Figures & data
Figure 1. Ensemble vesicle fusion assays. (A) Vesicle–vesicle fusion assay using fluorescently labeled lipid analogs (Struck et al., Citation1981). Vesicles containing a high concentration of NBD-PE and Rh-PE are characterized by quenching of NBD fluorescence due to FRET between NBD and rhodamine. Upon fusion to unlabeled vesicles the concentration of NBD and rhodamine decreases, resulting in less FRET efficiency and thus a consequent increase in NBD fluorescence (dequenching). (B) Application of the ensemble lipid-mixing vesicle fusion assay to proteoliposomes containing SNARE proteins (Weber et al., Citation1998). NBD-PE and Rh-PE containing vesicles reconstituted with v-SNARES (synaptobrevin shown in blue) fuse with unlabeled vesicle containing t-SNAREs (syntaxin shown in red and SNAP-25 shown in green), resulting in the dequenching of NBD fluorescence.
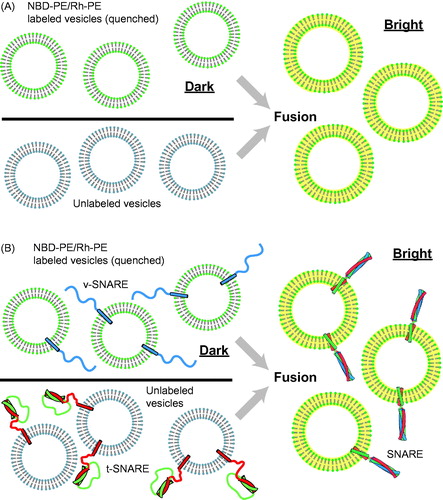
Figure 2. Single-vesicle bilayer fusion assays. (A) Lipid labeled (Fix et al., Citation2004) or (B) content labeled vesicles (Bowen et al., Citation2004) are monitored as they fuse to a lipid bilayer formed on a glass imaging surface. Fusion is indicated by the sudden appearance of fluorescence at the bilayer surface followed by a slow decay as the molecules diffuse. Reconstituted proteins are synaptobrevin (blue), syntaxin (red), and SNAP-25 (green). (C) To minimize the possible influence of the glass surface on the mobility of the lipid bilayer, a method with tethered membrane patches has been devised (Rawle et al., Citation2011).
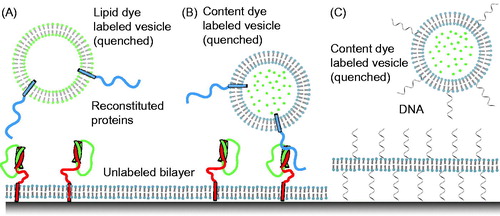
Figure 3. Single vesicle–vesicle lipid-mixing assays with tethered vesicles. (A) “Acceptor” vesicles containing v-SNAREs (synaptobrevin shown in blue) and labeled with DiD (red phospholipid head groups) and biotin (yellow) are immobilized to a PEG-biotin coated surface through linkage with neutravidin (navy blue). Donor vesicles containing t-SNAREs (syntaxin shown in red and SNAP-25 shown in green) and labeled with DiI (green phospholipids) are added and fusion is measured by measuring FRET using TIR microscopy (Yoon et al., Citation2006). (B) Synaptotagmin (light purple) was added in the lipid-mixing study by Lee et al. (Citation2010).
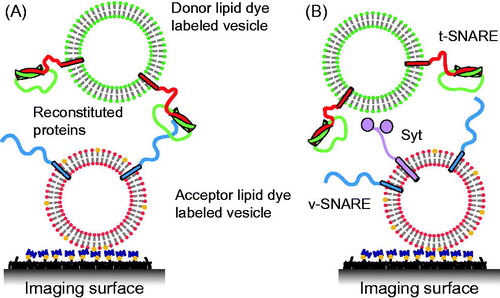
Figure 4. Single vesicle–vesicle lipid-mixing assays with freely diffusing vesicles. (A) Schematic representation of the ALEX method (Choi et al., Citation2013). Dilute mixtures of DiI labeled t-vesicles (donor) are mixed with DiD labeled v-vesicles (acceptor) and imaged using confocal microscopy with alternating laser excitation. The green area represents the confocal detection volume. (B) Representative data collected on individual vesicles diffusing through the confocal volume. Three data channels are recorded as shown: donor emission when directly excited (Ex: D, Em: D); acceptor emission when directly excited (Ex: A, Em: A); and acceptor emission resulting from FRET when the donor is excited (Ex: D, Em: A). The vesicles can be distinguished by their representative fluorescence signals as: t-vesicles only, emission of donor when directly excited, but no acceptor signal; v-vesicle only, no donor signal and emission of acceptor only when directly excited; docked vesicles, emission of both donor and acceptor when excited with the respective excitation sources but no FRET; and fused vesicles resulting in FRET (emission of both donor and acceptor when excited with the green laser). Reproduced with permission from Choi et al. (Citation2013).
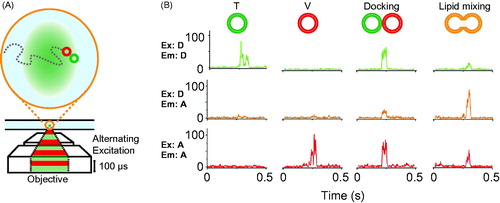
Figure 5. Single vesicle–vesicle content/lipid-mixing assays. (A) Setup of the single vesicle–vesicle assay that simultaneously monitors content and lipid mixing and that allows studies of Ca2+ triggered fusion (Diao et al., Citation2012; Kyoung et al., Citation2011, Citation2013; Lai et al., Citation2014). “Acceptor” vesicles containing syntaxin (red) and SNAP-25 (green) are immobilized via biotinylated lipids (yellow) to a glass surface that has been passivated with biotin-PEG and coated with neutravidin (dark purple). “Donor” vesicles containing high concentrations (and thus self-quenched) of sulforhodamine B in the lumen and DiD lipids are reconstituted with synaptobrevin (blue) and synaptogamin (light purple) molecules. The donor vesicles are incubated with the immobilized acceptor vesicles. Fusion is initiated by flowing buffer containing Ca2+ into the imaging chamber. (B) Docking, hemifusion, and complete fusion are monitored by measuring the fluorescence dequenching of the lipid and content dyes that result from dilution. (C) Representative fluorescence intensity time traces that result from fusion of a single donor vesicle to a single acceptor vesicle illustrating the detection of the individual fusion states shown in panel B. (D) A single vesicle content mixing assay with a large content probe, dye-labeled DNA hairpins (Diao et al., Citation2010). Upon fusion unlabeled DNA molecules complimentary to the FRET labeled hairpin bind and destabilize the hairpin leading to a decrease in FRET.
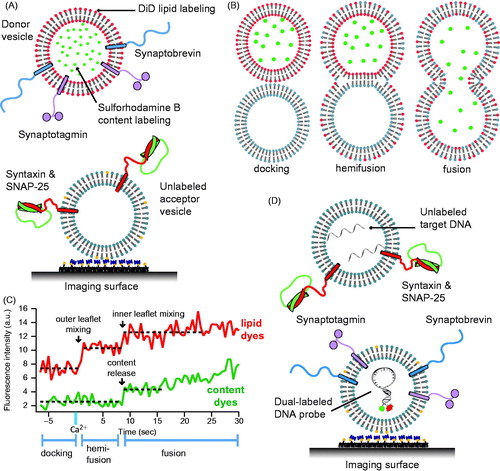
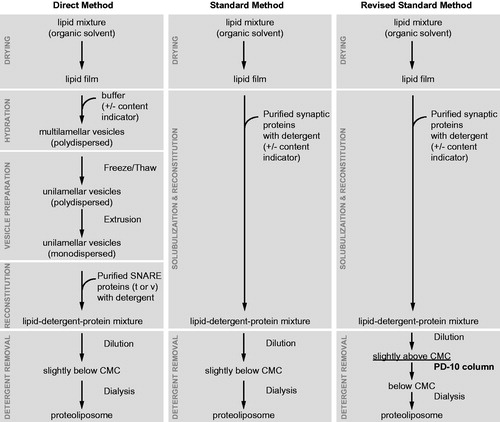
Figure 6. Proteoliposome reconstitution schemes. Outlined are three methods to create proteoliposomes with reconstituted purified synaptic proteins and various reporter fluorescent dyes. The modified standard method (Kyoung et al., Citation2011, Citation2013) produces vesicles with uniform size and protein distribution. This method has been used for the single vesicle content/lipid-mixing assay described in .