Figures & data
Figure 1. Proposed reaction cycle for the bacterial KdpFABC complex. Binding of a potassium ion to the E2 state enzyme promotes high-affinity ATP binding to the N-domain of KdpB. Subsequently, the KdpFABC complex is transformed to its E1 state, and the potassium ion is able to enter the now energetically favorable binding site in the center of the selectivity filter of KdpA. Upon reaching the transition state (TSI) followed by phosphorylation, the potassium ion becomes occluded. The subsequent E1 to E2 transition, accompanied by major conformational changes in KdpB, alters the dipole formed by D583 and K586 in the transmembrane domain of KdpB. This makes the position of the potassium ion in the center of the selectivity filter of KdpA unfavorable and, consequently, pushes the ion toward the cytoplasmic side of the membrane, where it is finally released. Dephosphorylation of the complex regenerates the E2 state.
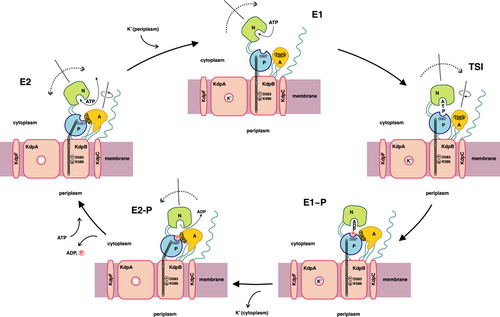
Figure 2. Schematic topology of P-type ATPases. The proposed topology of KdpB (type IA) with its seven transmembrane helices (TMH) is shown in dark gray. Heavy metal-transporting ATPases (type IB) comprise an N-terminal extension (depicted in yellow) with various metal-binding clusters and eight TMH. P-type ATPases of the type II-IV class comprise 10 TMH. The putative type V of P-type ATPases (with unknown substrate specificity) might have 12 TMH. Beside differences in the number of transmembrane helices all P-type ATPases have two large cytoplasmic domains. The first loop between TMH 2 and TMH 3 is called actuator (A-) domain. The second cytoplasmic loop is composed of two modules: The phosphorylation (P-) domain contains the conserved aspartate residue that becomes reversibly phosphorylated (circle). The nucleotide-binding (N-) domain is inserted into the P-domain and contains the ATP binding site (illustrated by ATP) (modified according to Figure 1 of Haupt et al. Citation2004).
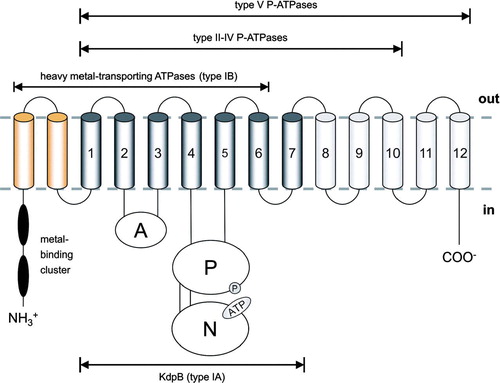
Figure 3. Comparison of the nucleotide-binding domain structures of various P-type ATPases. A comparative view on the structures of different P-type ATPases displays the similar core structures, but also reveals the insertions found in the eukaryotic members of the P-type ATPase family. Structures of nucleotide-binding domains of KdpB [KdpBN, 2A29] (Haupt et al. Citation2006), CopA [CopA_N, 2B8E] (Sazinsky et al. Citation2006b), Wilson disease protein [WDP_N, 2ARF] (Dmitriev et al. Citation2006), Na+,K+-ATPase [NaK_N, 1Q31] (Håkansson Citation2003), and Ca2 + -ATPase [SERCA_N, 1VFP] (Toyoshima & Mizutani Citation2004) are shown side by side (panel A). KdpBN exhibits a minimal scheme of a nucleotide binding domain with a central six-stranded, anti-parallel β-sheet, flanked by two α-helices on either side. This core motif is found in all N-domains. The comparison between prokaryotic (KdpBN, CopA_N) and eukaryotic (WDP-N, NaK_N, and SERCA_N) N-domains shows that additional amino acid residues are inserted in a way that leaves the central ATP binding pocket unaffected. The different sets of amino acids that are employed by type IB heavy metal-transporting ATPases (WDP_N) on the one hand and KdpB (type IA) as well as type II ATPases (Ca2 + -ATPase) on the other hand are highlighted in panel B. Amino acid residues that interact with the nucleotide are shown in green, and the nucleotidesare depicted in red.
![Figure 3. Comparison of the nucleotide-binding domain structures of various P-type ATPases. A comparative view on the structures of different P-type ATPases displays the similar core structures, but also reveals the insertions found in the eukaryotic members of the P-type ATPase family. Structures of nucleotide-binding domains of KdpB [KdpBN, 2A29] (Haupt et al. Citation2006), CopA [CopA_N, 2B8E] (Sazinsky et al. Citation2006b), Wilson disease protein [WDP_N, 2ARF] (Dmitriev et al. Citation2006), Na+,K+-ATPase [NaK_N, 1Q31] (Håkansson Citation2003), and Ca2 + -ATPase [SERCA_N, 1VFP] (Toyoshima & Mizutani Citation2004) are shown side by side (panel A). KdpBN exhibits a minimal scheme of a nucleotide binding domain with a central six-stranded, anti-parallel β-sheet, flanked by two α-helices on either side. This core motif is found in all N-domains. The comparison between prokaryotic (KdpBN, CopA_N) and eukaryotic (WDP-N, NaK_N, and SERCA_N) N-domains shows that additional amino acid residues are inserted in a way that leaves the central ATP binding pocket unaffected. The different sets of amino acids that are employed by type IB heavy metal-transporting ATPases (WDP_N) on the one hand and KdpB (type IA) as well as type II ATPases (Ca2 + -ATPase) on the other hand are highlighted in panel B. Amino acid residues that interact with the nucleotide are shown in green, and the nucleotidesare depicted in red.](/cms/asset/94c1c9ed-53ef-4c42-af46-46e4bccd538d/imbc_a_241777_f0003_b.jpg)
Figure 4. MPM-type K+ channel model of KdpA. The four MPM motifs of KdpA were modeled pairwise together with the connecting loop regions against the coordinates of the S. lividans KcsA K+ channel [1K4C] (Zhou et al. Citation2001). (A) Residues effecting ion selectivity upon mutagenesis are indicated in red (significant residues) and blue (essential residues). (B) Tetrameric assembly of A, with the color code ranging from yellow to blue with increasing effect on ion selectivity upon mutagenesis.
![Figure 4. MPM-type K+ channel model of KdpA. The four MPM motifs of KdpA were modeled pairwise together with the connecting loop regions against the coordinates of the S. lividans KcsA K+ channel [1K4C] (Zhou et al. Citation2001). (A) Residues effecting ion selectivity upon mutagenesis are indicated in red (significant residues) and blue (essential residues). (B) Tetrameric assembly of A, with the color code ranging from yellow to blue with increasing effect on ion selectivity upon mutagenesis.](/cms/asset/98acac83-a284-4a81-970c-39bcde6d029d/imbc_a_241777_f0004_b.jpg)