
Figures & data
Figure 1. Pathophysiology of Sickle Cell Disease (SCD). SCD is characterized by the production of hemoglobin S (HbS) in red blood cells (RBC). Hb S polymerizes when deoxygenated, causing the cells to become rigid and adopt a sickled state, as well as making cells more fragile and susceptible to rupture (hemolysis). Increasing levels of fetal hemoglobin (HbF) in RBC can reduce hemoglobin polymerization and therefore RBC sickling and hemolysis. Intravascular hemolysis has significant consequences; hemoglobin (Hb) released into the plasma results in endothelium-derived nitric oxide (NO) depletion in the blood vessel and the release of the toxic Hb product, hemin. Red cell lysis also increases plasma arginase, consequently limiting the availability of the substrate for NO synthesis, L-arginine. Oxidative Hb reactions, as well as reduced NO bioavailability and hemin production, contribute significantly to vascular oxidative stress and endothelial cell activation. Activated endothelium produces inflammatory cytokines and expresses adhesion molecules on its surface, resulting in leukocyte capture, and RBC and platelet adhesion to the vascular wall. Inflammatory mediator production from activated platelets, leukocytes and endothelial cells results in the inflammatory state that is associated with SCD and that drives recurrent vaso-occlusive processes, which result from leukocyte and RBC adhesion to the endothelium, occasioning reduced blood flow, diminished oxygen concentrations, consequent red cell sickling and eventually the occlusion of vessels. In a vicious cycle, chronic vascular inflammation and recurrent vaso-occlusive processes occur, causing hypoxia/reperfusion injury and resulting in further oxidative stress. Additionally, hypoxia triggers the synthesis of pro-angiogenic factors that results in the upregulation of angiogenesis and further endothelial activation and inflammation. Currently early investigational therapies under development for SCD use a pathophysiological-based approach to abolish or reduce one or more of the mechanisms that contribute to this disease’s complex pathophysiology.
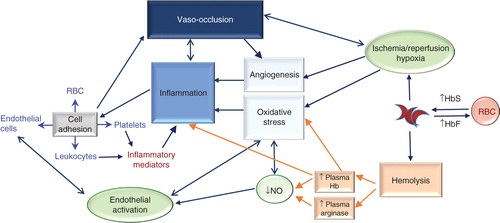
Table 1. Early investigational therapies for sickle cell disease and their pathophysiological targets.