNA damage. We and othersCitation3 hypothesize that these responses are crucial in preventing the emergence of mutant cell clones. This is of particular relevance for transcribed growth-controlling genes in mammalian dermal stem cells as they prevent the oncogenic derailment of these cells. Further research using defined mouse models for UV-TAM should bear this out.
Figures and Tables
Figure 1 Model for the roles of TCR and transcription arrest-associated apoptosis in protection against UV-TAM. (A) TCR is initiated by stalling of the elongating RNA polymerase and results in removal of the transcription-blocking photolesion, thereby allowing transcription recovery. In case the load of photolesions exceeds the capacity of TCR or when TCR is impaired, arrested transcription complexes induce apoptosis when the cell enters S phase. (B) Persistent stalling of the transcription complex at a deoxycytidine-containing-CPD at the template strand results in its increased spontaneous deamination frequency. In case the lesion is not timely removed by TCR, or when the cell escapes from apoptosis, it will result in a C to T transition mutation during replication. Alternatively, persistent stalling of the transcription complex at a photolesion may lead to collapse of an approaching replication fork, resulting in DSBs and genome instability.
Acknowledgements
This work was supported by the Dutch Cancer Society (grant RUL 2002-2736).
References
- Jacquier A. The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat Rev Genet 2009; 10:833 - 844
- Svejstrup JQ. The interface between transcription and mechanisms maintaining genome integrity. Trends Biochem Sci 2010; http://dx.doi.org/10.1016/j.tibs.2010.02.001
- Helleday T. Mutagenesis: Mutating a gene while reading it. Curr Biol 2010; 20:57 - 58
- Hendriks G, Calleja F, Vrieling H, Mullenders LH, Jansen JG, de Wind N. Gene transcription increases DNA damage-induced mutagenesis in mammalian stem cells. DNA Repair 2008; 7:1330 - 1339
- Hendriks G, Calleja F, Besaratinia A, Vrieling H, Pfeifer GP, Mullenders LH, et al. Transcription-dependent cytosine deamination is a novel mechanism in ultraviolet light-induced mutagenesis. Curr Biol 2010; 20:170 - 175
- Frederico LA, Kunkel TA, Shaw BR. A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and the activation energy. Biochemistry 1990; 29:2532 - 2537
- Gottipati P, Cassel TN, Savolainen L, Helleday T. Transcription-associated recombination is dependent on replication in Mammalian cells. Mol Cell Biol 2008; 28:154 - 164
- Deng WP, Nickoloff JA. Preferential repair of UV damage in highly transcribed DNA diminishes UV-induced intrachromosomal recombination in mammalian cells. Mol Cell Biol 1994; 14:391 - 399
- Tuduri S, Crabbe L, Conti C, Tourriere H, Holtgreve-Grez H, Jauch A, et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol 2009; 11:1315 - 1324
- Selby CP, Drapkin R, Reinberg D, Sancar A. RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res 1997; 25:787 - 793
- Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 2008; 9:958 - 970
- Takasawa K, Masutani C, Hanaoka F, Iwai S. Chemical synthesis and translesion replication of a cis-syn cyclobutane thymine-uracil dimer. Nucleic Acids Res 2004; 32:1738 - 1745
- van der Wees C, Jansen J, Vrieling H, van der Laarse A, Van Zeeland A, Mullenders L. Nucleotide excision repair in differentiated cells. Mutat Res-Fund Mol M 2007; 614:16 - 23
- Nouspikel T. DNA Repair in Mammalian Cells. Cell Mol Life Sci 2009; 66:965 - 967
- van Zeeland AA, Vreeswijk MP, de Gruijl FR, van Kranen HJ, Vrieling H, Mullenders LF. Transcription-coupled repair: impact on UV-induced mutagenesis in cultured rodent cells and mouse skin tumors. Mutat Res 2005; 577:170 - 178
- Ljungman M. The transcription stress response. Cell Cycle 2007; 6:2252 - 2257
- Wijnhoven SWP, Hoogervorst EM, de Waard H, van der Horst GTJ, van Steeg H. Tissue specific mutagenic and carcinogenic responses in NER defective mouse models. Mutat Res-Fund Mol M 2007; 614:77
- Pines A, Hameetman L, de Wilde J, Alekseev S, de Gruijl FR, Vrieling H, et al. Enhanced global genome nucleotide excision repair reduces UV carcinogenesis and nullifies strand bias in p53 mutations in Csb-/- mice. J Invest Dermatol 2010; 130:1746 - 1749
- van Oosten M, Rebel H, Friedberg EC, van Steeg H, van der Horst GTJ, van Kranen HJ, et al. Differential role of transcription-coupled repair in UVB-induced G2 arrest and apoptosis in mouse epidermis. P Natl Acad Sci USA 2000; 97:11268 - 11273
- Derheimer FA, O'Hagan HM, Krueger HM, Hanasoge S, Paulsen MT, Ljungman M. RPA and ATR link transcriptional stress to p53. P Natl Acad Sci USA 2007; 104:12778 - 12783
- Ljungman M, Lane DP. Transcription—guarding the genome by sensing DNA damage. Nat Rev Cancer 2004; 4:727 - 737
- Reefman E, Limburg PC, Kallenberg CG, Bijl M. Apoptosis in human skin: role in pathogenesis of various diseases and relevance for therapy. Ann N Y Acad Sci 2005; 1051:52 - 63
- Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair 2009; 8:1038 - 1046