Abstract
The eukaryotic chromatin structure is essential in correctly defining transcription units. Impairing this structure can activate cryptic promoters, and lead to the accumulation of aberrant RNA transcripts. Here we discuss critical pathways that are responsible for the repression of cryptic transcription and the maintenance of genome integrity.
Eukaryotic genomic DNA is packaged and organized into the chromatin structure. Chromatin compacts and protects the genome but also restricts access to the DNA. Chromatin modifying activities can control the accessibility of the DNA by opening or closing the chromatin structure, thereby playing an essential role in the regulation of gene expression. The correct chromatin conformation is also critical for establishing functional units in the genome, such as promoter- and coding-regions of transcription units. Disturbing the chromatin structure can lead to transcription initiation events outside of canonical promoter regions and to the accumulation of aberrant, non-coding RNA (ncRNA) transcripts. In this Point-of-View article, we summarize recent findings concerning the role of chromatin in the correct definition of transcription units and the mechanisms that prevent non-specific transcription initiation in eukaryotic cells. In addition, we discuss some surprising new developments and controversies in this field. Our point of view is particularly based on our recently published results on this topic.Citation1 Studying the mechanisms repressing cryptic transcription is crucial to understanding the role of the chromatin structure in establishing and maintaining genome integrity.
Genome-wide mapping of nucleosomes and histone modifications in diverse species has revealed that the characteristics of chromatin organization in transcription units are evolutionarily highly conserved from yeast to humans. Promoter regions typically possess a particularly open chromatin structure, which allows the binding of transcription factors and the transcription initiation machinery to these DNA regions. A characteristic feature of the chromatin at promoter regions is the presence of a nucleosome-free DNA region (NFR) just upstream of the transcription start site (TSS). The NFR is generally flanked by nucleosomes containing the histone variant H2A.Z. Typical histone modifications at promoter regions include trimethylated histone H3 at lysine 4 (H3K4me3) and high levels of acetylation of H3 and H4 at various lysine residues, a feature that is also a hallmark of an open chromatin state. In addition, promoter proximal nucleosomes show a high turnover rate; they are continuously evicted and re-incorporated into the chromatin, causing a rapid exchange between the chromatin-bound and soluble histone pool. Gene coding regions, on the other hand, possess a more closed chromatin structure. Here, the nucleosomes are tightly organized with roughly uniform spacing, avoiding the random appearance of larger nucleosome-free DNA segments. The turnover of these nucleosomes is slower relative to promoter regions. A characteristic histone modification of gene coding regions is the methylated form of histone H3 at lysine 36 (H3K36me), while H3K4me3 and acetylation levels are low compared with promoters (reviewed in Rando et al.Citation2). Since the majority of the aforementioned histone modifications show a significant correlation with transcriptional activity, it was anticipated that these chromatin marks play an important regulatory role in the transcription process. Surprisingly, the loss of most of these histone modifications in yeast does not cause a genome-wide effect on gene expression. Deletion of H2A.Z or elimination of H3K4 methylation does not significantly influence transcription initiation at the vast majority of genes.Citation3,Citation4 Similarly, abolishing H3K36 methylation or increasing the histone acetylation levels in gene coding regions doesn’t have a major impact on transcription elongation.Citation1,Citation4
These results have raised an important question about the physiological relevance of these evolutionarily highly conserved chromatin modifications. However, recent developments in genomic techniques have provided new clues about the role of these modifications by allowing the detection of transcripts from non-coding or antisense portions of the genome, which was simply not monitored in earlier studies. These studies have revealed that many mutations affecting the chromatin structure induce severe, genome-wide changes in the non-coding transcriptome, while the level of coding transcripts is only slightly affected.Citation1,Citation3,Citation5-Citation8 Most of these mutants accumulate aberrant non-coding sense or antisense (AS) transcripts by activating cryptic promoters throughout the genome. The requirement of specific sequences in eukaryotic promoter regions is rather vague and mostly substituted by a characteristic, open chromatin structure.Citation9 Disturbing the closed chromatin structure in intergenic or coding regions can lead to the activation of cryptic promoter sequences, and severely perturb genome integrity. In this article we will focus on the identified factors and chromatin features, whose main function is to suppress cryptic transcription and thereby play an essential role in the correct definition of transcription units.
Different techniques have been used to monitor non-specific transcription initiation events. Reporter genes combined with known cryptic promoter sequences were successfully used in genetic screens, that resulted in the identification of a large number of cryptic promoter-activating mutants.Citation10,Citation11 Northern-blot experiments showed the appearance of transcripts with irregular length, suggesting inaccurate transcription initiation and/or termination in these mutants.Citation5,Citation7,Citation12 Monitoring AS transcript levels is also a very sensitive tool for identifying cryptic transcript-accumulating mutants. Cryptic promoters in gene coding regions might initiate transcription on the forward or on the reverse DNA strand. Depending on the orientation of the gene, these cryptic transcripts can be aberrant sense or AS transcripts. Detection of aberrant sense transcripts is challenging, because they are mostly masked by the more abundant full-length mRNA transcripts. AS transcripts, on the other hand, can be very sensitively detected, since the level of these transcripts in wild type (WT) cells is low. In addition, AS transcripts can be easily monitored genome-wide using modern genomic techniques, allowing more robust conclusions and meaningful comparisons between mutants. With the help of these techniques, several mutations that lead to genome-wide activation of cryptic promoters have been identified.Citation10 Based on their mechanisms of action, most of these mutations can be sorted into four major groups:
1) Mutations causing decreased nucleosome occupancy
The levels of histone proteins are tightly controlled in eukaryotic cells, resulting in soluble histone proteins comprising less than 1% of the total histone content of the cell.Citation13,Citation14 Depletion of histones concomitantly leads to a decrease in the overall number of nucleosomes incorporated into the genome. Several mutations have been identified that significantly decrease the histone content of the cell mostly by disturbing the proper recycling of nucleosomes during transcription elongation. Nucleosomes must be temporarily evicted from the DNA during transcription, and re-incorporated after the passage of the RNA polymerase II (RNAP II) machinery, to re-establish the closed chromatin structure. The FACT complex, together with other histone chaperones and transcription elongation factors, such as Asf1, HIRA complex and Spt6, have a well-characterized role in this process.Citation15-Citation17 Mutations in these factors lead to severe depletion of histone proteins and strong activation of cryptic promoters throughout the genome.Citation10,Citation11,Citation18-Citation20
2) Disturbed nucleosome positioning in gene coding regions
Nucleosomes in gene coding regions are regularly positioned, resulting in compact arrays with nearly identical spacing between nucleosomes. This organized nucleosome pattern plays an important role in the repression of cryptic promoter activity. S. cerevisiae chromatin remodeling enzymes, Chd1 and Isw1, were reported to cooperate in the proper positioning of nucleosomes in gene coding regions.Citation6 Deletion of Chd1 and Isw1 leads to a strong, genome-wide increase in cryptic promoter activity.Citation7,Citation10,Citation21 Similar results were reported in the distantly related S. pombe, indicating an evolutionarily conserved mechanism. ISWI-type chromatin remodeling factors are missing in S. pombe, but deletion of the Chd1-type remodelers, Hrp1 and Hrp3, leads to irregular nucleosome positioning in gene coding regions, and to genome-wide activation of cryptic transcription.Citation1,Citation8,Citation22
3) Increased histone acetylation in gene coding regions
Nucleosomes in gene coding regions show low acetylation levels compared with promoter proximal nucleosomes. This acetylation pattern is mainly controlled by a histone deacetylase (HDAC) complex, Rpd3S complex in S. cerevisiae or Clr6 complex II (Clr6-CII) in S. pombe, which is specifically recruited to the coding regions of transcription units.Citation5,Citation23-Citation25 Mutations in this complex lead to increased bulk H3 and H4 acetylation levels in the cell.Citation24 The acetylation pattern in transcription units is inverted in these mutants, resulting in significantly higher acetylation levels in coding regions compared with promoter regions.Citation1 This drastic change in the acetylation pattern of transcription units leads to a massive activation of cryptic transcription, but only marginally affects the expression levels of coding genes. Recently, another HDAC complex, the Set3C complex, was reported to play a role in the deacetylation of the 5′end of gene coding regions, and thereby repress cryptic transcription.Citation26
4) Loss of H3K36 methylation
The Set2 histone methyltransferase is recruited to the elongating RNA polymerase and methylates H3K36 in gene coding regions.Citation27,Citation28 Deletion of Set2 abolishes any form of H3K36 methylation in yeast and leads to the activation of cryptic promoters. H3K36me was suggested to serve as a recruitment signal for the Rpd3S HDAC complex by the chromodomain-containing subunit Eaf3.Citation5 According to this model, elimination of H3K36me would inhibit the recruitment of the Rpd3S HDAC complex to coding regions and thereby increase histone acetylation and trigger cryptic transcription. However, more recent studies have shown that the Rpd3S complex is recruited directly to gene coding regions by the C-terminal domain (CTD) of RNAP II,Citation29,Citation30 and deletion of Set2 does not change the localization of the Rpd3S complex.Citation30 Our recent study in S. pombe is also inconsistent with a significant role for H3K36me in the recruitment or activity of the Clr6-CII HDAC complex.Citation1 Cells carrying a mutant allele of Clr6-CII are especially defective in the removal of the acetyl groups present on H3K9 and H3K14 and, to a lesser extent, H4K5 and H4K8.Citation24 We assayed bulk H3K9ac and H3K14ac levels, and the genomic distribution of these histone modifications in WT and mutant cells. As previously reported, deletion of the Clr6-CII subunit Alp13 (homolog of S. cerevisiae Eaf3) resulted in dramatically increased H3K9ac and H3K14ac levels in coding regions. However, we could detect only a minor increase in H3 acetylation levels in the set2∆ strain, which was not comparable to the increase detected in the alp13∆ strain.Citation1 Furthermore, combination of set2∆ and alp13∆ show an additive effect on cryptic transcription activity, indicating that these genes contribute to the repression of cryptic transcription mainly via parallel pathways.Citation25 A recent study suggests that H3K36me suppresses nucleosome turnover at gene coding regions.Citation7,Citation31 Nucleosomes at promoter regions are rapidly exchanged while coding regions show a slower kinetic in nucleosome turnover. In the absence of H3K36me, the nucleosome turnover rate in coding regions slightly increases compared with WT cells. Since soluble histones are hyper-acetylated, the increased histone turnover rate could explain the slightly increased acetylation levels in the set2∆ strain.
The exact mechanism for how H3K36me represses nucleosome turnover is not clear. However, in S. cerevisiae H3K36me can specifically recruit the Isw1b chromatin remodeling complex,Citation7 which was reported to reduce nucleosome turnover in gene-coding regions. An additional layer of complexity was revealed by data suggesting that deletion of the Chd1 chromatin remodeler also increases nucleosome turnover rates in coding regions.Citation7,Citation32-Citation34 A possible model is that chromatin remodeling factors, such as Chd1 and Isw1b, might not only be responsible for the correct positioning of nucleosomes, but they might also prevent trans-histone exchange by histone chaperone complexes, thereby decreasing the rate of nucleosome turnover in gene coding regions.Citation35
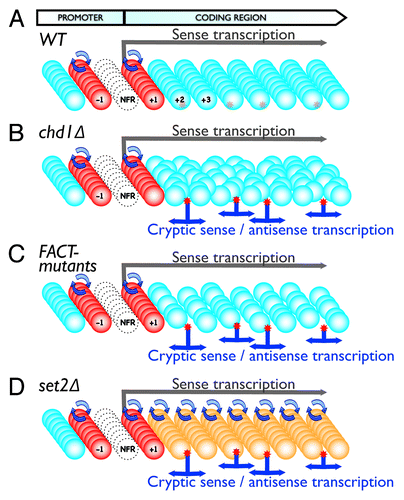
Interestingly, the previously described mutations lead to a nearly identical phenotype in cryptic transcript accumulation, despite the fact that their effect on chromatin organization is remarkably different. Although these chromatin features might be strongly interconnected, our results showed that histone acetylation or H3K36 methylation does not detectably influence nucleosome positioning or occupancy.Citation1 Similarly, impaired nucleosome positioning or occupancy did not significantly change histone acetylation patterns. How do these seemingly very different mechanisms give rise to such a similar aberrant cryptic transcription phenotype? A possible explanation is that all of these mutations lead to the temporary appearance of NFRs in gene coding regions, which can expose cryptic promoter sequences. The presence of a NFR seems to be one of the most conserved and essential features of eukaryotic promoters, and several lines of evidence show that an artificial NFR can act as a minimal promoter.Citation9,Citation36-Citation38 In contrast, gene-coding regions possess a chromatin structure that prevents the occurrence of NFRs (). Mutations in the Chd1-type chromatin remodeling factors lead to irregular nucleosome positioning in gene coding regions, and therefore to the random appearance of NFR-like regions (). Genome-wide nucleosome depletion, such as that observed for the FACT complex mutants or mutations in histone chaperones, do not significantly change the positions of the nucleosomes, but they do increase the time that certain nucleosome positions remain in an unoccupied state,Citation39 creating transient NFRs (). Increased nucleosome turnover, such as that reported in the set2∆ strain, leads to more frequent assembly and disassembly of nucleosomes, thereby temporarily creating NFRs, which could ultimately be responsible for transcription initiation from these DNA regions (). Increased acetylation in gene coding regions, as observed for HDAC mutants, might also result in elevated nucleosome turnover, probably by weakening histone-DNA interactions and recruiting bromodomain-containing chromatin remodeling complexes, such as the RSC complex. The crosstalk between histone modifications, turnover, histone occupancy, and nucleosome positioning is poorly understood, and further studies are necessary to better understand this highly regulated network.
Figure 1. Model of key mechanisms controlling cryptic transcription. Panels show schematics representing transcription units in WT (A) or mutant cells (B-D). Colored balls represent nucleosomes (Red: “hot” nucleosomes with a high turnover rate; Blue: “cold” nucleosomes with a low turnover rate; Orange: nucleosomes with an elevated turnover rate). Each row of nucleosomes indicates the chromatin structure of a single cell within the cell population (multiple rows). Red stars represent cryptic promoter sequences, which can either be shielded by the nucleosomes (A) or exposed when chromatin structure is impaired, resulting in cryptic transcription initiation (B-D). The empty circles indicate NFRs in the promoter region. Overall, the model demonstrates: Cryptic promoters are shielded in WT cells with proper chromatin structure (A); Cryptic promoters are exposed leading to cryptic transcription in cells with impaired chromatin structure due to altered nucleosome positioning (B), nucleosome depletion (C) or increased nucleosome turnover rate in gene coding regions (D)
Uncovering the mechanisms that repress cryptic promoter activity will also help to further understand the chromatin organization of canonical promoters. Some promoter-specific chromatin modifications, such as H2A.Z or H3K4me3 nucleosomes, turned out to be the consequences of promoter regions rather than determining factors for promoter activity.Citation40 Lessons from cryptic promoter-activating mutants suggest that the key chromatin features in determining promoter regions are the presence of an NFR flanked by hyperacetylated nucleosomes with high turnover rates. Eukaryotic cells have established a sophisticated machinery that prohibits these chromatin features in gene coding regions, thus preventing unwanted transcriptional initiation events. Interestingly, impairing these mechanisms and thereby opening the chromatin structure in gene coding regions does not significantly influence transcription elongation, as demonstrated by the relatively minor changes in mRNA levels in these mutants. Moreover, the closed chromatin structure is rather inhibitory for transcription elongation, and requires additional complexes to open the chromatin at each transcription cycle. Consequently, the above-mentioned mutants are viable in yeast systems and show only slight growth defects. These observations raise the question: Why are these cryptic transcription-repressing mechanisms so important that they are evolutionarily highly conserved from yeast to humans? Excessive cryptic transcription and accumulation of ncRNAs impair genomic stability, as evidenced by the increased sensitivity of these mutants against genotoxic agents.Citation24,Citation25 One of the major functions of the chromatin structure is the maintenance of the integrity and stability of the eukaryotic genome. Mechanisms that impair genome integrity are also a major cause of cancer development. Further understanding of the molecular mechanisms leading to genomic instability might provide important insights into the mechanisms underlying tumorigenesis.
| Abbreviations: | ||
| AS | = | antisense |
| Clr6-CII | = | Clr6 complex II |
| CTD | = | C-terminal domain |
| HDAC | = | histone deacetylase |
| H3K36me | = | methylated form of histone H3 at lysine 36 |
| ncRNA | = | non-coding RNA |
| NFR | = | nucleosome-free DNA region |
| RNAP II | = | RNA polymerase II |
| S. cerevisiae | = | Saccharomyces cerevisiae |
| S. pombe | = | Schizosaccharomyces pombe |
| TSS | = | transcription start site |
| H3K4me3 | = | trimethylated histone H3 at lysine 4 |
| WT | = | wild type |
Acknowledgments
We would like to thank Emmalene Bartlett, Ke Zhang and Martin Kos for proofreading the manuscript. B.P.H. and T.F. were supported by a grant from the Ministry of Science, Research and the Arts of Baden-Wuerttemberg.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Hennig BP, Bendrin K, Zhou Y, Fischer T. Chd1 chromatin remodelers maintain nucleosome organization and repress cryptic transcription. EMBO Rep 2012; 13:997 - 1003; http://dx.doi.org/10.1038/embor.2012.146; PMID: 23032292
- Rando OJ, Chang HY. Genome-wide views of chromatin structure. Annu Rev Biochem 2009; 78:245 - 71; http://dx.doi.org/10.1146/annurev.biochem.78.071107.134639; PMID: 19317649
- Zofall M, Fischer T, Zhang K, Zhou M, Cui B, Veenstra TD, et al. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 2009; 461:419 - 22; http://dx.doi.org/10.1038/nature08321; PMID: 19693008
- Lenstra TL, Benschop JJ, Kim T, Schulze JM, Brabers NA, Margaritis T, et al. The specificity and topology of chromatin interaction pathways in yeast. Mol Cell 2011; 42:536 - 49; http://dx.doi.org/10.1016/j.molcel.2011.03.026; PMID: 21596317
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 2005; 123:581 - 92; http://dx.doi.org/10.1016/j.cell.2005.10.023; PMID: 16286007
- Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, et al. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science 2011; 333:1758 - 60; http://dx.doi.org/10.1126/science.1206097; PMID: 21940898
- Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, et al. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol 2012; 19:884 - 92; http://dx.doi.org/10.1038/nsmb.2312; PMID: 22922743
- Shim YS, Choi Y, Kang K, Cho K, Oh S, Lee J, et al. Hrp3 controls nucleosome positioning to suppress non-coding transcription in eu- and heterochromatin. EMBO J 2012; 31:4375 - 87; http://dx.doi.org/10.1038/emboj.2012.267; PMID: 22990236
- Sekinger EA, Moqtaderi Z, Struhl K. Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast. Mol Cell 2005; 18:735 - 48; http://dx.doi.org/10.1016/j.molcel.2005.05.003; PMID: 15949447
- Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, et al. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol 2008; 6:e277; http://dx.doi.org/10.1371/journal.pbio.0060277; PMID: 18998772
- Silva AC, Xu X, Kim HS, Fillingham J, Kislinger T, Mennella TA, et al. The replication-independent histone H3-H4 chaperones HIR, ASF1, and RTT106 co-operate to maintain promoter fidelity. J Biol Chem 2012; 287:1709 - 18; http://dx.doi.org/10.1074/jbc.M111.316489; PMID: 22128187
- Pattenden SG, Gogol MM, Workman JL. Features of cryptic promoters and their varied reliance on bromodomain-containing factors. PLoS One 2010; 5:e12927; http://dx.doi.org/10.1371/journal.pone.0012927; PMID: 20886085
- Gunjan A, Paik J, Verreault A. The emergence of regulated histone proteolysis. Curr Opin Genet Dev 2006; 16:112 - 8; http://dx.doi.org/10.1016/j.gde.2006.02.010; PMID: 16510276
- Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, et al. Elevated histone expression promotes life span extension. Mol Cell 2010; 39:724 - 35; http://dx.doi.org/10.1016/j.molcel.2010.08.015; PMID: 20832724
- Formosa T, Ruone S, Adams MD, Olsen AE, Eriksson P, Yu Y, et al. Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: polymerase passage may degrade chromatin structure. Genetics 2002; 162:1557 - 71; PMID: 12524332
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science 2003; 301:1090 - 3; http://dx.doi.org/10.1126/science.1085703; PMID: 12934006
- Formosa T. FACT and the reorganized nucleosome. Mol Biosyst 2008; 4:1085 - 93; http://dx.doi.org/10.1039/b812136b; PMID: 18931784
- Kaplan CD, Laprade L, Winston F. Transcription elongation factors repress transcription initiation from cryptic sites. Science 2003; 301:1096 - 9; http://dx.doi.org/10.1126/science.1087374; PMID: 12934008
- Mason PB, Struhl K. The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Mol Cell Biol 2003; 23:8323 - 33; http://dx.doi.org/10.1128/MCB.23.22.8323-8333.2003; PMID: 14585989
- Celona B, Weiner A, Di Felice F, Mancuso FM, Cesarini E, Rossi RL, et al. Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol 2011; 9:e1001086; http://dx.doi.org/10.1371/journal.pbio.1001086; PMID: 21738444
- Quan TK, Hartzog GA. Histone H3K4 and K36 methylation, Chd1 and Rpd3S oppose the functions of Saccharomyces cerevisiae Spt4-Spt5 in transcription. Genetics 2010; 184:321 - 34; http://dx.doi.org/10.1534/genetics.109.111526; PMID: 19948887
- Pointner J, Persson J, Prasad P, Norman-Axelsson U, Strålfors A, Khorosjutina O, et al. CHD1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in S. pombe. EMBO J 2012; 31:4388 - 403; http://dx.doi.org/10.1038/emboj.2012.289; PMID: 23103765
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 2005; 123:593 - 605; http://dx.doi.org/10.1016/j.cell.2005.10.025; PMID: 16286008
- Nakayama J, Xiao G, Noma K, Malikzay A, Bjerling P, Ekwall K, et al. Alp13, an MRG family protein, is a component of fission yeast Clr6 histone deacetylase required for genomic integrity. EMBO J 2003; 22:2776 - 87; http://dx.doi.org/10.1093/emboj/cdg248; PMID: 12773392
- Nicolas E, Yamada T, Cam HP, Fitzgerald PC, Kobayashi R, Grewal SI. Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nat Struct Mol Biol 2007; 14:372 - 80; http://dx.doi.org/10.1038/nsmb1239; PMID: 17450151
- Kim T, Xu Z, Clauder-Münster S, Steinmetz LM, Buratowski S. Set3 HDAC mediates effects of overlapping noncoding transcription on gene induction kinetics. Cell 2012; 150:1158 - 69; http://dx.doi.org/10.1016/j.cell.2012.08.016; PMID: 22959268
- Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol 2003; 23:4207 - 18; http://dx.doi.org/10.1128/MCB.23.12.4207-4218.2003; PMID: 12773564
- Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, et al. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev 2003; 17:654 - 63; http://dx.doi.org/10.1101/gad.1055503; PMID: 12629047
- Govind CK, Qiu H, Ginsburg DS, Ruan C, Hofmeyer K, Hu C, et al. Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol Cell 2010; 39:234 - 46; http://dx.doi.org/10.1016/j.molcel.2010.07.003; PMID: 20670892
- Drouin S, Laramée L, Jacques PÉ, Forest A, Bergeron M, Robert F. DSIF and RNA polymerase II CTD phosphorylation coordinate the recruitment of Rpd3S to actively transcribed genes. PLoS Genet 2010; 6:e1001173; http://dx.doi.org/10.1371/journal.pgen.1001173; PMID: 21060864
- Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S, et al. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 2012; 489:452 - 5; http://dx.doi.org/10.1038/nature11326; PMID: 22914091
- Radman-Livaja M, Quan TK, Valenzuela L, Armstrong JA, van Welsem T, Kim T, et al. A key role for Chd1 in histone H3 dynamics at the 3′ ends of long genes in yeast. PLoS Genet 2012; 8:e1002811; http://dx.doi.org/10.1371/journal.pgen.1002811; PMID: 22807688
- Lee JS, Garrett AS, Yen K, Takahashi YH, Hu D, Jackson J, et al. Codependency of H2B monoubiquitination and nucleosome reassembly on Chd1. Genes Dev 2012; 26:914 - 9; http://dx.doi.org/10.1101/gad.186841.112; PMID: 22549955
- Zentner GE, Tsukiyama T, Henikoff S. ISWI and CHD chromatin remodelers bind promoters but act in gene bodies. PLoS Genet 2013; 9:e1003317; http://dx.doi.org/10.1371/journal.pgen.1003317; PMID: 23468649
- Smolle M, Workman JL, Venkatesh S. reSETting chromatin during transcription elongation. Epigenetics 2012; 8:10 - 5; http://dx.doi.org/10.4161/epi.23333; PMID: 23257840
- Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, et al. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 2005; 123:233 - 48; http://dx.doi.org/10.1016/j.cell.2005.10.002; PMID: 16239142
- Venters BJ, Pugh BF. A canonical promoter organization of the transcription machinery and its regulators in the Saccharomyces genome. Genome Res 2009; 19:360 - 71; http://dx.doi.org/10.1101/gr.084970.108; PMID: 19124666
- Raveh-Sadka T, Levo M, Shabi U, Shany B, Keren L, Lotan-Pompan M, et al. Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat Genet 2012; 44:743 - 50; http://dx.doi.org/10.1038/ng.2305; PMID: 22634752
- Gossett AJ, Lieb JD. In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet 2012; 8:e1002771; http://dx.doi.org/10.1371/journal.pgen.1002771; PMID: 22737086
- Hartley PD, Madhani HD. Mechanisms that specify promoter nucleosome location and identity. Cell 2009; 137:445 - 58; http://dx.doi.org/10.1016/j.cell.2009.02.043; PMID: 19410542