Abstract
B cells utilize three DNA alteration strategies—V(D)J recombination, somatic hypermutation (SHM) and class switch recombination (CSR)—to somatically mutate their genome, thereby expressing a plethora of antibodies tailor-made against the innumerable antigens they encounter while in circulation. Of these three events, the single-strand DNA cytidine deaminase, Activation Induced cytidine Deaminase (AID), is responsible for SHM and CSR. Recent advances, discussed in this review article, point toward various components of RNA polymerase II “stalling” machinery as regulators of AID activity during antibody diversification and maintenance of B cell genome integrity.
Antibody diversification mechanisms in B-lymphocytes
Ever since Frank Macfarlane Burnet proposed the clonal selection theory to account for antibody diversification,Citation1 questions have been raised about the molecular mechanism(s) by which the vast reservoir of genetically distinct antibody expressing genes are generated as part of the adaptive immune system. B cells that express these antigen-specific antibodies are generated by multiple steps of selection as they arise in the bone marrow and subsequently mature in peripheral germinal centers.Citation2 In this review we concentrate on the molecular mechanism(s) by which mutations are incorporated in the variable (V) genes and constant region switch sequences (IgS), leading to B cells that express antigen-recognizing and/or effector function capable antibodies.
Upon exposure to antigen, B cells undergo two types of controlled DNA mutation. Through somatic hypermutation (SHM) B cells incorporate point mutations in their variable gene exons to encode antigen-specific antibodies, while class switch recombination (CSR) rearranges the constant region genes, permitting them to express constant region exons downstream of IgM (e.g., the IgG series, IgE, IgA). Immunoglobulins are composed of two distinct polypeptides, the heavy chain and the light chain. The heavy chain is encoded by the immunoglobulin heavy chain locus (IgH), the light chain (IgL) by one of two separate loci, Igκ and Igλ. IgH undergoes CSR as well as SHM whereas the IgL loci only undergo SHM. In this review, we focus on genetic alterations in IgH, predominantly because more analysis has been performed on these sequences with respect to the mutagenesis process involving DNA single-strand and double-strand break formation and repair during SHM and CSR, respectively.Citation2-Citation5 A schematic representation of the antibody-expressing IgH locus is shown in .
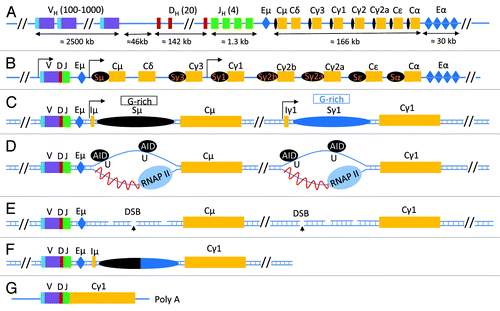
Figure 1. class switch recombination at the Immunoglobulin Heavy Chain locus. (A) The configuration of the unrearranged immunoglobulin heavy chain locus in immature B cells according to NG_005838.1. Here, VH, DH and JH represent the various unrearranged gene segments that will generate the VDJ exon following V(D)J recombination and are followed by the various constant regions genes (Cμ-α, yellow boxes). Each constant region gene is preceded by a switch sequence (Sμ-α, black oval); switch sequences are non-coding regions transcribed by their own transcriptional regulatory promoter elements. Two important regions that have enhancer functions and influence various recombination events in the IgH locus are shown in a blue box and are labeled as Eμ and Eα (also known as 3′ regulatory region, 3′RR). (B) Following V(D)J recombination, various B cell signaling pathways induce transcription at switch sequence promoter regions. Transcription at the upstream switch sequence Sμ is constitutive whereas transcription at the downstream switch sequence (in this case, Sγ1) is induced due to activation of its promoter elements by various signaling pathways. (C) A schematic of a simplified IgH locus that is poised to undergo CSR to IgG1 following transcription activation at Sμ and Sγ1. A region of the switch sequences, known as the core switch region (G-rich on the non-template strand), is capable of forming stable RNA/DNA hybrids that lead to ssDNA structure “R-loop” formation. (D) Transcription at switch sequences induces formation of R-loops which become targets for AID activity. AID converts cytidine residues to uracils, that are then recognized by the base excision pathway uracil DNA deglycosylase (UNG). (E) UNG activity induces generation of abasic residues that are then cleaved by the apurinic endonuclease family of proteins (APE1/2) to generate DNA double strand breaks (DSBs) at both upstream (Sμ) and downstream (Sx, in this case, Sγ1) switch sequences. (F) Recognition of these two DSBs by two cellular DNA damage repair pathways known as non homologous end-joining (NHEJ) and alternative end joining (AEJ) leads to joining of the two distant switch sequences that have DSBs leading to the completion of CSR. (G) The final configuration of the antibody heavy chain molecule coding mRNA is shown.
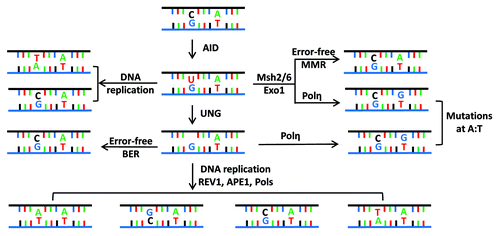
Following antigen exposure in the germinal center, B cells proliferate and undergo CSR and SHM.Citation2 Transcriptional activation of various regions of the IgH locus leads to epigenetic and structural changes of genes in the coding and non-coding regions of the variable and constant regions. Such changes are required for the B cell mutator Activation Induced cytidine Deaminase (AID) to access these substrate DNA sequences.Citation6,Citation7 Various lines of evidence, previously summarized,Citation7 have established that AID mutates single-stranded (ss) DNA in vitro and potentially in vivo as well. These studies have demonstrated that AID deaminates deoxycytidine residues (dC) to deoxyuridines (dU) which are then either repaired as deoxycytidines, replicated through during DNA replication to introduce a deoxythymidine (dT) in one daughter cell, or converted to dA, dG or dT by the coordinated actions of the cellular base excision repair and mis-match repair machinery. A schematic representation of the mutagenesis of an AID target dC residue is shown in , where it is also explain how neighboring residues of AID-deaminated dCs can be subjected to mutagenesis due to the action of the error-prone DNA polymerase, polηCitation8. Other published reviews can provide a better and more detailed overview of the mechanisms that govern the specifications and behavior of AID-induced mutagenesis shown in .Citation9-Citation11
Figure 2. Fate of cytidine residues following AID mediated DNA deamination. (A) AID deaminates cytidine residues to uracils that are identified by the cellular base excision repair pathway (UNG) or the mismatch repair pathway (MSH2/MSH6) for repair. Neighboring residues (A/T based pairs) may be mutated in this process of DNA lesion repair that depends upon the DNA polymerase η. (B) Multiple possibilities exist explaining how a lesion could be repaired. Based on the activity of error prone DNA polymerases (DNA pol η), change of the neighboring A/T based pair could be to T/A or G/C or C/G base pairs. The nascently formed uracil residues are substrates of the apurinic endonucleases (APE1/2), and this reaction eventually leads to creation of single-strand DNA (ssDNA) nicks. ssDNA nicks on both strands of switch sequences can generate DNA double strand breaks that are intermediates during CSR.
AID will mutagenize DNA only when the DNA is stabilized in a single stranded conformation (ssDNA). In this context, recent advances in genome-sequencing technologies have permitted a better understanding of potential AID targets in the IgH locus and the remainder of the genome of normal and malignant B cells, thereby permitting the investigation of how ssDNA AID substrates are generated at such a wide variety of DNA sequences that do not have any obvious characteristics identified with ssDNA-generating structures.Citation5,Citation12 It is likely that AID utilizes various transcription-associated events and co-factors to identify and generate its ssDNA substrates. In mouse models in which ssDNA generating R-loop structures are perturbed in the switch regions of the IgH locusCitation13,Citation14 or where recruitment of ssDNA structure stabilizing protein (such as Replication protein A (RPA)) is affected,Citation15 CSR is significantly reduced. In this review we explore the postulated roles of RNA polymerase II-associated AID cofactors in the generation of ssDNA substrates for AID.
DNA motifs that influence AID’s activity
AID deamination hotspot RGYW motif
Two non-exclusive signature motifs in the switch sequences of the IgH locus have been identified as influencing AID activity. Ig switch sequences (collectively referred as IgS) contain RGYW motifs (where R is a purine, G is a guanine, Y is a pyrimidine, and W is A or T), and have R-loop (a secondary, stable DNA structure) forming sequences caused by G-rich motifs on the non-template strand. The RGYW motif rich sequences are found to be embedded in secondary structure forming G-rich IgS region DNA sequences. As an approximate estimate, the mouse Sμ region contains 3.5kb of repetitive sequence and the downstream switch regions Sγ1, Sγ3, Sγ2b, Sε, and Sα have 6.5kb, 2.0 kb, 2.7kb, 2.0kb, and 4.0kb of repetitive elements, respectively, all rich in the RGYW motif. Other species, from humans to amphibians (xenopus) to birds (chicken) also have repetitive sequences in their switch regions. Switch sequences in these species are either comprised of the RGYW motif (as in Xenopus) or contain large R-loop structure generating sequences that are embedded and flanked with RGYW-rich motifs (as in human and mouse).Citation16 While switch sequences have high RGYW density, oddly, variable region sequences are not particularly enriched for RGYW content compared with the remainder of the genome.Citation16 Even so, AID prefers to mutate transcribed DNA substrates rich in RGYW motifs in vitro, indicating that this motif is a preferred AID target.Citation7 The reason(s) why this motif attracts and/or stimulates AID activity is not understood beyond some correlative studies based on AID activity at RGYW motif dense DNA sequences.Citation14,Citation17-Citation19 Limited knowledge has been obtained from cell-free extract experiments that have demonstrated robust AID activity on RGYW-rich sequences, having employed naked DNA transcribed by a viral T7 promoter or a bacterial RNA polymerase.Citation7,Citation14,Citation17,Citation20 Other experiments using chimeric AID proteins (generated with amino acid substitutions from APOBEC active sites) have revealed a role of amino acids neighboring the active site of AID (amino acids 115–123) in recognizing RGYW motifs.Citation21-Citation23 In the future, the crystal structure of AID bound with RGYW motifs will conclusively illuminate how resident motifs in AID generate RGYW-specificity.
Single-stranded DNA structures that attract AID
DNA secondary structures that are generated co-transcriptionally are potential genome wide substrates for AID, specially if they are stabilized due to their sequence inherent properties (eg., G-richness) or due to associated DNA binding proteins. It was recently observed that AID-initiated DNA double-strand breaks are predominantly located in proximity to transcription start sites genome wide.Citation24-Citation26 Thus, from small transcription bubbles that accompany the RNA polymerase II (RNAP II) transcription complex to large ssDNA structures such as R-loops and G-quadruplex structures, all DNA secondary structures that are generated by transcription-dependent mechanisms are potential targets of AID.Citation13,Citation27 DNA secondary structures can be influenced by various factors such as ion concentrations in the cell, binding of proteins that stabilize DNA secondary structures, purine and pyrimidine distribution that permits formation of R-loops or the four stranded DNA structure called “G-quadruplex” or an i-motif.Citation28 G-quadruplex structures have various functions including protection of chromosome ends and regulation of gene expression through their influence on transcription initiation. The AID target c-Myc locus forms such structures to recruit transcriptional activator and non-duplex DNA binding factors NH23-H2 and hnRNP-K, which are known to stimulate c-Myc transcription.Citation29,Citation30
Unlike G-quadruplexes, R-loops are well known secondary DNA structures that can be targeted by AID. R-loops can be of various types. Transcriptionally active RNAP II complexes can generate R-loops of approximately 8–10 base pairs within the transcription bubble. On their own, these small R-loops may not induce robust somatic hypermutation. However, a low frequency of AID-induced mutagenesis could possibly occur at these transcription complex coupled DNA bubbles. It is also possible that negative DNA supercoils generated preceding the transcribing RNAP II are a source of ssDNA structures that are converted to targets of AID.Citation19,Citation31 Negatively supercoiled DNA bubbles may utilize additional co-factors at canonical AID target sequences to stabilize ssDNA structures and stimulate robust AID activity. Deviations in RNA processing, RNA splicing, replication and/or RNAP II transcription pre-termination pathways can also induce spreading of the transcription bubble R-loop into a larger ssDNA structure which can then act as a better AID substrate. As a model example in a heterologous system, it has been demonstrated that depletion of the THO complex, a co-transcriptional RNA processing pathway component, increases the levels of AID-mediated mutations on the single stranded non-template DNA strand at R-loops in S. cerevisiae.Citation32,Citation33 These mutations are generated due to the slow kinetics of ribonucleoprotein complex (RNP) formation that associate with the transcription complex-coupled nascent transcript, which is then able to hybridize with the template strand of the transcribed DNA to facilitate stable R-loop formation on the DNA. In B cells, similar R-loops are generated in immunoglobulin switch sequences, although here the transcription associated small transcriptional bubble is converted to large R-loop structures by the inherent nature of the DNA sequence that contains large stretches of G-rich sequences. Some of these AID target switch sequences, IgS, now have been extensively characterized via in vitro and in vivo studies and shown to form R-loops due to the G-richness of the sequence.Citation13,Citation27,Citation34 Specifically, the largest switch sequence, IgSγ1, has been experimentally demonstrated to generate long stretches of ssDNA R-loop structures using sodium bisulphite crosslinking/DNA sequencing based assays; furthermore, these regions are direct targets of AID and found to be recombined at high frequency with downstream switch sequences during CSR.Citation27,Citation34 In support of these observations, genetic inversion of IgSγ1 sequence leads to loss of co-transcriptional R-loop formation and subsequent defects in CSR efficiency in mouse B cells.Citation13 Taken together, these observations provide compelling evidence that ssDNA R-loop structures are robust AID targets sequences in B cells. Given that DNA/RNA hybrids present in R-loops may also alter the landscape with respect to histone position and status and also control transcription rates,Citation35 it is likely that R-loops play additional roles beyond generating ssDNA to support AID activity in the Ig locus. In the next sections, we will discuss the many means by which the transcriptional machinery promotes formation of DNA secondary structures and regulates AID activity in B cells.
AID regulation due to association with the RNA polymerase II complex
Before the advent of genome sequencing technology, it was assumed that AID only mutates the variable region genes and switch sequences of the immunoglobulin loci. This assumption existed due to the argument that AID-generated mutations are deleterious to the genomic integrity of cells and thus there should be a factor that only promotes AID mutations in the Ig locus. However, recent studies utilizing genome sequencing technologies have generated a detailed map of AID target sequences genome wide and these studies have also correlated AID association with levels of mutagenesis at transcribed genes in the IgH locus as well as at other genes.Citation25,Citation36 Chromatin immunoprecipitation studies of AID and components of transcribing RNAP II in B cells indicate that RNAP II, in addition to generating secondary DNA structures for AID, may directly contribute toward AID recruitment. Many protein factors and AID modification events have been postulated to generate secondary DNA structures and/or target AID to regions of the B cell genome. A summary of these AID regulatory events is schematically represented in and and described in . Detailed descriptions of direct AID regulatory events have already been extensively discussed previously.Citation37,Citation38 In the following sections we discuss AID’s interaction with various states of RNAP II with which AID may bind and how these AID/ RNAP II complexes determine somatic mutagenesis in the B cell genome.
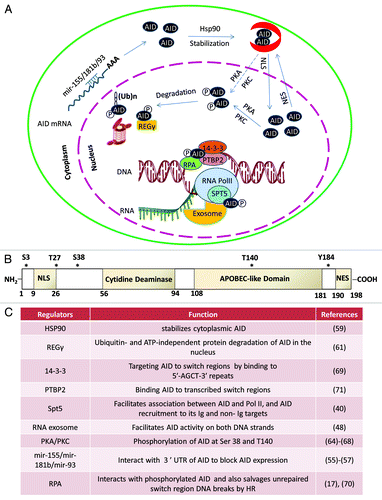
Figure 3. Subcellular control of AID activity by various post-transcriptional regulatory pathways. AID mRNA is recognized by two miRNAs (miR-155, miR-181) that bind to its 3′UTR to regulate AID mRNA translational efficiency and prevent AID hyperactivity caused by its overexpression.Citation55-Citation58 One proposed mode of cytoplasmic AID protein regulation is the chaperone complex HSP90Citation59 and EF1a,Citation60 controlling AID’s ability to translocate into the nucleus of B cells. AID uses its nuclear localization signal (NLS) to translocate into the nucleus where its steady-state nuclear protein levels are further controlled by another chaperone, REGγ Citation61. AID translocated in the nucleus can have multiple fates that include its ubiquitinationCitation62,Citation63 and its phosphorylation at various serine, threonine, or tyrosine residues (see (B) for details of known phosphorylation sitesCitation64-Citation68). Phosphorylated AID forms a complex with its cofactors 14–3-3Citation69 and RPACitation17,Citation70 and PTPBP2Citation71 and binds to the stalled RNA polymerase II complex (marked by RNAP II stalling marker Spt5)Citation40 at AID target sequences, where it interacts with the DNA/RNA hybrid and the 3′-5′ RNA exonuclease, RNA exosome.Citation48 It is postulated that the macromolecular complex RNA exosome provides AID the ability to deaminate both strands of its DNA substrate by processing the RNA present in the RNA/DNA hybrid associated with the transcription complex. The DNA DSBs in the immunoglobulin switch regions are intermediates that ultimately are utilized by the cellular DNA DSB response factors to complete CSR. (B) Schematic representation of AID phosphorylation sites along with AID’s cytidine deaminase domain, nuclear localization signal (NLS), APOBEC-like region, and nuclear export signal (NES) motif. (C) A detailed chart of various regulatory elements of AID that are known to directly control its activity. The protein factors and AID modifications are schematized in (A). The mRNA stability of AID is regulated by various miRNAs, as indicated in (C).Citation55-Citation58
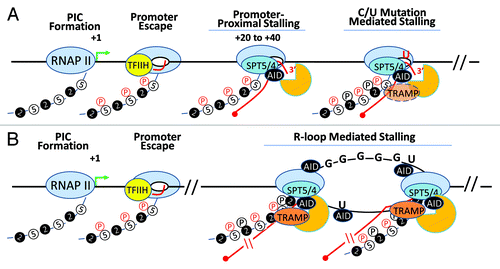
Promoter proximal stalling of RNA Polymerase II
Two related methods have demonstrated that AID can bind to various transcribed genes genome wide and induce mutagenesis in some of these sequences. Chromatin immunoprecipitation (ChIP) of AID from CSR stimulated B cells followed by high throughput sequencing of AID-associated DNA fragments (ChIP-seq) demonstrated that AID can bind to various regions of the B cell genome. Moreover, AID bound sequences have high occupancy by RNA polymerase II, some of this RNAP II is in the elongation phase with the remainder in the paused or stalled conformation. Consistent with these observations, it has been reported that RNAP II is enriched at various regions of IgS sequencesCitation39 and these IgS sequences are enriched with AID and the RNAP II “stalling” factor Spt5.Citation36,Citation40 For a deeper interpretation of RNAP II occupancy results at AID target sequences one requires an understanding of the transcription machinery and the various mechanisms that regulate the transcription complex.
The RNAP II transcription complex initiates transcription from transcription start sites and undergoes “promoter escape” mostly at +1 nucleotide downstream of the transcription start site (TSS). Promoter escape is dependent upon the RNAP II associated factor TFIIH that rides the RNAP II complex into the elongation phase, and on the phosphorylation of the C-terminal tail of RNAP II at residue serine-5, S5. Following promoter escape, RNAP II encounters a second rate-limiting step before entering transcription elongation, this step is known as “ RNAP II stalling” and more accurately termed as “promoter-proximal transcription stalling (PPTS).” Promoter-proximal transcription stalling is known to “poise” the transcription complex in such a way that following release of the stalling state the transcription complex rapidly can enter the elongation phase.Citation41 The physiological signals that induce release of the paused state of the transcription complex include stress, environment-induced signaling cascades, and development. At a molecular level, the poised transcription complex contains additional cofactors, namely the NELF complex and the transcription stalling factor DSIF (composed of Spt4 and Spt5). Following phosphorylation of NELF, DSIF and the C-terminal tail of RNAP II at serine residue S2 by positive transcription elongation factor P-TEFb and release of phosphorylated NELF, the transcription complex enters the elongation phase of transcription.Citation41-Citation43
Multiple fates of the RNA pol II and the associated transcript are possible when it is in the stalled state. Most often the RNA pol II readjusts itself to keep the RNA transcript aligned with the transcribing RNAP II via a process known as RNAP II “backtracking.” RNAP II undergoing backtracking requires activity of transcript cleavage factor TFIIS to induce internal cleavage of the RNA by the polymerase active site, create a new 3′-end that is properly aligned, and continue transcription. RNAP II stalling may provide regulatory advantages during rapid gene expression. One evolving hypothesis is that paused RNAP II has been optimized by evolution to promote rapid activation of RNAP II at genes that are developmentally regulated and require instantaneous “off-on” switches. The mechanisms behind RNAP II stall-state release are still being unraveled but chromatin modifications and chromosomal positioning are factors that are reported to control these fates.Citation44-Citation46 Secondary DNA structures like R-loops, G-quartets or repetitive DNA sequences may also cause RNA pol II stalling. However, secondary DNA structure-induced RNAP II stalling, as would be expected in IgS sequences, may not occur at promoter proximal regions of the transcribed DNA element but rather at sequences that are substantially downstream of the transcription start site. Thus, these stalled transcription complexes may attract the RNA pol II pre-termination complex that facilitates premature transcription termination. It can be postulated that pretermination stalled RNAP II recruits AID at regions that are distal from the promoter of the target sequences and initiate mutagenesis. In the next section, we discuss the RNAP II pretermination complex and how it provides AID with its necessary co-factor RNA exosome to stimulate DNA deamination.
RNA polymerase II premature termination
Transcription complexes “stall” at regions proximal to the promoter or during the elongation phase but resolve the paused phase to continue transcribing (); however, at times due to various factors including environmental stress, DNA sequence context, or other reasons a stalled RNAP II does not continue transcribing further and undergoes premature transcription termination (). The factors that determine RNAP II elongation competence and prevent premature termination following polII’s stalling are not completely understood. However, efficiency of RNAP II backtracking resolution or its ability to undergo “ RNAP II bubble expansion” are some mechanisms that may allow it to bypass termination and continue on the path of transcription elongation. The mechanism of RNAP II termination at protein coding genes, at non-coding genes, and on sequences undergoing premature termination are very different. In these three scenarios the mechanism of 3′ end processing of the nascent transcript determines whether transcription termination will occur.
Figure 4. AID in the transcription complex. AID, RNA exosome and Spt5 can associate with transcribing RNA polymerase II in two distinct transcription complexes. (A) Following transcription initiation, RNA polymerase II at many transcribed genes enters a phase called “transcription stalling.” The association of RNAP II co-factors NELF and Spt5 and TFIIH leads to the resolution of RNAP II stalling and promotes RNAP II to transition into its transcription elongation phase. AID, by virtue of its association with Spt5, accesses this complex and its associated DNA. Moreover, RNA exosome is recruited to this complex and can associate with AID and Spt5-associated RNAP II to promote DNA deamination during somatic hypermutation in the first 100–200 base pairs downstream of the transcription start site. However, during SHM as well as in CSR, it is possible that the RNAP II is able to overcome the promoter proximal stalling and enter the elongation phase. Here, if it encounters dU residues (incorporated by preceding RNAP II-associated AID complexes), it may stall and recruit RNA exosome and catalyze robust cytidine deamination at DNA regions kilobases downstream from the transcription start sites. (B) If the transcription complex does continue into the elongation phase, it may generate stable DNA secondary structures like R-loops depending upon the physical properties of the transcribed DNA. For example, transcription of switch sequences is proposed to generate stable large R-loop structures due to the presence of G-richness on the template strand. If R-loops or other transcription complex impedance factors are recruited (or preexist), elongating RNAP II molecules that are loaded on the DNA secondary structure containing templates may undergo a second “stalling” event that is analogous to RNAP II pre-termination. In this pre-termination complex RNA exosome actively degrades the nascent transcript to prevent continuity of abortive transcription and aberrant DNA/RNA hybrids that can initiate genomic instability. This RNA exosome-associated RNAP II pretermination complex can potentially associate with AID and provide another hub for AID and its associated co-factors to catalyze DNA deamination. This scenario parallels the transcription stalling associated AID DNA deamination activity observed during class switch recombination.
In contrast to conventional termination, premature transcription termination requires a separate set of proteins that promote the removal of the nascent transcript from the template DNA associated RNAP II complex. In yeast, the TRAMP (Trf4/5, Air1/2, and Mtr4) complex utilizes its co-factor Nrd1 complex (Nrd1-Nab3-Sen1) to bind RNAP II-CTD and recruit the 3′-5′ RNA exonuclease known as the RNA exosome complex that has the ability to degrade the prematurely terminated nascent RNA. RNA exosome is a part of the RNA surveillance machinery of eukaryotic cells. The function of the TRAMP complex is to add short polyadenylation signals to RNA exosome substrate nascent RNA and/or recognize secondary RNA structures that facilitate RNA exosome recruitment. Recent developments, as discussed below, postulate that the pre-termination state of RNAP II may be responsible for recruiting AID co-factors and facilitating DNA deamination activity in B cells. These possibilities are schematized in . For more details of the various mechanisms that control transcription termination, please see the reviews by Manley et al.Citation43,Citation47
RNA polymerase II associated RNA degradation
In a recent study it was reported that the RNA exosome complex binds with AID in B cells. Moreover, RNA exosome recruitment to the IgH locus depends upon the activity of AID, since AID deficient B cells demonstrate reduced RNA exosome recruitment to transcribed switch sequences.Citation48 Analysis of B cell lines that can inducibly undergo CSR demonstrates a significant decrease in CSR levels following stable small hairpin RNA (shRNA) induced knockdown of RNA exosome subunits. In in vitro assays, purified mammalian RNA exosome complex or recombinant RNA exosome complex is able to stimulate AID mediated DNA deamination activity on the template and the non-template strands of transcribed DNA substrates, thus providing evidence that the RNA exosome complex is a functional co-factor of AID activity.Citation48 In light of these observations, it can be postulated that AID utilizes the RNA exosome associated RNAP II complex to induce genome mutations. Given that AID-generated mutations can occur at varying distances from the transcription start site, it is likely that the composition of the RNAP II complex is different when AID is mutating its target sequence within the first 100 base pairs (stalled due to promoter proximal ”pausing/stalling”) vs. kilobases downstream of the transcription start site (stalled following pre-termination pausing events). A simplified model would predict that in IgH switch sequences, where AID mutates its targets at regions kilobases downstream of the transcription start sites, transcriptional paused complexes are not promoter proximal and are potentially induced by transcription pre-termination caused by the presence of secondary DNA structures (R-loops), repetitive sequences (RGYW-motifs in the Xenopus switch sequence) and/or the presence of chromatin bound protein factors (). Indeed, AID deamination motif-rich sequences are embedded in R-loop structures that pause RNAP II.Citation35 Thus an RNAP II complex located in switch regions may have two distinct functions. First, it transcribes switch sequences to generate DNA secondary structures such as R-loops (first function). Once an R-loop is stabilized, subsequent RNAP II complexes tend to stall and recruit associated factors such as RNA exosome (second function) (for details of pre-termination complex composition, please seeCitation43,Citation47). This stalled complex may now recruit AID and its co-factors to facilitate DNA deamination induced double-strand breaks. In , we have schematically represented the various cofactors of AID that ultimately support its binding in the RNAP II complex but a complete understanding of how all these factors collectively will control AID activity is emerging.Citation38,Citation49
Mutations in the variable region sequences show a different distribution, they are either promoter proximal or distributed between 100 base pairs and two kilobases downstream of the IgV transcription start site. How does such a distribution occur, given that IgV sequences do not have any particular sequence motif, similar to R-loops in heavy chain switch sequences, that can induce RNA polymerase II stalling? It is possible that promoter proximal stalling is the mechanism that leads to accumulation of RNAP II at variable region sequences in the first 50 to 200 base pairs downstream of the transcription start site. However, the mutations that occur in IgV sequences or at other genomic sequences far beyond promoter proximal distances require a more satisfying explanation for AID targeting. Multiple possibilities exist including AID binding and traveling with the stalled RNAP II to downstream DNA sequences (where the complex may encounter pre-termination events that recruit RNA exosome and promote DNA deamination) or the induction of low levels of DNA deamination events, induced by mechanisms such as DNA supercoiling, that lead to incorporation of deoxyuracils in the DNA in a distributive fashion downstream of the promoter proximal stalling sites. It can be speculated that the presence of these AID-generated deoxyuracils in the DNA template strand may produce stalling of the subsequent transcription complex as it attempts to utilize these non-canonical dU bases as a transcription template at regions that are not promoter proximal AID targets (). The stalled RNAP II may now recruit RNA exosome and, if associated with AID, catalyze further mutagenesis of the V genes. Thus, RNAP II stalling and AID mediated DNA deamination may stimulate one another, possibly forming a regulatory feedback loop to promote robust mutagenesis at Ig loci in B cells.
Concluding remarks
AID mutations genome-wide are a source of genomic instability and chromosomal translocations. Various studies that addressed single gene translocations or more recent ones that have evaluated chromosomal translocations genome-wide have consistently established the role of AID in causing genomic instability.Citation24-Citation26,Citation50-Citation54 All these studies clearly establish a relationship between AID, transcription, RNA pol II regulation proximal to or distal from transcription start sites, and genome maintenance. Future studies should now focus on revealing the environmental factors in the germinal center, epigenetic regulation of AID target genes and nuclear organization into transcription control regions, and AID mutation repair factories in prevention of initiation of oncogenesis. With evolving cutting edge technologies that can sequence vast genomes at high resolution or microscopically monitor nuclear organization in minute detail, following AID generated mutations in B cells may provide answers to questions that have a direct implication on many poorly understood aspects of mammalian gene expression biology.
Acknowledgments
We thank Dr. Rushad Pavri (IMP, Vienna) for critical input on the manuscript. This work was supported by funding from the National Institutes of Health Director’s office (1DP2OD008651–01) and the National Institute of Allergy and Infectious Diseases (1R01AI099195–01A1).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Burnet FM. A modification of Jerne's theory of antibody formation using the concept of clonal selection. J Sci 1957; 20:67 - 9
- Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol 2012; 30:429 - 57; http://dx.doi.org/10.1146/annurev-immunol-020711-075032; PMID: 22224772
- Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol 2006; 6:573 - 83; http://dx.doi.org/10.1038/nri1896; PMID: 16868548
- Neuberger MS. Antibody diversification by somatic mutation: from Burnet onwards. Immunol Cell Biol 2008; 86:124 - 32; http://dx.doi.org/10.1038/sj.icb.7100160; PMID: 18180793
- Robbiani DF, Nussenzweig MC. Chromosome Translocation, B Cell Lymphoma, and Activation-induced Cytidine Deaminase. Annu Rev Pathol 2012; PMID: 22974238
- Rush JS, Liu M, Odegard VH, Unniraman S, Schatz DG. Expression of activation-induced cytidine deaminase is regulated by cell division, providing a mechanistic basis for division-linked class switch recombination. Proc Natl Acad Sci U S A 2005; 102:13242 - 7; http://dx.doi.org/10.1073/pnas.0502779102; PMID: 16141332
- Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol 2007; 94:157 - 214; http://dx.doi.org/10.1016/S0065-2776(06)94006-1; PMID: 17560275
- Delbos F, De Smet A, Faili A, Aoufouchi S, Weill JC, Reynaud CA. Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse. J Exp Med 2005; 201:1191 - 6; http://dx.doi.org/10.1084/jem.20050292; PMID: 15824086
- Reynaud CA, Delbos F, Faili A, Guéranger Q, Aoufouchi S, Weill JC. Competitive repair pathways in immunoglobulin gene hypermutation. Philos Trans R Soc Lond B Biol Sci 2009; 364:613 - 9; http://dx.doi.org/10.1098/rstb.2008.0206; PMID: 19010770
- Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 2007; 76:1 - 22; http://dx.doi.org/10.1146/annurev.biochem.76.061705.090740; PMID: 17328676
- Neuberger MS, Di Noia JM, Beale RC, Williams GT, Yang Z, Rada C. Somatic hypermutation at A.T pairs: polymerase error versus dUTP incorporation. Nat Rev Immunol 2005; 5:171 - 8; http://dx.doi.org/10.1038/nri1553; PMID: 15688043
- Pavri R, Nussenzweig MC. AID targeting in antibody diversity. Adv Immunol 2011; 110:1 - 26; http://dx.doi.org/10.1016/B978-0-12-387663-8.00005-3; PMID: 21762814
- Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, Alt FW. The influence of transcriptional orientation on endogenous switch region function. Nat Immunol 2003; 4:435 - 41; http://dx.doi.org/10.1038/ni918; PMID: 12679811
- Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, et al. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat Immunol 2004; 5:1275 - 81; http://dx.doi.org/10.1038/ni1137; PMID: 15531884
- Cheng HL, Vuong BQ, Basu U, Franklin A, Schwer B, Astarita J, et al. Integrity of the AID serine-38 phosphorylation site is critical for class switch recombination and somatic hypermutation in mice. Proc Natl Acad Sci U S A 2009; 106:2717 - 22; http://dx.doi.org/10.1073/pnas.0812304106; PMID: 19196992
- Hackney JA, Misaghi S, Senger K, Garris C, Sun Y, Lorenzo MN, et al. DNA targets of AID evolutionary link between antibody somatic hypermutation and class switch recombination. Adv Immunol 2009; 101:163 - 89; http://dx.doi.org/10.1016/S0065-2776(08)01005-5; PMID: 19231595
- Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature 2004; 430:992 - 8; http://dx.doi.org/10.1038/nature02821; PMID: 15273694
- Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 2003; 424:103 - 7; http://dx.doi.org/10.1038/nature01760; PMID: 12819663
- Shen HM, Storb U. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc Natl Acad Sci U S A 2004; 101:12997 - 3002; http://dx.doi.org/10.1073/pnas.0404974101; PMID: 15328407
- Besmer E, Market E, Papavasiliou FN. The transcription elongation complex directs activation-induced cytidine deaminase-mediated DNA deamination. Mol Cell Biol 2006; 26:4378 - 85; http://dx.doi.org/10.1128/MCB.02375-05; PMID: 16705187
- Kohli RM, Abrams SR, Gajula KS, Maul RW, Gearhart PJ, Stivers JT. A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J Biol Chem 2009; 284:22898 - 904; http://dx.doi.org/10.1074/jbc.M109.025536; PMID: 19561087
- Wang M, Rada C, Neuberger MS. Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID. J Exp Med 2010; 207:141 - 53; http://dx.doi.org/10.1084/jem.20092238; PMID: 20048284
- Wang M, Yang Z, Rada C, Neuberger MS. AID upmutants isolated using a high-throughput screen highlight the immunity/cancer balance limiting DNA deaminase activity. Nat Struct Mol Biol 2009; 16:769 - 76; http://dx.doi.org/10.1038/nsmb.1623; PMID: 19543289
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 2011; 147:95 - 106; http://dx.doi.org/10.1016/j.cell.2011.07.048; PMID: 21962510
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 2011; 147:107 - 19; http://dx.doi.org/10.1016/j.cell.2011.07.049; PMID: 21962511
- Gostissa M, Alt FW, Chiarle R. Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu Rev Immunol 2011; 29:319 - 50; http://dx.doi.org/10.1146/annurev-immunol-031210-101329; PMID: 21219174
- Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 2003; 4:442 - 51; http://dx.doi.org/10.1038/ni919; PMID: 12679812
- Bochman ML, Paeschke K, Zakian VA. DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet 2012; 13:770 - 80; http://dx.doi.org/10.1038/nrg3296; PMID: 23032257
- Brooks TA, Hurley LH. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010; 1:641 - 9; http://dx.doi.org/10.1177/1947601910377493; PMID: 21113409
- Brooks TA, Kendrick S, Hurley L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J 2010; 277:3459 - 69; http://dx.doi.org/10.1111/j.1742-4658.2010.07759.x; PMID: 20670278
- Shen HM, Ratnam S, Storb U. Targeting of the activation-induced cytosine deaminase is strongly influenced by the sequence and structure of the targeted DNA. Mol Cell Biol 2005; 25:10815 - 21; http://dx.doi.org/10.1128/MCB.25.24.10815-10821.2005; PMID: 16314506
- Gómez-González B, Aguilera A. Activation-induced cytidine deaminase action is strongly stimulated by mutations of the THO complex. Proc Natl Acad Sci U S A 2007; 104:8409 - 14; http://dx.doi.org/10.1073/pnas.0702836104; PMID: 17488823
- Ruiz JF, Gómez-González B, Aguilera A. AID induces double-strand breaks at immunoglobulin switch regions and c-MYC causing chromosomal translocations in yeast THO mutants. PLoS Genet 2011; 7:e1002009; http://dx.doi.org/10.1371/journal.pgen.1002009; PMID: 21383964
- Roy D, Yu K, Lieber MR. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol Cell Biol 2008; 28:50 - 60; http://dx.doi.org/10.1128/MCB.01251-07; PMID: 17954560
- El Hage A, French SL, Beyer AL, Tollervey D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev 2010; 24:1546 - 58; http://dx.doi.org/10.1101/gad.573310; PMID: 20634320
- Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol 2011; 12:62 - 9; http://dx.doi.org/10.1038/ni.1964; PMID: 21113164
- Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol 2012; 12:517 - 31; http://dx.doi.org/10.1038/nri3216; PMID: 22728528
- Vuong BQ, Chaudhuri J. Combinatorial mechanisms regulating AID-dependent DNA deamination: interacting proteins and post-translational modifications. Semin Immunol 2012; 24:264 - 72; http://dx.doi.org/10.1016/j.smim.2012.05.006; PMID: 22771392
- Wang L, Whang N, Wuerffel R, Kenter AL. AID-dependent histone acetylation is detected in immunoglobulin S regions. J Exp Med 2006; 203:215 - 26; http://dx.doi.org/10.1084/jem.20051774; PMID: 16418396
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 2010; 143:122 - 33; http://dx.doi.org/10.1016/j.cell.2010.09.017; PMID: 20887897
- Cheung AC, Cramer P. A movie of RNA polymerase II transcription. Cell 2012; 149:1431 - 7; http://dx.doi.org/10.1016/j.cell.2012.06.006; PMID: 22726432
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR 3rd, et al. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell 2009; 33:365 - 76; http://dx.doi.org/10.1016/j.molcel.2008.12.028; PMID: 19217410
- Richard P, Manley JL. Transcription termination by nuclear RNA polymerases. Genes Dev 2009; 23:1247 - 69; http://dx.doi.org/10.1101/gad.1792809; PMID: 19487567
- Core LJ, Waterfall JJ, Gilchrist DA, Fargo DC, Kwak H, Adelman K, et al. Defining the status of RNA polymerase at promoters. Cell Rep 2012; 2:1025 - 35; http://dx.doi.org/10.1016/j.celrep.2012.08.034; PMID: 23062713
- Adelman K, Rogatsky I. RNA polymerase II stalling mediates cytokine gene expression. Cell Cycle 2010; 9:630 - 1; http://dx.doi.org/10.4161/cc.9.4.10841; PMID: 20107307
- Gilchrist DA, Dos Santos G, Fargo DC, Xie B, Gao Y, Li L, et al. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 2010; 143:540 - 51; http://dx.doi.org/10.1016/j.cell.2010.10.004; PMID: 21074046
- Hsin JP, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 2012; 26:2119 - 37; http://dx.doi.org/10.1101/gad.200303.112; PMID: 23028141
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell 2011; 144:353 - 63; http://dx.doi.org/10.1016/j.cell.2011.01.001; PMID: 21255825
- Orthwein A, Di Noia JM. Activation induced deaminase: how much and where?. Semin Immunol 2012; 24:246 - 54; http://dx.doi.org/10.1016/j.smim.2012.05.001; PMID: 22687198
- Ramiro A, Reina San-Martin B, McBride K, Jankovic M, Barreto V, Nussenzweig A, et al. The role of activation-induced deaminase in antibody diversification and chromosome translocations. Adv Immunol 2007; 94:75 - 107; http://dx.doi.org/10.1016/S0065-2776(06)94003-6; PMID: 17560272
- Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature 2006; 440:105 - 9; http://dx.doi.org/10.1038/nature04495; PMID: 16400328
- Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, et al. AID is required for c-myc/IgH chromosome translocations in vivo. Cell 2004; 118:431 - 8; http://dx.doi.org/10.1016/j.cell.2004.08.006; PMID: 15315756
- Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Küppers R, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001; 412:341 - 6; http://dx.doi.org/10.1038/35085588; PMID: 11460166
- Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 2012; 484:69 - 74; PMID: 22314321
- de Yébenes VG, Belver L, Pisano DG, González S, Villasante A, Croce C, et al. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med 2008; 205:2199 - 206; http://dx.doi.org/10.1084/jem.20080579; PMID: 18762567
- Borchert GM, Holton NW, Larson ED. Repression of human activation induced cytidine deaminase by miR-93 and miR-155. BMC Cancer 2011; 11:347; http://dx.doi.org/10.1186/1471-2407-11-347; PMID: 21831295
- Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity 2008; 28:630 - 8; http://dx.doi.org/10.1016/j.immuni.2008.04.002; PMID: 18455451
- Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, et al. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity 2008; 28:621 - 9; http://dx.doi.org/10.1016/j.immuni.2008.03.015; PMID: 18450484
- Orthwein A, Patenaude AM, Affar B, Lamarre A, Young JC, Di Noia JM. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med 2010; 207:2751 - 65; http://dx.doi.org/10.1084/jem.20101321; PMID: 21041454
- Häsler J, Rada C, Neuberger MS. Cytoplasmic activation-induced cytidine deaminase (AID) exists in stoichiometric complex with translation elongation factor 1α (eEF1A). Proc Natl Acad Sci U S A 2011; 108:18366 - 71; http://dx.doi.org/10.1073/pnas.1106729108; PMID: 22042842
- Uchimura Y, Barton LF, Rada C, Neuberger MS. REG-γ associates with and modulates the abundance of nuclear activation-induced deaminase. J Exp Med 2011; 208:2385 - 91; http://dx.doi.org/10.1084/jem.20110856; PMID: 22042974
- Aoufouchi S, Faili A, Zober C, D’Orlando O, Weller S, Weill JC, et al. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med 2008; 205:1357 - 68; http://dx.doi.org/10.1084/jem.20070950; PMID: 18474627
- Delker RK, Zhou Y, Strikoudis A, Stebbins CE, Papavasiliou FN. Solubility-based genetic screen identifies RING finger protein 126 as an E3 ligase for activation-induced cytidine deaminase. Proc Natl Acad Sci U S A 2013; 110:1029 - 34; http://dx.doi.org/10.1073/pnas.1214538110; PMID: 23277564
- Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, et al. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature 2005; 438:508 - 11; http://dx.doi.org/10.1038/nature04255; PMID: 16251902
- McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, et al. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci U S A 2006; 103:8798 - 803; http://dx.doi.org/10.1073/pnas.0603272103; PMID: 16723391
- McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med 2008; 205:2585 - 94; http://dx.doi.org/10.1084/jem.20081319; PMID: 18838546
- Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, et al. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat Immunol 2009; 10:420 - 6; http://dx.doi.org/10.1038/ni.1708; PMID: 19234474
- Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci U S A 2006; 103:395 - 400; http://dx.doi.org/10.1073/pnas.0509969103; PMID: 16387847
- Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, et al. 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nat Struct Mol Biol 2010; 17:1124 - 35; http://dx.doi.org/10.1038/nsmb.1884; PMID: 20729863
- Yamane A, Robbiani DF, Resch W, Bothmer A, Nakahashi H, Oliveira T, et al. RPA accumulation during class switch recombination represents 5′-3′ DNA-end resection during the S-G2/M phase of the cell cycle. Cell Rep 2013; 3:138 - 47; http://dx.doi.org/10.1016/j.celrep.2012.12.006; PMID: 23291097
- Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol 2011; 12:160 - 6; http://dx.doi.org/10.1038/ni.1977; PMID: 21186367