Abstract
Stable isotope labeling by amino acids combined with mass spectrometry is a widely used methodology to quantitatively examine metabolic and signaling pathways in yeast, fruit flies, plants, cell cultures and mice. However, only metabolic labeling using 15N has been applied to examine such events in the nematode Caenorhabditis elegans. We have recently shown that C. elegans can be completely labeled with heavy-labeled lysine by feeding worms on prelabeled lysine auxotroph Escherichia coli for just one generation. We applied this methodology to examine the organismal response to functional loss or RNAi mediated knock down of the transcription factor NHR-49, and found numerous proteins involved in lipid metabolism to be downregulated, which is consistent with its previously proposed function as a transcriptional regulator of fatty acid metabolism. The combined use of quantitative proteomics and selective gene knockdown by RNAi provides a powerful tool with broad implications for C. elegans biology.
Quantitative proteomics is increasingly being applied to examine and understand how cells and organisms regulate their metabolism in order to support growth, proliferation, differentiation, development and survival.Citation1 Several different strategies for quantitative proteomics have been developed including label-free quantification, chemical labeling e.g., iTRAQ and dimethyl labeling or metabolic labeling using heavy isotopes.Citation2-Citation5 The simplest strategy is label-free quantification, where signal intensities of a given peptide in a number of spectra are used as an estimate of the abundance of the sample protein.Citation3 Such an approach does not require any sample preparation prior to analysis, thus this approach is applicable to all kinds of samples and an indefinite number of experiments can be compared. However, this methodology suffers from sensitivity to variations in sample composition that can easily affect ionization of the relevant peptide and hence alter its apparent abundance. Consequently, as sample complexity grows the utility of such strategy declines. Instead, several labeling approaches have been developed that benefit from stable isotopes having the same physicochemical properties except from their masses, making distinguishable by mass spectrometry. In vitro labeling includes incorporation of 18O by enzymatic hydrolysis in heavy water and chemical labeling of amino acids containing certain reactive groups.Citation3 The latter features isotope-coded affinity tags to facilitate purification and isobaric tags for relative and absolute quantitation (iTRAQ). iTRAQ are isobaric tags that, upon fragmentation, release reporter ions of unique masses. The advantage of these in vitro approaches is that any sample or tissue can be labeled. However, the samples undergo a number of preparation steps before labeling and mixing, which increases the risk of introducing a quantitative bias in the samples and thereby decreasing the quantitative accuracy.
Particularly, metabolic labeling with stable isotopes has become the prevailing strategy to quantitatively compare proteomes of cells and organisms. C. elegans has during the past decade proven to be a powerful model in identification and characterization of novel genes and signaling pathways regulating cell division, development, aging, apoptosis and metabolism.Citation1 However, quantitative proteomics has only to a limited extent been applied to study signaling events governing metabolism in C. elegans. In 2003 Krijgsveld et al. showed that C. elegans animals can be metabolically labeled with 15N by feeding them on 15N-labeled E. coli for two generations,Citation6 that, when combined with an 14N-labeled worm population, could be used to determine the relative protein abundance among the two populations by mass spectrometry. Henceforth, they applied this strategy to identify differentially expressed proteins in glp-4 animals compared with wild type animals.Citation6 Analogously, Yates and coworkers examined how loss of functional insulin receptors affects the proteome of C. elegans, and identified novel key regulators of insulin regulated metabolic outputs.Citation7 Recently, Simonsen et al. used a similar approach to identify differentially expressed proteins in C. elegans in response to short- and long-term infection by a pathogenic adherent-invasive strain of E. coli, that were isolated from patients suffering from the inflammatory bowel disease Crohn disease.Citation8 However, compared with metabolic labeling with 15N, stable isotope labeling by amino acids in cell cultureCitation9,Citation10 (SILAC) provides a more precise mass spectrometry-based quantitative strategy, as it provides a defined number of labels per identified peptide and therefore enables easier and more comprehensive peptide identification and data analysis. Such methodologies have proven to be a highly valuable tool for studies in in vivo systems like the yeast Saccharomyces cerevisiae,Citation9,Citation11 the fruit fly Drosophila melanogaster,Citation12 the plant Arabidopsis thalianaCitation13 and mice.Citation14 Recently, our laboratory and others added C. elegans to the SILAC zoo (see below).Citation15,Citation16
We have shown that C. elegans can be completely labeled by stable isotope labeled lysine by feeding animals on a lysine auxotroph E. coli strain for a single generation.Citation15 Moreover, following protein extraction from light and heavy labeled C. elegans we showed that peptides can be identified and quantified with high accuracy (standard deviation of log2 = 0.22), which is comparable to similar approaches applied on the fruit fly Drosophila melanogaster.Citation12 SILAC based quantitative proteomics is typically based on labeling with both arginine and lysine, which provides one label per peptide after trypsin digestion, and hence improved proteome coverage. Although we only labeled C. elegans with lysine, and subsequently digested with lysyl endopeptidase (Lys-C) resulting in longer peptides, we were able to identify and quantify a vast number of proteins due to intensive peptide fractionation prior to mass spectrometry analysis. Moreover, arginine to proline conversion imposes a major challenge in peptide identification and quantification. To this end, Larance et al. recently showed that up to 20% of the proline become labeled when C. elegans is labeled with arginine, which can be prevented by RNAi mediated knock down of ornithine transaminase, orn-1, required for the conversion.Citation16 However, labeling with lysine combined with extensive peptide fractionation may be advantageous as orn-1 knock down may interfere with the metabolism of the nematode, and prevents that other genes are efficiently knocked down by RNAi.Citation17
One of the major advantages by C. elegans as a model organism is the unprecedented applicability of RNA interference to systematically study gene functions. We therefore rendered the lysine auxotroph E. coli strain RNAi compatible by modifying it to express the T7 RNA polymerase from an IPTG-inducible promoter and by eliminating RNaseIII to prevent degradation of dsRNA in E. coli. Subsequently, to validate the use of lysine labeling of C. elegans in quantitative proteomics studies we aimed to identify differentially expressed proteins in response to functional loss or RNAi mediated knock down of the nuclear hormone receptor NHR-49. The expression of genes involved in lipid metabolism in C. elegans is coordinately controlled by a number of transcription factors including the NHR-49, which is a hepatocyte nuclear factor (HNF)-4α ortholog and has a function analogous to that of peroxisome proliferator-activated receptor α, PPARα, in mammals. By quantitative proteomics we identified 3949 and 4627 proteins, respectively, of which 143 and 330 proteins were differentially expressed in response to disruption or knock down of NHR-49 function.Citation15 Among the downregulated proteins we identified proteins involved in lipid metabolism to be significantly overrepresented. In particular, we found that the Δ9 fatty acid desaturases FAT-5 and FAT-6, which previously have been shown to be controlled by NHR-49,Citation18 were significantly downregulated in response to functional loss or knockdown of nhr-49. Moreover, we found FAT-1 and FAT-2, an ω3 fatty acid desaturase and a Δ12 fatty acid desaturase, respectively, to be downregulated. This observation supports the notion that loss of NHR-49 function impedes on fatty acid desaturation leading to accumulation of saturated fatty acids.Citation18 Besides fat-5, fat-6 and fat-7, van Gilst et al. also found three genes involved in mitochondrial β-oxidation of fatty acids (ech-1, cpt-5 and acs-2), three genes involved in peroxisomal β-oxidation (two putative acyl-CoA oxidases and ech-9), two genes involved in fatty acid binding/transport (lbp-7 and lbp-8) and two genes involved in the glyoxylate pathway (gei-7 and sdha-1), arguing that NHR-49 is required for fatty acid degradation in C. elegans.Citation18 Accordingly, nhr-49 animals have enlarged lipid stores.Citation18 Consistently, we find an array of proteins ( and ) to be downregulated in nhr-49 animals that either have been shown, or based on sequence similarities to functionally characterized gene products from other model organisms like Saccharomyces cerevisiae and mice, are predicted to be involved in β-oxidation of fatty acids or metabolism of acetyl-CoA. Since these proteins contain a C-terminal peroxisomal targeting signal or that their mammalian counterpart previously has been identified to the peroxisomes, our observations suggest that NHR-49 primarily regulates peroxisomal β-oxidation rather than mitochondrial β-oxidation, as suggested by van Gilst et al.Citation18 The inability to degrade long-chain fatty acids would consequently result in increased intracellular levels of unbound fatty acids and fatty acyl-CoA esters. Consistent with this notion, we find the predicted fatty acid binding protein LBP-3 and acyl-CoA binding protein ACBP-1 to be upregulated in response to loss of NHR-49 function. These binding proteins may bind, sequester and hence protect cells from detrimental effects of large increases in free fatty acid and acyl-CoA levels, respectively. The fact that we also find glutathione and xenobiotic metabolism to be upregulated may also reflect an increased cellular stress response in response to loss of NHR-49 function.
Figure 1. Downregulation of nhr-49 by RNAi affects the abundance of proteins involved in fatty acid metabolism. Stable amino acid labeling and quantitative proteomics was use to identify the differentially expressed proteins in L4 stage nematodes treated with nhr-49 RNAi compared with empty vector controls. Among the regulated proteins, enzymes involved in fatty acid metabolism, especially peroxisomal β-oxidation, are significantly overrepresented. The indicated protein is known or predicted, based on sequence homology to yeast or mouse orthologs, to be involved in the indicated biochemical pathway. Green and red indicate proteins that become less and more abundant, respectively, in response to RNAi mediated knock down of nhr-49.
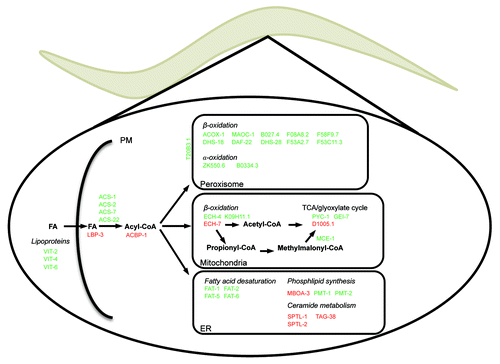
Table 1. NHR-49 affects abundance of metabolic enzymes. Quantitative proteomics was use to identify the differentially expressed proteins in L4 stage nematodes treated with nhr-49 RNAi compared to empty vector controls. Among the total number of identified regulated proteins a subset is shown. The log2 ratios indicate less or more abundant proteins after RNAi against NHR-49. See Fredens et al. for details.Citation15
Gene expression levels are often interpreted based on mRNA levels, yet, it is increasingly recognized that the mRNA and protein levels may not correlate.Citation19,Citation20 While the abundance of the majority of the proteins, we identified to be regulated upon impaired NHR-49 function, correlated well with the mRNA levels previously reported by van Gilst et al.,Citation18 the level of some proteins did not correlate with the mRNA level.Citation15 This may be due to alternative RNA splicing, differences in the half-lives of mRNAs and proteins, as well as rates of transcription and translation.Citation21
Conclusion
Stable isotope labeling of C. elegans with lysine and/or arginine provides a simple and straightforward approach for in vivo incorporation of a label into proteins for mass spectrometry-based quantitative proteomics. We anticipate that the recently described labeling methodologiesCitation15,Citation16 greatly will facilitate characterization of gene functions in the multicellular organism C. elegans and become a widely used technique in all areas of C. elegans biology. In contrast to metabolic labeling with 15N, stable isotope labeling with amino acids provides an in vivo strategy to label proteins with different stable isotopic forms of the amino acids (e.g., lys0, lys4, lys8, Arg4 or Arg10), making it possible to monitor differences at the protein level between multiple conditions or over time in a quantitative manner. Thus, stable isotope labeling with amino acids can be used to monitor how genetic, chemical or environmental perturbations affect the proteome of C. elegans over time. Combined with enrichment of posttranslational modified peptides, e.g., phosphopeptides, this approach can also delineate how various signaling cascades are affected in response to a specific perturbation.
References
- Corsi AK. A biochemist's guide to Caenorhabditis elegans.. Anal Biochem 2006; 359:1 - 17; http://dx.doi.org/10.1016/j.ab.2006.07.033; PMID: 16942745
- Thingholm TE, Jensen ON, Larsen MR. Analytical strategies for phosphoproteomics. Proteomics 2009; 9:1451 - 68; http://dx.doi.org/10.1002/pmic.200800454; PMID: 19235172
- Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem 2007; 389:1017 - 31; http://dx.doi.org/10.1007/s00216-007-1486-6; PMID: 17668192
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 2004; 3:1154 - 69; http://dx.doi.org/10.1074/mcp.M400129-MCP200; PMID: 15385600
- Hsu JL, Huang SY, Chow NH, Chen SH. Stable-isotope dimethyl labeling for quantitative proteomics. Anal Chem 2003; 75:6843 - 52; http://dx.doi.org/10.1021/ac0348625; PMID: 14670044
- Krijgsveld J, Ketting RF, Mahmoudi T, Johansen J, Artal-Sanz M, Verrijzer CP, et al. Metabolic labeling of C. elegans and D. melanogaster for quantitative proteomics. Nat Biotechnol 2003; 21:927 - 31; http://dx.doi.org/10.1038/nbt848; PMID: 12858183
- Dong MQ, Venable JD, Au N, Xu T, Park SK, Cociorva D, et al. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans.. Science 2007; 317:660 - 3; http://dx.doi.org/10.1126/science.1139952; PMID: 17673661
- Simonsen KT, Moller-Jensen J, Kristensen AR, Andersen JS, Riddle DL, Kallipolitis BH. Quantitative proteomics identifies ferritin in the innate immune response of C. elegans.. Virulence 2011; 2:120 - 30; http://dx.doi.org/10.4161/viru.2.2.15270; PMID: 21389771
- Jiang H, English AM. Quantitative analysis of the yeast proteome by incorporation of isotopically labeled leucine. J Proteome Res 2002; 1:345 - 50; http://dx.doi.org/10.1021/pr025523f; PMID: 12645890
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 2002; 1:376 - 86; http://dx.doi.org/10.1074/mcp.M200025-MCP200; PMID: 12118079
- Gruhler A, Olsen JV, Mohammed S, Mortensen P, Faergeman NJ, Mann M, et al. Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol Cell Proteomics 2005; 4:310 - 27; http://dx.doi.org/10.1074/mcp.M400219-MCP200; PMID: 15665377
- Sury MD, Chen JX, Selbach M. The SILAC fly allows for accurate protein quantification in vivo. Mol Cell Proteomics 2010; 9:2173 - 83; http://dx.doi.org/10.1074/mcp.M110.000323; PMID: 20525996
- Gruhler A, Schulze WX, Matthiesen R, Mann M, Jensen ON. Stable isotope labeling of Arabidopsis thaliana cells and quantitative proteomics by mass spectrometry. Mol Cell Proteomics 2005; 4:1697 - 709; http://dx.doi.org/10.1074/mcp.M500190-MCP200; PMID: 16088002
- Krüger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, et al. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 2008; 134:353 - 64; http://dx.doi.org/10.1016/j.cell.2008.05.033; PMID: 18662549
- Fredens J, Engholm-Keller K, Giessing A, Pultz D, Larsen MR, Hojrup P, et al. Quantitative proteomics by amino acid labeling in C. elegans.. Nat Methods 2011; 8:845 - 7; http://dx.doi.org/10.1038/nmeth.1675; PMID: 21874006
- Larance M, Bailly AP, Pourkarimi E, Hay RT, Buchanan G, Coulthurst S, et al. Stable-isotope labeling with amino acids in nematodes. Nat Methods 2011; 8:849 - 51; http://dx.doi.org/10.1038/nmeth.1679; PMID: 21874007
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. . Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biology2001; 2.
- Van Gilst MR, Hadjivassiliou H, Jolly A, Yamamoto KR. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans.. PLoS Biol 2005; 3:e53; http://dx.doi.org/10.1371/journal.pbio.0030053; PMID: 15719061
- Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet 2011; 7:e1001393; http://dx.doi.org/10.1371/journal.pgen.1001393; PMID: 21695224
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature 2011; 473:337 - 42; http://dx.doi.org/10.1038/nature10098; PMID: 21593866
- Muers M. Gene expression: Transcriptome to proteome and back to genome. Nat Rev Genet 2011; 12:518; http://dx.doi.org/10.1038/nrg3037; PMID: 21709688