
Abstract
Coeliac disease (CD) is an immune-mediated disorder triggered by the ingestion of gluten in genetically susceptible individuals. However, only a small proportion of subjects harbouring CD-related genetic risk develop the disease. Among the environmental factors that may influence CD risk, pre- and perinatal factors, delivery methods, parental lifestyle, infant feeding practices, seasonality, dietary factors, drug use, childhood infections and variability in gut microbiota are those most widely studied regarding the risk to develop CD. Although for many of these external factors the exact mechanism of action is unknown, most of them are thought to act by disrupting the intestinal barrier, facilitating contact between potential antigens and the immune system effector cells. Management of CD is relatively easy in patients with a definite diagnosis and requires a strict, lifelong, gluten-free diet. Better knowledge of environmental exposures apart from gluten can facilitate understanding of the pathogenesis of the disorder and the wide heterogeneity of its clinical spectrum. The purpose of this review is to discuss current knowledge on environmental CD risk factors, as well as possible interaction between them, on the grounds of the reliable scientific evidence available.
The risk of developing CD is influenced not only by gluten ingestion but also by a number of environmental factors including childhood infections and variability in gut microbiota, pre- and perinatal factors, infant feeding practices, delivery methods, parental lifestyle, seasonality, dietary factors and drug use, acting mainly by disrupting intestinal permeability. Better knowledge of exposure to these factors can facilitate their identification, and subsequent elimination, in the individual patient.
Key messages
1. Introduction
Coeliac Disease (CD) is a chronic immune-mediated disorder of the small intestinal mucosa that occurs in genetically predisposed individuals following the ingestion of gluten, the major protein fraction contained in cereals [Citation1,Citation2]. In Western countries, its prevalence is around 1%, with great local variability [Citation3], while in Asian countries it is rarer [Citation4]. In populations of Caucasian origin, CD is primarily diagnosed during childhood, although the disease may become manifest at practically any age. After the 1960–1970s its clinical presentation became highly polymorphic and characterized by a myriad of signs and/or symptoms [Citation5].
Several epidemiological studies have claimed that CD incidence has risen in recent decades including in developing countries [Citation6]. However, in some Northern European countries like Finland, CD incidence appears to have leveled off during the 21st century after a period of increase [Citation7]. This rise in incidence of CD to a previously unknown extent cannot so much be attributed to greater awareness of the disease among physicians, or to improved non-invasive diagnostic tools, as to the abrupt change in lifestyle and diet in Western countries, with the additional influence of use of drugs and other external factors acting as precipitating events.
The importance of environmental exposures other than gluten is further strengthened by studies on migrants from low CD risk regions who acquire the higher prevalence of the receiving country [Citation8,Citation9]. In recent years, considerable efforts have been made to disentangle the complex interplay between genetic factors, gluten (and other prolamin- or glutenin-containing dietary proteins), and the gut mucosal immune system.
Despite this recent evidence, the relative importance of these factors in CD occurrence remains largely unknown. Deeper understanding may help in the prevention of the onset of the disease in exposed age groups or categories at risk [Citation10]. The purpose of this review is to discuss current knowledge on the environmental risk factors involved in CD pathogenesis and their possible interrelation, with the ultimate aim of improving strategies for disease prevention by identifying and eliminating these aetiologically relevant exposures.
After a brief summary of the current knowledge on the significance of genetics and gluten in CD, a field that is beyond the scope of this article, the review continues with sections dealing with environmental factors in terms of relevance and strength of evidence; the next two sections describe, respectively, the interaction between different environmental factors, and between genes and environmental factors. The last section is devoted to the final remarks. A 5-step rating system adapted from Hadorn has been used to summarize the level of scientific evidence for each of the risk factors screened [Citation11]. It consists, in descending order, of a meta-analysis of randomized studies (Level 1); a single study (Level 2); non-randomized studies (Level 3); retrospective studies (Level 4), and a series of cases without controls (Level 5).
2. The role of genetics and gluten in coeliac disease
Among the genetic factors, the main contribution to heritability of the disease is conferred by the human leukocyte antigen HLA-DQ2 or HLA-DQ8 haplotypes [Citation12] which drive an inflammatory T helper 1 (TH1) immune response against gluten proteins. In addition, more than 40 non-HLA associated loci have been described in relation to CD risk by large genome-wide association (GWA) studies [Citation13,Citation14], making CD a typical polygenic trait. However, studies on twins only revealed 70% concordance rate for CD amongst monozygotic twins [Citation12], suggesting that additional environmental factors must be considered aetiologically as important as genetic ones.
Exposure to dietary gluten is the most important environmental factor involved in CD pathogenesis. After deamination by tissue transglutaminase [Citation15], gluten peptides become immunogenic and bind to DQ2/8 molecules of antigen-presenting cells, thus triggering an adaptive immune response via the CD4+ T lymphocytes located in the intestinal mucosa [Citation16]. These cells migrate into the subepithelial mucosa and release several cytokines, such as interferon gamma, interleukin 2, interleukin 4 and TNF-α [Citation17], causing lymphocyte hyperproliferation and apoptosis of enteric cells, eventually resulting in intestinal mucosal atrophy [Citation18].
It has been observed that the rise in CD incidence corresponds with major temporal changes in population nutrition, known as nutrition transition [Citation19]. The so-called “Westernization of diet” had, as a consequence, the introduction of more varied, challenging antigens [Citation20] that could alter the permeability of the gut membrane leading to excess stimulation of the immune system. However, different studies on dietary risk factors for CD have yielded conflicting results. Data from epidemiological and nutritional studies indicate considerable heterogeneity as regards dietary constituents potentially triggering autoimmunity. Some studies have attempted to test the hypothesis that modern cereals may express higher levels of CD-triggering gluten proteins than cereals consumed in the past [Citation20]. During several recent decades, new strains of wheat produced using modern hybridization techniques have been introduced into the human diet: it is theoretically possible that the higher protein content of these new hybrids (which correlated well with the gluten content) may be associated with an increase in CD risk.
3. Dietary factors
3.1. Early feeding practices
The possible significance of breastfeeding duration on later development of CD, as well as the amount of gluten in the diet, and age at gluten introduction have been the subject of several retrospective and prospective studies, including observational retrospective studies conducted in Sweden [Citation21–23], Germany [Citation24] and Italy [Citation25,Citation26] (reviewed in the meta-analysis by Akobeng et al. [Citation27]). Despite their limited sample size, these studies indicate overall, that breastfeeding may protect against the later occurrence of CD in childhood [pooled odds ratio (OR): 0.48, 95% confidence interval (CI): 0.40– 0.59]. In contrast, two carefully conducted randomized trials, the Prevent Coeliac Disease study [Citation28] and the CELIPREV study [Citation29], which enrolled biopsy-proven CD cases for testing the breastfeeding hypothesis, did not find evidence for the influence of duration of breastfeeding on CD risk occurrence. Similarly, the Generation R prospective study that examined Dutch children with genetic susceptibility for CD failed to find any association between breastfeeding duration and CD [Citation30]. However, the recent Environmental Determinants of Diabetes in the Young (TEDDY) study, a population-based Swedish prospective study which analyzed 6327 singleton infants with CD, born in the period 1987–1997, detected an increased risk of developing CD later (adjusted hazard ratio 2.02, 95%CI: 1.37–2.96) but only for infants breastfed for less than 4 months [Citation31].
The possible role of the amount of gluten introduced into infant formulas was investigated by the Prevent Coeliac Disease study which enrolled two cohorts of Swedish children with higher and lower risk of CD [Citation32]. In this study the average daily flour consumption from milk- and cereal-based formulas was lower in children at low risk of CD (24 vs. 38 g/child/d), and the proportion of children in whom breastfeeding was prolonged after gluten introduction was significantly larger in the low-risk cohort (70% vs. 78%, p < .001). However, in a further study of the same cohort, the consumption of 200 mg/day of gluten at 4–6 months of age did not have any effect on CD risk at 3 years of age compared with no intake of gluten [Citation28].
Apart from the amount of gluten introduced, the time of gluten introduction was also tested as a CD risk factor. The largest systematic review available on the effect of time of gluten introduction is the meta-analysis by Szajewska et al. [Citation33]. The comparison of any breastfeeding with no breastfeeding, pooling the results of five observational studies, showed no effect on the risk of developing CD (OR: 0.69, 95%CI: 0.30–1.59). The effect of breastfeeding at the time of gluten introduction was protective when considering only the case-control studies (OR: 0.51, 95%CI: 0.34–0.77); it ceased to be a significant risk factor when considering the pooled observational studies (OR: 0.88, 95%CI: 0.52– 1.51). Based on the randomized, controlled trials, an increased CD risk at 2 years was observed only for the introduction of gluten at 6 months of age, compared with gluten at 12 months of age (OR: 2.36, 95%CI: 1.27–4.36), whereas no association was detected when comparing gluten at 4–6 months vs.>6 months of age or gluten at 6 months vs. 12 months of age, at 5 years.
To summarize the results of these studies, it must be acknowledged that the protective effect of breastfeeding hypothesized in early observational studies has not been confirmed by subsequent randomized, controlled trials and meta-analyses (level 1), so it must be concluded that only short breastfeeding duration may be associated with significant risk, whereas the time of gluten introduction does not affect the risk of CD occurrence in infancy. In addition, the evidence that high amount of gluten at weaning or type of gluten are CD risk factors is weak, though sufficient to recommend keeping the intake of gluten low in the first year of life [Citation34].
3.2. Additional dietary factors
The significance of the overall diet in the risk to develop CD autoimmunity was addressed in the study of Barroso et al. in children around the age of 1 year within the Generation R prospective study [Citation35]. After adjustments for potential confounders, a high adherence to a dietary pattern rich in vegetables; vegetable oils; pasta, rice, and other grains and poor in simple sugars, was significantly associated with lower odds of developing CD (OR: 0.67; 95% CI: 0.53–0.84) whereas the consumption of snacks and processed foods was associated with a non-significant increased risk. This finding suggests a possible protective role against CD of dietary patterns usually associated with healthy eating habits.
There are few studies examining the association between micronutrient intake and the risk of CD. Vitamin D has been examined as a mediator of CD risk [Citation36–38]. Apart from its role in calcium metabolism, it has been recognized as essential for the maintenance of an innate immune system function [Citation37]. By binding to the vitamin D receptor (VDR), the active form of vitamin D, 1α,25-dihydroxy-vitamin D3, decreases the production of interleukin-17 by TH17 cells, increases the activity of regulatory T cells and enhances cytokine production by natural killer T cells [Citation38].
The Norwegian Mother and Child Cohort Study (MoBa) did not gain support for the hypothesis that maternal or neonatal vitamin D status is related to the risk of CD in childhood [Citation36]. Apart from anecdotal reports, no prospective studies are available inquiring into the association between plasma vitamin D levels and CD risk, thus making the level of evidence weak (Level 2).
Hydrolyzed gluten is used during the wine clarification process therefore, gluten residues can be found in the final product and may represent a risk for subjects genetically predisposed to CD [Citation39]. With the new technologies, however, immunoreactive proteins are almost completely removed from red wine [Citation40].
An unexpected myriad of foods may contain gluten and can potentially precipitate CD in subjects at risk [Citation41]. However, scientific evidence to incriminate these alleged culprits is virtually absent (Level 5).
4. Infections
Viral, bacterial or protozoan infections in the first year of life, or later in childhood, have been implicated in CD pathogenesis. Infections may act directly by altering the intestinal mucosal barrier, or indirectly by modifying the gut microbiota and maturation of the immune system. Several studies have tested the hypothesis that the intestinal microflora of CD patients may have a different composition compared with that of non-CD subjects. In fact, the most popular mechanism to explain gluten-induced autoimmunity is the presence of intestinal pathogens able to induce local inflammation directly or indirectly by means of molecular mimicry [Citation42]. Although the hypothesis of a causal role of infections seems to be corroborated by robust findings, the level of evidence for specific microbial species is quite variable and often weak. For several bacterial or viral species an anti-inflammatory rather than pro-inflammatory role has been suggested [Citation42], but at the moment their exact role is unknown.
4.1. Viral infections
The association between viral infections and CD was evaluated in several studies [Citation43–Citation63]. In a large Norwegian cohort study the relationship between the influenza virus and later CD was recently addressed [Citation43]. The HR after seasonal influenza was 1.26 (95%CI: 1.17–1.36) and was similar (HR: 1.29, 95%CI: 1.15–1.44) following pandemic influenza after adjustment for the other covariates. Furthermore, after pandemic vaccination, the HR for CD was 1.15 (95%CI: 1.09–1.21) [Citation43]. Overall, these findings support the hypothesis of a positive association with CD development, though a causal relationship cannot be determined [Citation43].
The role of respiratory infections was investigated in 373 Italian newborns with one first-degree relative diagnosed with CD, recruited in the PreventCD study [Citation44]: the incidence of respiratory infections was two-fold higher in the cases compared with controls in the second year only (OR: 2.23, 95%CI: 1.04–4.77) and not in the first or third, and in this study the variance explained by respiratory infections was higher than that explained by sex or genetic factors. A positive association between a prior diagnosis of Respiratory Syncytial Virus (RSV) infection and later CD was found in a large study of Swedish children, with an OR of 1.46 (95%CI: 1.03–2.07) but the risk was not significantly increased beyond the first year after RSV infection [Citation45].
Both cross-sectional and prospective studies have investigated the role of specific viral infections of the gastrointestinal tract in CD [Citation46–53]. In general, molecular mimicry between viral/bacterial antigens and intestinal antigens is frequently used to explain the triggering effect of viral infections on the CD process, although the exact underlying molecular mechanisms are not clear. In one prospective study, 1931 children from the Denver Metropolitan Area carrying HLA risk alleles were recruited and followed for the emergence of rotavirus infections and the detection of tissue transglutaminase (tTG) autoantibodies [Citation46]. Unadjusted CD risk was higher after one (OR: 1.94, 95%CI: 0.39–9.56) and two (OR: 3.76, 95%CI: 0.76–18.7) infections, and the rate ratio for trend per increase in number of infections was 1.94, 95% CI: 1.04–3.61 (p = .037) adjusting for gender, ethnic group, maternal education, breastfeeding and day care. However, the wide confidence intervals indicate a modest evidence of increased risk [Citation46]. Moreover, molecular studies aimed at demonstrating specific immune responses to rotavirus antigenic peptides did not find any difference between untreated CD patients and controls [Citation47]. Adenovirus serotype 12 may also frequently be isolated from the human gut and has been associated with the development of CD. An α-gliadin region shares the amino acid sequence homology with the E1b protein of human adenovirus 12 [Citation48], representing the basis of the hypothesized cross-reactivity between host antibodies and α-gliadin [Citation42,Citation49]. Increased titres of serum antibodies cross-reacting with adenovirus type 12 have been found in CD patients following a free diet (89%) and gluten-free diet (30–33%), compared with non-CD subjects (0–12.8%), suggesting past infections as a trigger [Citation49]. However, subsequent studies were not able to confirm these findings, and in particular, failed to demonstrate the presence of specific antibodies to the viral E1b-58-kDa protein [Citation50]. Specific molecular studies aimed at identifying rotavirus-related particles directly in duodenal biopsies from children with active CD were negative [Citation51–Citation53].
Several studies investigated the relationship between HCV infection and the development of CD autoimmunity [Citation54–Citation57]. A cross-sectional study on 244 patients with chronic HCV infection, 121 patients with HCV-negative liver disease and 1230 healthy blood donors recruited as controls, found significantly increased titres of CD-related antibodies in HCV-infected patients compared with blood donors (OR: 12.8; 95%CI: 2.4–66) [Citation55], whereas other studies did not find any association [Citation54,Citation56]. Since hepatitis C virus is involved in the pathogenesis of extrahepatic inflammatory diseases by eliciting a HLA-DQ2-restricted cell-mediated inflammatory response [Citation57,Citation58], the same mechanism might be associated with CD autoimmunity [Citation56]. Interestingly, positivity for anti-tTG antibodies was observed during interferon therapies, formerly used to treat HCV-related chronic liver disease [Citation59,Citation60]. Transient infections are able to activate inflammatory cells in the lamina propria which, in turn, may produce types I and II interferons and other pro-inflammatory cytokines capable to increase epithelial permeability, enabling gluten peptides to cross the intestinal mucosa. The role of interferons in mucosal permeability is supported by studies where the occurrence of CD was observed during the course of hepatitis C treatment with interferon [Citation59,Citation61]. As for HBV-related chronic liver disease, a metanalysis of retrospective and prospective studies found that CD patients have lower rate of protective HBV antibodies [in the retrospective studies relative risk (RR): 0.732, 95%CI: 0.664–0.808; in the prospective studies, RR: 0.777, 95%CI: 0.629–0.960] [Citation62]. However, the lack of randomized, controlled trials and the low number of participants – making it difficult to draw definitive conclusions - were acknowledged by the authors of this analysis. As for the specific type of infection, a distinct increase in CD risk was reported for early gastrointestinal infections in the TEDDY study (HR: 1.33, 95%CI: 1.11–1.59) [Citation31]. Considering the evidence available from these carefully conducted prospective studies, it must be concluded that gastrointestinal (Level 1), and, to a lesser extent, respiratory infections (Level 3), increase the risk of subsequent CD. On the other hand, recent investigations aimed at highlighting the presence of species-specific antibodies gave negative results, except that they might even have a protective role [Citation63].
4.2. Bacterial infections
The immune system is still immature at birth and protection against various infectious agents relies on antibodies transferred by the mother through the placenta or maternal milk. A significantly increased risk conferred by early infections was reported in a study based on the Swedish Medical Birth Register after adjusting for multiple maternal variables (OR: 1.52, 95%CI: 1.19–1.95) [Citation64], as well as in the Swedish National Childhood Coeliac Disease Register which reported that three or more infections of any kind during the first six months of life significantly increased the risk of subsequent CD (OR: 1.5; 95%CI: 1.1–2.0) [Citation65].
Infection due to Helicobacter pylori is acquired early in childhood. Risk factors for infection acquisition are sibship size, household crowding, poor sanitary facilities, contact with farm animals, especially sheep and social class [Citation66]. Prior investigations found a positive association between H. pylori and duodenal intra-epithelial lymphocytes (IELs), a finding consistent with the early intestinal mucosa damage in CD [Citation67,Citation68]. On the other hand, Villanacci et al., observed a less severe degree of villous atrophy among CD patients with H. pylori infection [Citation69]. Colonization by H. pylori has been associated with protection from autoimmunity by increasing the expression of genes (e.g. FOXP3) involved in T-regulatory cell function [Citation70]. However, investigations aimed at addressing the relationship between H. pylori and CD yielded conflicting results [Citation71–73]. A recent prospective, observational study performed in patients positive for HLA-DQ2 and/or DQ8 haplotypes with or without CD, undergoing upper endoscopy for any reason, failed to find any significant relationship between H. pylori and CD risk, even taking into account potential confounders [Citation74]. For these reasons, an association between H. pylori infection and CD must still be considered inconclusive.
4.3. Helminths
Several studies have claimed that helminths may have a role in CD pathogenesis [Citation75–77]. These infestations, mostly determined by pinworms (Enterobius vermicularis) and roundworms (Ascaris lumbricoides), may cause a TH2-response in the immune system and prevent an autoimmune reaction by increasing the number of regulatory T-cells [Citation76]. In addition, several anti-inflammatory cytokines like IL-10 and TGF–β may increase as a result of helminth infestation [Citation77]. Paradoxically, the reduced rate of chronic helminth infections among children of Western countries may have resulted in an abnormal maturation of the immune system in infancy, and possibly in increased susceptibility to autoimmune disorders including CD. Considerable evidence supports this “hygiene hypothesis”, which postulates that the improved socio-economic status of most Western populations in past decades determined a lack of childhood exposure to enteric organisms and worms, thus impairing colonization of the gut after birth [Citation78]. This, in turn, might predispose a subject to inappropriate mucosal immune responses that could damage the epithelial barrier and further modify the gut microbiota, by generating a self-reinforcing loop. However, current evidence does not support an association between worm infestation and CD [Citation75] and a randomized, placebo-controlled trial that tested the use of helminths in CD patients did not show any improvement in symptoms [Citation79].
5. Drugs
A list of drugs that have been suspected of playing a role in CD risk [Citation80] is provided in . Antibiotic use in the first year of life was associated with increased CD risk in children (incidence rate ratio: 1.24, 95%CI: 1.07–1.43), with a dose-response relationship for a number of antibiotic courses (P-trend <0.01) [Citation81]. On the contrary, the TEDDY study reported a weak association between the most common antibiotics in early life (HR: 1.11, 95%CI: 1.00–1.22; p=.05) although statistical significance disappeared after correction for multiple testing (HR: 1.00; 95% CI: 0.98–1.02 for positivity of anti-transglutaminase autoantibody) [Citation82]. Therefore, evidence for an association between the use of antibiotics in pregnancy and the risk of CD in the offspring obtained by prospective studies is lacking.
Table 1. Level of evidence for the association of environmental factors with coeliac disease.
Nonsteroidal Anti-inflammatory Drugs (NSAIDs), including aspirin, have been suspected of causing intestinal mucosal damage as a primary consequence of COX-1 inhibition in the upper gastrointestinal tract [Citation83].
The use of proton pump inhibitors has been associated with CD [Citation84]. The pathogenetic mechanism has been linked with the suppression of acid secretion in the stomach and subsequently altered protein digestion, and increased exposure to gluten-like antigens, as well as increased permeability of the gastrointestinal mucosa which could enable gluten peptides to reach the gut immune system.
Some sartans such as olmesartan may induce sprue-like enteropathy [Citation85]. Although the exact pathogenetic mechanism is unclear, impaired angiotensin II signalling in the proximal gut together with tight junction disruption has been implicated in this condition.
Vaccinations have been investigated as possible triggers for CD. We have already mentioned that a large randomized, prospective study conducted in Sweden did not find a causal association for pandemic vaccination against influenza [Citation43], and a study in Finnish children showed that exposure to live oral rotavirus vaccination reduced, though not significantly, the risk of CD in the following 4–6 years (RR: 0.87, 95%CI: 0.65–1.17) [Citation86]. Protection against CD induced by rotavirus vaccination was also mentioned in a recent editorial in Clinical Gastroenterology and Hepatology [Citation87]. A possible decreased risk by vaccination against tuberculosis (BCG) was suggested in the Swedish study based on the National Childhood Coeliac Disease Register (CD register) (adjusted OR: 0.54; 95%CI: 0.31–0.94), but the evidence remains weak [Citation88]. In summary, the level of evidence is high enough only for the use of antibiotics early in life (level 2), but not for other drugs (level 3 for NSAIDs, PPIs and sartans), including vaccinations. The presence of confounding variables, including undiagnosed maternal CD, may generate spurious association without there necessarily being direct causation.
6. Birth-related factors
6.1. Delivery mode
The influence of delivery mode on CD, especially the Caesarean section, was investigated by observational studies, with mixed results [Citation89–91]. The pathogenetic mechanism was related to an altered composition of the gut microbiota, to be precise to delayed acquisition of Bacteroides, Bifidobacteria, and Escherichia coli in infants born by Caesarean section, resulting from lack of contact with the mother’s vaginal flora [Citation34]. A large Swedish population-based case-control study found a modest, but statistically significant excess risk for later CD after elective (OR: 1.15, 95%CI: 1.04–1.26), though not emergency (OR: 1.02, 95%CI: 0.92–1.13) Caesarean delivery [Citation91]. Among randomized trials, the CELIPREV Study at 5 years of age did not find any difference in CD autoimmunity risk between children born by Caesarean or vaginal delivery (24% and 19%, p = .2; 19% and 14%, p = .2, respectively) [Citation92]. Similarly, a recent, very large observational cohort study in Denmark and Norway found no association between mode of delivery and risk of diagnosed CD (OR: 1.20, 95%CI: 1.00–1.43 and 0.96, 95%CI: 0.79–1.17 for the Danish and Norwegian cohorts, respectively) [Citation93]. As for prospective studies, the Norwegian Mother and Child (MoBa) Cohort Study did find a non-significant reduction in disease risk (OR: 0.84, 95%CI: 0.65–1.09) following Caesarean delivery [Citation94]. The TEDDY study that followed up a birth cohort of 8676 children in the U.S. and Europe with HLA-DR-DQ genotypes associated with type 1 diabetes and/or CD, even reported a lower risk estimate for Caesarean compared with vaginal delivery in the univariate analysis (HR: 0.75; 95%CI: 0.58–0.98), though in the multivariate analysis statistical significance disappeared [Citation31,Citation95]. Therefore, on the basis of current evidence (Level 1), it seems unlikely that the mode of delivery may affect CD risk.
6.2. Seasonality and region of birth
The relationship between seasonality and CD risk was investigated in several studies [Citation31,Citation46,Citation96–99]. CD seasonality suggests the existence of factors fluctuating throughout the year and may be related to three mechanisms: (i) exposure to gluten more likely in children born in summer months than in winter months, when concurrent viral infections are more probable [Citation97]; (ii) lower level of vitamin D found in the diet in winter [Citation100] that may predispose to immune-system imbalance [Citation98]; (iii) incidence of some enteric and gastrointestinal viral pathogens associated with the annual variability in the immune suppression function usually observed in mammals [Citation101,Citation102]. In the Lebwohl et al. study, after stratification according to age at diagnosis, summer births were not associated with a CD diagnosis in later childhood (age 2–18 years: OR: 1.02, 95%CI: 0.97–1.08) but had a marginal effect on the risk of CD in adulthood (age ≥18 years: OR: 1.04, 95%CI: 1.01–1.07) [Citation96]. A study based on the Swedish Medical Birth Register found that the risk of developing CD was significantly higher for children born during the summer compared with winter (RR: 1.4, 95%CI: 1.2–1.7) [Citation97]. The results of two prospective studies seem to suggest a very weak association: a prospective longitudinal study conducted in Sweden, based on national registers, showed a significantly increased risk in summer and autumn at multivariable analysis (HR: 1.10, 95%CI: 1.03–1.18) [Citation103]; in the TEDDY study the hazard ratio for CD was significantly lower in the winter/autumn seasons compared with the summer/spring seasons (HR: 0.86; 95%CI: 0.74–0.99), although statistical significance disappeared in the multivariate analysis (HR: 0.89; 95%CI: 0.77–1.03) [Citation31].
Despite the high level of evidence available (Level 1), the strength of association is weak and, in general, the role of yearly fluctuating factors is not likely to be a major cause of CD.
The risk of CD has also been investigated in relation to the region of birth. The prospective Swedish study mentioned above provided evidence that children born in southern Sweden showed higher CD risk compared with children born in northern Sweden (HR: 1.53, 95%CI: 1.41–1.67), p < .001) [Citation103]. The explanation may partly be related to variation in sunlight and dietary vitamin D or summer duration, but further evidence is necessary.
6.3. Birth weight
Early investigations based on case-control studies hypothesized that a low birth weight might be a risk factor for the development of CD autoimmunity, but the findings have been controversial. The rationale is that, premature children may have specifically altered gut cell-mediated immunological development although, it is likely that more than one factor contributes to the alleged increased CD risk. A study based on the Swedish Medical Birth Register showed that low birth weight (<2.5 kg) is associated with increased CD risk (OR: 1.27, 95%CI: 1.07–1.52), after adjusting for potential confounders [Citation64]. In contrast, another Swedish study that enrolled 11,749 infants with biopsy-proven CD did not find any association with low birth weight (OR: 0.82, 95%CI: 0.66–1.03) [Citation91]. Subsequent North European studies did not find a statistically significant association; for example, the Norwegian Mother and Child (MoBa) Cohort Study reported an adjusted OR for children to develop CD of 0.99 (95%CI: 0.91–1.08) for infants with low birth weight [Citation94]. Similarly, the recent study by Kuja-Halkola et al. [Citation104], based on the Swedish Twin Registry, found no overall association between low birth weight and future CD although gender differences were found. In conclusion, this association should be considered weak or non-existent, with a high level of evidence (level 2).
7. Gut microbiota
In the last two decades, alterations in the intestinal microbiota have been extensively studied, as potential culprits of CD occurrence in predisposed individuals. More specifically, the modulation of the epithelial barrier, exacerbation of the gliadin-specific immune response and activation of the innate immune system may be the mechanisms by which the intestinal microbiota prompt the development of CD [Citation105].
7.1. Dysbiosis and CD
An alteration in the intestinal microbiota (“dysbiosis”) was reported in subjects carrying the fucosyltransferase 2 gene (FUT 2), a gene involved, with others, in the regulation of mucin secretion in the gut [Citation106]. In the non-secretor individual a lower number of Bifidobacteria spp, especially B. bifidum, B. adolescentis and B. catenulatum/pseudocatenulatum was observed [Citation107]. Fucosyltransferase 2 could contribute to the genetic variance of CD, estimated in around 14% of cases [Citation108]. To assess the importance of dysbiosis among the environmental factors involved in CD, several studies have been carried out on children with genetic susceptibility for CD. The PROFICEL study reported that in faecal samples of infants with increased CD genetic risk a higher number of B. fragilis and Staphylococcus spp. and a lower number of Bifidobacterium spp. and B. longum were detected [Citation109]. Moreover, a sub-analysis revealed that among CD susceptible infants those who were breastfed, harboured an increased Clostridium leptum population, while in formula-fed infants increased Staphylococcus spp. and B. fragilis populations were found [Citation110]. More recently, the same research group reported, in infants at family risk, that HLA-DQ2 haplotypes influence early gut microbiota composition. For instance, infants with a higher genetic risk for CD, as well as those showing gastrointestinal symptoms, had their intestine colonized by higher proportions of Firmicutes and Proteobacteria, while lower-risk infants had lower proportions of Actinobacteria [Citation111]. An additional study confirmed the tendency to harbour more Firmicutes/Bacteroides in high-risk infants [Citation112]. It is worthy of note that this faecal signature was confirmed in the microbiota of duodenal mucosa. In coeliac patients a reduction in Bifidobacterium spp. and B. longum was reported compared with controls [Citation113]. Finally, a very elegant study demonstrated that gut microbiota trajectory in early life may predict the risk of developing CD. A group of infants enrolled in the PROFICEL study was followed for five years; a significant decline in microbiota diversity was observed in children who developed CD between 4 and 6 months compared with controls. To be more precise, at the genus level, an increased proportion of B. breve and Enterococcus spp. was associated with CD development, while an increased relative abundance of B. longum was detected in children without CD [Citation114]. In conclusion, alterations in normal microbiota maturation early in life could probably be implicated in CD development [Citation115].
7.2 Gut microbiota and gluten
Gluten-degrading microbiota, especially if located in the upper gastrointestinal tract, could have a role in the pathophysiology of gluten-related disorders. Rothia strains, in particular, R. mucilaginosa and R. aeria, have the ability to produce gliadin-degrading enzymes that cleave immunogenic gluten [Citation116]. Additional microbial species capable of cleaving gluten, such as Actinomyces odontolyticus and Streptococcus spp. were isolated in the upper gastrointestinal tract [Citation117]. In an experimental model, Bifidobacteria spp. were able to attenuate the pro-inflammatory effect of gliadin on Caco-2 cells, together with a significant reduction in secretion of inflammatory mediators like NF-kB, TNF–α and IL–1β [Citation118]. Furthermore, Bifidobacteria spp. also protected the tight junctions of Caco-2 cells against the effects of gliadin, enhancing Zonulin-1 (ZO-1) expression [Citation119]. Moreover, Shigella, E. coli and Bifidobacteria spp. influence the production of cytokines by monocytes [Citation120] and, Bifidobacteria spp., in particular, modulate the phenotypic and functional maturation of dendritic cells [Citation121]. Although a growing number of studies are reporting statistically significant differences between the faecal microbiota in CD patients and that of normal subjects, doubts remain as to whether CD patient microbiota is distinct since birth and has a causative role, or the alteration is merely a consequence of the intestinal inflammation triggered by the disease.
8. Parental factors
Use of antibiotics during pregnancy has been linked with CD in children because these drugs can modify the gut microbiota and alter the permeability of intestinal mucosa [Citation122]. In the Swedish All Babies in Southeast Sweden (ABIS) prospective study an increased risk (unadjusted HR: 1.33; 95%CI: 0.69–2.56) of CD was observed for infants of mothers exposed to antibiotics during pregnancy compared with unexposed children, even after adjustment for breastfeeding, age at gluten introduction and infection load in the child’s first year of life (HR: 1.28; 95%CI: 0.66–2.48) [Citation123]. Although these findings are statistically non-significant due to low statistical power, in this prospective study (Level 2) the authors did not rule out a weak risk of association for children whose mothers were treated with antibiotics.
Early reports dating back to the past century have hinted that maternal age may be a risk factor for CD [Citation124]. In 1951 Thompson reported that maternal age at the birth of a coeliac child was significantly later than that of mothers with non-coeliac children [Citation125]. The author also observed that later birth rank was associated with an increased risk of CD. On the basis of these findings, it has been hypothesized that older or multiparous mothers may have an unfavourable uterine environment that might impair the development of the immune system of their infants. Interestingly, these early associations have been confirmed by more recent investigations. For example, Canova et al. reported a significantly greater risk for children born of aged mothers (OR: 1.30, 95%CI: 1.02–1.77 for those aged 30–34; OR: 1.33, 95%CI: 1.01–1.74 for those aged 35–39) [Citation81].
Family size has been claimed to influence the chance of acquiring enteric infections during childhood and, in turn, CD risk. Households with a larger number of siblings are often overcrowded and children may experience a greater probability of acquiring infections able to stimulate the gut immune system to react appropriately to bowel infections later in life. However, prospective studies such as the TEDDY study failed to find differences in CD risk according to sibship size [Citation31].
The lifestyle of parents, especially the mother, has been called into question as a risk factor for CD. Higher maternal education was found to increase CD risk [Citation31,Citation81]. Canova et al. reported a risk greater than 30% of CD developing in children born of better-educated mothers (OR: 1.32, 95%CI: 1.14–1.53 for those who attended university vs. eighth-grade school as a reference frame) [Citation81]. It is possible that educated mothers are more sensitive to their children’s health, thus facilitating an early CD diagnosis. A cleaner domestic environment, usually pursued by more educated mothers, may negatively affect maturation of the intestinal immune system, according to the “hygiene hypothesis” mentioned previously [Citation78].
The impact of socio-economic status (SES) on the risk of CD was evaluated in several studies, with contrasting results [Citation126–131]. Some of these studies seem to hint that CD is somewhat more common in children with high SES. Intriguingly, the study based on the Swedish National Childhood Coeliac Disease Register reported a reverse association, i.e. children from families of lower SES compared with high-medium SES, had an increased CD risk (OR: 1.5, 95%CI: 1.2–2.0) [Citation65]. Overall, the results of these randomized studies (Level 1) indicate that perinatal risk factors play a minor role in CD autoimmunity.
9. Cigarette smoking
When dealing with the potential association of cigarette smoking with CD, a distinction must be made between passive exposure to parental smoking and the active smoking of patients themselves. In a small-scale association study on biopsy-proven CD, Snook et al. observed a protective effect of cigarette smoking on the onset of the disease (OR: 0.15, 95%CI: 0.06–0.38) [Citation132], while Roberts et al. found a significantly increased cumulative incidence rate (55.5 vs. 26.1, p = .009) [Citation89]. The effect of parental smoking was explored in the study of Patel et al. who did not find any significant association (OR: 1.5, 95%CI: 0.5–4.3) [Citation133] and again in the TEDDY study with negative results [Citation95]. However, only case-control studies are available and no prospective study provided evidence that an association really does exist [Citation134–136].
The apparent protective effect of cigarette smoking on CD risk is puzzling since it is known that, in general, smoking enhances the probability of developing autoimmunity [Citation137,Citation138]. Cigarette smoking might act by suppressing the CD8+ T cells that have a major role in CD mucosal damage [Citation139]. Overall, evidence linking smoking and adult-onset CD is weak (Level 3).
10. Interaction between different environmental factors
In general, any external factor, such as antibiotic use or agents like NSAIDs that potentially alter the commensal flora and the intestinal mucosa may contribute to increasing the risk of the disease by facilitating the passage of antigenic peptide fragments involved in CD pathogenesis [Citation140]. Gender-specific interaction has been reported between seasonality and CD incidence in children below 2 years if they were born during the summer/spring season as compared with the winter/autumn season [Citation31]. It has been suggested that children born in the summertime had intrauterine life during the winter period, when the rate of maternal infections is higher.
11. Interaction between genes and environmental factors
One interesting recent research topic is the possible interplay between genes and environmental factors in CD pathogenesis. A particular genetic makeup, especially related to HLA, may enhance the effect of several external factors. For example, the increased risk of CD related to a short breastfeeding period may depend on HLA susceptibility. This risk is more pronounced among children carrying the HLA-DQ8/8 or HLA-DQ4/8 genotypes (adjusted HR: 9.76; 95%CI: 3.87–24.80) [Citation31]. However, in the PREVENTCD study different HLA types did not influence the effect of gluten or placebo on the development of CD [Citation33], therefore caution should be used in drawing conclusions.
The possible relationship between milk and the gut microbial colonization process in full-term healthy infants with a family history of CD was investigated by De Palma et al. [Citation120,Citation121]. The authors observed strong interaction with HLA-dependent genetic risk: the newborns with the lowest genetic risk showed an increased number of Bifidobacterium spp. and B. longum, while those with the highest genetic CD risk showed increased numbers of Staphylococcus spp. and B. fragilis. The TEDDY study also reported a CD risk twice as strong among infants breastfed for less than 4 months compared with those breastfed longer, and this risk was dependent on HLA genotypes [Citation82]. Collectively, these findings could represent classical gene-environment interaction, and suggest that both milk-feeding type and HLA-DQ genotype may modulate the bacterial colonization pattern of the newborn’s intestine after birth and, in turn, the risk of developing CD later in life. In addition, Sanchez et al. observed that breastfeeding may influence the diversity of Bacteroides in the gut [Citation141]. This study revealed that infants with CD-predisposing HLA genotypes showed an increased prevalence of B. vulgatus and reduced prevalence of B. ovatus, B. plebeius and B. uniformis compared with infants with low genetic risk. Interestingly, the polysaccharide of B. fragilis may link with HLA class II molecules during a process similar to antigen recognition by T cells. This recent evidence (Level 2) seems to indicate that in addition to immune-related genetic susceptibility, the HLA may confer an additional risk for CD via a selection of bacterial strains from the microbiota.
12. Role of environmental factors in the pathogenesis of coeliac disease
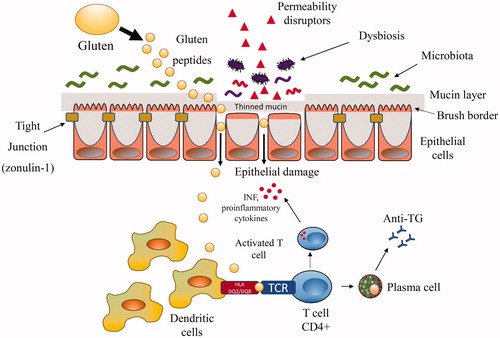
Overall, the mechanisms by which the aetiological factors outlined above are implicated in mucosal damage have been preponderantly attributed to immune system alterations and are illustrated in . The gluten-containing proteins or the proteins derived from pathogens (virus and bacteria) showing structural similarities with self-epitopes may trigger activation of autoreactive T cells in a specific TH1 response. However, this “molecular mimicry” mechanism requires high molecular homology which, being a statistically rare event, does not fully explain the high proportion of genetically predisposed subjects who develop inflammatory lesions. Furthermore, intact intestinal mucosa should contrast the passage of foreign molecules and their contact with cells of the mucosal immune system. Attention has recently been focussed on mechanisms able to alter the barrier function of the intestinal mucosa, facilitating the possible trafficking of damaging macromolecules between the environment and the host. The mechanism studied much more frequently in intestinal permeability concern the tight junction, although other mechanisms are possible. Tight junction competency may be altered by a large number of agents facilitating the contact of molecules structurally similar to self-antigens with T cells: this hypothesis, which makes mucosal permeability one of the central mechanisms, implies that a great number of potential environmental barrier disruptors may be at play [Citation142,Citation143]. Empirical evidence showing increased circulating levels of the main regulator of the tight junctions, zonulin, in CD subjects is available [Citation144]. The process is self-perpetuating because the inflammation resulting from disrupting the intestinal barrier is able per se to increase mucosal leaking. Animal models of autoimmune diseases are known in which the administration of a zonulin inhibitor reduces the onset of the disease. However, studies that correlate zonulin levels with the effect of a putative CD factor are scant [Citation140]. A better understanding of zonulin signalling could potentially lead to two desirable effects: (i) widening the list of environmental factors acting through this pathway and potentially capable of starting self-perpetuating mucosal damage; (ii) the possibility of developing drugs to prevent mucosal leakage, or blocking potential antigens to activate the immune T cells.
Figure 1. Mechanisms involved in the pathogenesis of coeliac disease. The figure shows the intestinal cells maintained in mutual contact by tight junctions. Luminal and early mucosal events able to alter intestinal barrier (e.g. mucine thinning) and/or to directly damage the intestinal mucosa (such as drugs, infections, changes in the microbiota) may facilitate macromolecules including gluten peptides to cross the leaking mucosa. Gluten not fully digested in the lamina propria is captured by tissue transglutaminase and deamidated. The more sticky fragments are bound to DQ2 or DQ8 antigen-presenting cells leading to the release of cytokines and activation of CD4+ T cells and, in turn, to tissue damage. INF: interferon; TCR: T cell receptor.
Conclusion
In this review, we have summarized the current knowledge of the association between several putative environmental exposures and CD development. Since it is currently easy to ascertain genetic susceptibility in individuals by molecular techniques, knowledge of the wide spectrum of causative environmental factors could enable elimination of them in predisposed subjects, to prevent the onset of intestinal lesions. The existence of CD cases in which symptoms appear in adulthood or even in advanced age, suggests that in such cases environmental exposures occurred, unbeknown to the patient, late in life. Change in the intestinal microbiota and consequent alterations of intestinal mucosa permeability are the most probable candidates, and suggest the need to monitor this aspect in subjects with HLA-predisposing genotypes. In addition, it should also be recognized that changes in the environment may have modified the epidemiology of CD.
Based on the studies outlined above, most environmental risk factors for CD can roughly be divided into three categories: those for which there is convincing evidence that they can actually increase the risk, those in which the evidence is currently insufficient, and those in which the possible association has been investigated but not confirmed by the results. They are summarized in according to the strength of scientific evidence.
Convincing evidence of a rise in risk for early breastfeeding, rotavirus and gastrointestinal infections, and modifications of gut microbiota shows strong evidence of association (Levels 1–2). This might be of practical importance since the extension of breastfeeding time and protection from infections during infancy would result in a decrease in CD cases. There are conflicting results, as for the association with the Caesarean section, for adenovirus infections, influenza and H. pylori, and subsequent CD risk. For some of these, their involvement has been suspected although the evidence is still weak due to the limited number of studies and the usually small size of cohorts. Further research is needed to confirm these conclusions.
Further studies are needed to clarify the complex interplay between genetics, immune cells, microbiota and intestinal mucosa in order to identify novel targets for therapy as well as lifestyle and behavioural recommendations to improve patient outcomes.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Ludvigsson JF, Leffler DA, Bai JC, et al. The Oslo definitions for coeliac disease and related terms. Gut. 2013;62:43–52.
- Catassi C, Gatti S, Fasano A. The new epidemiology of celiac disease. J Pediatr Gastroenterol Nutr. 2014;59:S7–S9.
- Dube C, Rostom A, Sy R, et al. The prevalence of celiac disease in average-risk and at-risk Western European populations: a systematic review. Gastroenterology. 2005;128:S57–S67.
- Cummins AG, Roberts-Thomson IC. Prevalence of celiac disease in the Asia-Pacific region. J Gastroenterol Hepatol. 2009;24:1347–1351.
- Dore MP, Cuccu M, Pes GM, et al. Clinical pattern of celiac disease in a population residing in North Sardinia (Italy). Recenti Prog Med. 2012;103:564–569.
- Aziz I, Branchi F, Sanders DS. The rise and fall of gluten! Proc Nutr Soc. 2015;74:221–226.
- Virta LJ, Saarinen MM, Kolho KL. Declining trend in the incidence of biopsy-verified coeliac disease in the adult population of Finland, 2005-2014. Aliment Pharmacol Ther. 2017;46:1085–1093.
- Nelson R, McNeish AS, Anderson CM. Coeliac disease in children of Asian immigrants. Lancet. 1973;1:348–350.
- Cataldo F, Montalto G. Celiac disease in the developing countries: a new and challenging public health problem. Wjg. 2007;13:2153–2159.
- Leonard MM, Fasano A. The microbiome as a possible target to prevent celiac disease. Expert Rev Gastroenterol Hepatol. 2016;10:555–556.
- Hadorn DC, Baker D, Hodges JS, et al. Rating the quality of evidence for clinical practice guidelines. J Clin Epidemiol. 1996;49:749–754.
- Greco L, Romino R, Coto I, et al. The first large population based twin study of coeliac disease. Gut. 2002;50:624–628.
- Dubois PC, Trynka G, Franke L, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302.
- Kuja-Halkola R, Lebwohl B, Halfvarson J, et al. Heritability of non-HLA genetics in coeliac disease: a population-based study in 107 000 twins. Gut. 2016;65:1793–1798.
- Di Sabatino A, Vanoli A, Giuffrida P, et al. The function of tissue transglutaminase in celiac disease. Autoimmun Rev. 2012;11:746–753.
- Halstensen TS, Brandtzaeg P. Activated T lymphocytes in the celiac lesion: non-proliferative activation (CD25) of CD4+ alpha/beta cells in the lamina propria but αproliferation (Ki-67) of alpha/beta and gamma/delta cells in the epithelium. Eur J Immunol. 1993;23:505–510.
- Beckett CG, DellʼOlio D, Shidrawi RG, et al. Gluten-induced nitric oxide and pro-inflammatory cytokine release by cultured coeliac small intestinal biopsies. Eur J Gastroenterol Hepatol. 1999;11:529–535.
- Ciccocioppo R, D’Alò S, Sabatino ADI, et al. Mechanisms of villous atrophy in autoimmune enteropathy and coeliac disease. Clin Exp Immunol. 2002;128:88–93.
- Caballero B, Popkin BM. The nutrition transition: diet and disease in the developing world. Amsterdam: Academic Press; 2002.
- de Lorgeril M, Salen P. Gluten and wheat intolerance today: are modern wheat strains involved? Int J Food Sci Nutr. 2014;65:577–581.
- Falth-Magnusson K, Franzen L, Jansson G, et al. Infant feeding history shows distinct differences between Swedish celiac and reference children. Pediatr Allergy Immunol. 1996;7:1–5.
- Ascher H, Krantz I, Rydberg L, et al. Influence of infant feeding and gluten intake on coeliac disease. Arch Dis Child. 1997;76:113–117.
- Ivarsson A, Hernell O, Stenlund H, et al. Breast-feeding protects against celiac disease. Am J Clin Nutr. 2002;75:914–921.
- Peters U, Schneeweiss S, Trautwein EA, et al. A case-control study of the effect of infant feeding on celiac disease. Ann Nutr Metab. 2001;45:135–142.
- Greco L, Mayer M, Grimaldi M, et al. The effect of early feeding on the onset of symptoms in celiac disease. J Pediatr Gastroenterol Nutr. 1985;4:52–55.
- Auricchio S, Follo D, de Ritis G, et al. Does breast feeding protect against the development of clinical symptoms of celiac disease in children? J Pediatr Gastroenterol Nutr. 1983;2:428–433.
- Akobeng AK, Ramanan AV, Buchan I, et al. Effect of breast feeding on risk of coeliac disease: a systematic review and meta‐analysis of observational studies. Arch Dis Child. 2005;91:39–43.
- Vriezinga SL, Auricchio R, Bravi E, et al. Randomized feeding intervention in infants at high risk for celiac disease. N Engl J Med. 2014;371:1304–1315.
- Lionetti E, Castellaneta S, Francavilla R, SIGENP (Italian Society of Pediatric Gastroenterology, Hepatology, and Nutrition) Working Group on Weaning and CD Risk, et al. Introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med. 2014;371:1295–1303.
- Jansen MA, Tromp II, Kiefte-de Jong JC, et al. Infant feeding and anti-tissue antitransglutaminase antibody concentrations in the Generation R Study. Am J Clin Nutr. 2014;100:1095–1101.
- Kemppainen KM, Lynch KF, Liu E, TEDDY Study Group, et al. Factors that increase risk of celiac disease autoimmunity after a gastrointestinal infection in early life. Clin Gastroenterol Hepatol. 2017;15:694–702.
- Ivarsson A, Myléus A, Norström F, et al. Prevalence of childhood celiac disease and changes in infant feeding. Pediatrics. 2013;131:e687–e694.
- Szajewska H, Shamir R, Chmielewska A, PREVENTCD Study Group, et al. Systematic review with meta-analysis: early infant feeding and coeliac disease–update 2015. Aliment Pharmacol Ther. 2015;41:1038–1054.
- Sarno M, Discepolo V, Troncone R, et al. Risk factors for celiac disease. Ital J Pediatr. 2015;41:57.
- Barroso M, Beth SA, Voortman T, et al. Dietary patterns after the weaning and lactation period are associated with celiac disease autoimmunity in children. Gastroenterology. 2018;154:2087–2096.
- Mårild K, Tapia G, Haugen M, et al. Maternal and neonatal vitamin D status, genotype and childhood celiac disease. PLoS One. 2017;12:e0179080.
- Kamen DL, Tangpricha V. Vitamin D and molecular actions on the immune system: modulation of innate and autoimmunity. J Mol Med. 2010;88:441–450.
- Yang CY, Leung PS, Adamopoulos IE, et al. The implication of vitamin D and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol. 2013;45:217–226.
- Cattaneo A, Ballabio C, Bertelli AA, et al. Evaluation of residual immunoreactivity in red and white wines clarified with gluten or gluten derivatives. Int J Tissue React. 2003;25:57–64.
- Simonato B, Mainente F, Tolin S, et al. Immunochemical and mass spectrometry detection of residual proteins in gluten fined red wine. J Agric Food Chem. 2011;59:3101–3110.
- O'Shea N, Arendt E, Gallagher E. State of the art in gluten-free research. J Food Sci. 2014;79:R1067–R1076.
- Plot L, Amital H. Infectious associations of Celiac disease. Autoimmunity Reviews. 2009;8:316–319.
- Kårhus LL, Gunnes N, Størdal K, et al. Influenza and risk of later celiac disease: a cohort study of 2.6 million people. Scand J Gastroenterol. 2018;53:15–23.
- Auricchio R, Cielo D, de Falco R, et al. Respiratory infections and the risk of celiac disease. Pediatrics. 2017;140:e20164102.
- Tjernberg AR, Ludvigsson JF. Children with celiac disease are more likely to have attended hospital for prior respiratory syncytial virus infection. Dig Dis Sci. 2014;59:1502–1508.
- Stene LC, Honeyman MC, Hoffenberg EJ, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterology. 2006;101:2333–2340.
- Ziberna F, De Lorenzo G, Schiavon V, et al. Lack of evidence of rotavirus-dependent molecular mimicry as a trigger of coeliac disease. Clin Exp Immunol. 2016;186:356–363.
- Kagnoff MF, Austin RK, Hubert JJ, et al. Possible role for a human adenovirus in the pathogenesis of celiac disease. J Exp Med. 1984;160:1544–1557.
- Kagnoff MF, Paterson YJ, Kumar PJ, et al. Evidence for the role of a human intestinal adenovirus in the pathogenesis of coeliac disease. Gut. 1987;28:995–1001.
- Diaconescu I, Alexandru G, Carpa R, et al. Too few studies provided a link between viral infections and celiac disease. Int J Celiac Dis. 2016;4:135–137.
- Howdle PD, Blair Zajdel ME, Smart CJ, et al. Lack of a serologic response to an E1B protein of adenovirus 12 in coeliac disease. Scand J Gastroenterol. 1989;24:282–286.
- Mahon J, Blair GE, Wood GM, et al. Is persistent adenovirus 12 infection involved in coeliac disease? A search for viral DNA using the polymerase chain reaction. Gut. 1991;32:1114–1116.
- Vesy CJ, Greenson JK, Papp AC, et al. Evaluation of celiac disease biopsies for adenovirus 12 DNA using a multiplex polymerase chain reaction. Mod Pathol. 1993;6:61–64.
- Thevenot T, Denis J, Jouannaud V, et al. Coeliac disease in chronic hepatitis C. A French multicentre prospective study. Aliment Pharmacol Ther. 2007;26:1209–1216.
- Ruggeri C, La Masa AT, Rudi S, et al. Celiac disease and non-organ-specific autoantibodies in patients with chronic hepatitis C virus infection. Dig Dis Sci. 2008;53:2151–2155.
- Casella G, Viganò D, Settanni CR, et al. Association between celiac disease and chronic hepatitis C. Gastroenterol Hepatol Bed Bench. 2016;9:153–157.
- Fine KD, Ogunji F, Saloum Y, et al. Celiac sprue: another autoimmune syndrome associated with hepatitis C. Am J Gastroenterology. 2001;96:138–145.
- Satta R, Pes GM, Quarta Colosso BM, et al. Skin manifestations in patients with hepatitis C virus-related chronic liver disease. J Eur Acad Dermatol Venereol. 2018.
- Bardella MT, Marino R, Meroni PL. Celiac disease during interferon treatment. Ann Intern Med. 1999;131:157–158.
- Durante-Mangoni E, Iardino P, Resse M, et al. Silent celiac disease in chronic hepatitis C: impact of interferon treatment on the disease onset and clinical outcome. J Clin Gastroenterol. 2004;38:901–905.
- Sjöberg K, Lindgren S, Eriksson S. Frequent occurrence of non-specific gliadin antibodies in chronic liver disease. Endomysial but not gliadin antibodies predict coeliac disease in patients with chronic liver disease. Scand J Gastroenterol. 1997;32:1162–1167.
- Opri R, Veneri D, Mengoli C, et al. Immune response to Hepatitis B vaccine in patients with celiac disease: a systematic review and meta-analysis. Hum Vaccin Immunother. 2015;11:2800–2805.
- Plot L, Amital H, Barzilai O, et al. Infections may have a protective role in the etiopathogenesis of celiac disease. Ann N Y Acad Sci. 2009;1173:670–674.
- Sandberg-Bennich S, Dahlquist G, Källén B. Coeliac disease is associated with intrauterine growth and neonatal infections. Acta Paediatr. 2007;91:30–33.
- Myléus A, Hernell O, Gothefors L, et al. Early infections are associated with increased risk for celiac disease: an incident case-referent study. BMC Pediatr. 2012;12:194.
- Dore MP, Bilotta M, Vaira D, et al. High prevalence of Helicobacter pylori infection in shepherds. Dig Dis Sci. 1999;44:1161–1164.
- Cabral VL, Patrício FR, Gabbay MA, et al. Intraepithelial lymphocytes in duodenum from Brazilian adolescents with type 1 diabetes. Influence of Helicobacter pylori. Pediatr Diabetes. 2009;10:316–320.
- Memeo L, Jhang J, Hibshoosh H, et al. Duodenal intraepithelial lymphocytosis with normal villous architecture: common occurrence in H. pylori gastritis. Mod Pathol. 2005;18:1134–1144.
- Villanacci V, Bassotti G, Liserre B, et al. Helicobacter pylori infection in patients with celiac disease. Am J Gastroenterol. 2006;101:1880–1885.
- Cho KY, Cho MS, Seo JW. FOXP3+ regulatory T cells in children with Helicobacter pylori infection. Pediatr Dev Pathol. 2012;15:118–126.
- Abenavoli L, Arena V, Giancotti F, et al. Celiac disease, primary biliary cirrhosis and helicobacter pylori infection: one link for three diseases. Int J Immunopathol Pharmacol. 2010;23:1261–1265.
- Narang M, Puri AS, Sachdeva S, et al. Celiac disease and Helicobacter pylori infection in children: is there any association? J Gastroenterol Hepatol. 2017;32:1178–1182.
- Basyigit S, Unsal O, Uzman M, et al. Relationship between Helicobacter pylori infection and celiac disease: a cross-sectional study and a brief review of the literature. Prz Gastroenterol. 2017;12:49–54.
- Dore MP, Salis R, Loria MF, et al. Helicobacter pylori infection and occurrence of celiac disease in subjects HLA-DQ2/DQ8 positive: a prospective study. Helicobacter. 2018;23:e12465.
- Ludvigsson J, Jones MP, Faresjö Å. Worm infestations and development of autoimmunity in children - The ABIS study. PLoS One. 2017;12:e0173988.
- Maizels RM, Pearce EJ, Artis D, et al. Regulation of pathogenesis and immunity in helminth infections. J Exp Med. 2009;206:2059–2066.
- Sher A, Fiorentino D, Caspar P, et al. Production of IL-10 by CD4+ T lymphocytes correlates with down-regulation of Th1 cytokine synthesis in helminth infection. J Immunol. 1991;147:2713–2716.
- Sotgiu S, Pugliatti M, Sotgiu A, et al. Does the "hygiene hypothesis" provide an explanation for the high prevalence of multiple sclerosis in Sardinia? Autoimmunity. 2003;36:257–260.
- Daveson AJ, Jones DM, Gaze S, et al. Effect of hookworm infection on wheat challenge in celiac disease–a randomised double-blinded placebo controlled trial. PLoS One. 2011;6:e17366.
- Freeman HJ. Drug-induced sprue-like intestinal disease. Int J Celiac Dis. 2014;2:49–53.
- Canova C, Zabeo V, Pitter G, et al. Association of maternal education, early infections, and antibiotic use with celiac disease: a population-based birth cohort study in northeastern Italy. Am J Epidemiol. 2014;180:76–85.
- Kemppainen KM, Vehik K, Lynch KF, Environmental Determinants of Diabetes in the Young (TEDDY) Study Group, et al. Association between early-life antibiotic use and the risk of islet or celiac disease autoimmunity. JAMA Pediatr. 2017;171:1217–1225.
- Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology. 1999;117:17–25.
- Lebwohl B, Spechler SJ, Wang TC, et al. Use of proton pump inhibitors and subsequent risk of celiac disease. Dig Liver Dis. 2014;46:36–40.
- Ianiro G, Bibbò S, Montalto M, et al. Systematic review: sprue-like enteropathy associated with olmesartan. Aliment Pharmacol Ther. 2014;40:16–23.
- Vaarala O, Jokinen J, Lahdenkari M, et al. Rotavirus vaccination and the risk of celiac disease or type 1 diabetes in finnish children at early life. Pediatr Infect Dis J. 2017;36:674–675.
- Silvester JA, Leffler DA. Is autoimmunity infectious? the effect of gastrointestinal viral infections and vaccination on risk of celiac disease autoimmunity. Clin Gastroenterol Hepatol. 2017;15:703–705.
- Myléus A, Stenlund H, Hernell O, et al. Early vaccinations are not risk factors for celiac disease. Pediatrics. 2012;130:e63–e70.
- Roberts SE, Williams JG, Meddings D, et al. Perinatal risk factors and coeliac disease in children and young adults: a record linkage study. Aliment Pharmacol Ther. 2009;29:222–231.
- Decker E, Engelmann G, Findeisen A, et al. Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Pediatrics. 2010;125:e1433–e1440.
- Mårild K, Stephansson O, Montgomery S, et al. Pregnancy outcome and risk of celiac disease in offspring: a nationwide case-control study. Gastroenterology. 2012;142:39–45.
- Lionetti E, Castellaneta S, Francavilla R, SIGENP Working Group of Weaning and CD Risk, et al. Mode of delivery and risk of celiac disease: risk of celiac disease and age at gluten introduction cohort study. J Pediatr. 2017;184:81–86.e2.
- Dydensborg Sander S, Hansen AV, Størdal K, et al. Mode of delivery is not associated with celiac disease. Clin Epidemiol. 2018;10:323–332.
- Emilsson L, Magnus MC, Størdal K. Perinatal risk factors for development of celiac disease in children, based on the prospective Norwegian Mother and Child Cohort Study. Clin Gastroenterol Hepatol. 2015;13:921–927.
- Koletzko S, Lee HS, Beyerlein A, TEDDY Study Group, et al. Cesarean section on the risk of celiac disease in the offspring: the TEDDY study. J Pediatr Gastroenterol Nutr. 2018;66:417–424.
- Lebwohl B, Green PH, Murray JA, et al. Season of birth in a nationwide cohort of coeliac disease patients. Arch Dis Child. 2013;98:48–51.
- Ivarsson A, Hernell O, Nyström L, et al. Children born in the summer have increased risk for coeliac disease. J Epidemiol Community Health. 2003;57:36–39.
- Lewy H, Meirson H, Laron Z. Seasonality of birth month of children with celiac disease differs from that in the general population and between sexes and is linked to family history and environmental factors. J Pediatr Gastroenterol Nutr. 2009;48:181–185.
- Daniel S, Kalansky A, Tsur A, et al. Seasonality of birth affects paediatric coeliac disease. Acta Paediatr. 2019;108:529–534.
- Andersen R, Mølgaard C, Skovgaard LT, et al. Teenage girls and elderly women living in northern Europe have low winter vitamin D status. Eur J Clin Nutr. 2005;59:533–541.
- Fisman D. Seasonality of viral infections: mechanisms and unknowns. Clin Microbiol Infect. 2012;18:946–954.
- Brodin P, Davis MM. Human immune system variation. Nat Rev Immunol. 2017;17:21–29.
- Namatovu F, Lindkvist M, Olsson C, et al. Season and region of birth as risk factors for coeliac disease a key to the aetiology? Arch Dis Child. 2016;101:1114–1118.
- Kuja-Halkola R, Lebwohl B, Halfvarson J, et al. Birth weight, sex, and celiac disease: a nationwide twin study. Clin Epidemiol. 2017;9:567–577.
- Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol. 2015;12:497–506.
- Parmar A, Alakulppi N, Paavola-Sakki P, et al. Association study of FUT2 (rs601338) with celiac disease and inflammatory bowel disease in the Finnish population. Tissue Antigens. 2012;80:488–493.
- Wacklin P, Mäkivuokko H, Alakulppi N, et al. Secretor genotype (FUT2 gene) is strongly associated with the composition of Bifidobacteria in the human intestine. PLoS ONE. 2011;6:e20113.
- Trynka G, Hunt KA, Bockett NA, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43:1193–1201.
- Pozo-Rubio T, de Palma G, Mujico JR, et al. Influence of early environmental factors on lymphocyte subsets and gut microbiota in infants at risk of celiac disease; the PROFICEL study. Nutr Hosp. 2013;28:464–473.
- De Palma G, Capilla A, Nova E, et al. Influence of milk-feeding type and genetic risk of developing coeliac disease on intestinal microbiota of infants: the PROFICEL study. PLoS ONE. 2012;7:e30791.
- Olivares M, Neef A, Castillejo G, et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut. 2015;64:406–417.
- Sellitto M, Bai G, Serena G, et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS ONE. 2012;7:e33387.
- Collado MC, Donat E, Ribes-Koninckx C, et al. Imbalances in faecal and duodenal Bifidobacterium species composition in active and non-active coeliac disease. BMC Microbiol. 2008;8:232.
- Olivares M, Benítez-Páez A, de Palma G, et al. Increased prevalence of pathogenic bacteria in the gut microbiota of infants at risk of developing celiac disease: the PROFICEL study. Gut Microbes. 2018;9:551–558.
- Olivares M, Walke AW, Capilla A, et al. Gut microbiota trajectory in early life may predict development of celiac disease. Microbiome. 2018;6:36.
- Zamakhchari M, Wei G, Dewhirst F, et al. Identification of Rothia bacteria as gluten-degrading natural colonizers of the upper gastro-intestinal tract. PLoS One. 2011;6:e24455.
- Fernandez-Feo M, Wei G, Blumenkranz G, et al. The cultivable human oral gluten-degrading microbiome and its potential implications in coeliac disease and gluten sensitivity. Clin Microbiol Infect. 2013;19:E386–E394.
- Laparra JM, Sanz Y. Bifidobacteria inhibit the inflammatory response induced by gliadins in intestinal epithelial cells via modifications of toxic peptide generation during digestion. J Cell Biochem. 2010;109:801–807.
- Lindfors K, Blomqvist T, Juuti-Uusitalo K, et al. Live probiotic Bifidobacterium lactis bacteria inhibit the toxic effects induced by wheat gliadin in epithelial cell culture. Clin Exp Immunol. 2008;152:552–558.
- De Palma G, Cinova J, Stepankova R, et al. Pivotal advance: Bifidobacteria and Gram-negative bacteria differentially influence immune responses in the proinflammatory milieu of celiac disease. J Leukoc Biol. 2010;87:765–778.
- De Palma G, Kamanova J, Cinova J, et al. Modulation of phenotypic and functional maturation of dendritic cells by intestinal bacteria and gliadin: relevance for celiac disease. J Leukoc Biol. 2012;92:1043–1054.
- Jacobson ED, Prior JT, Faloon WW. Malabsorptive syndrome induced by neomycin: morphologic alterations in the jejunal mucosa. J Lab Clin Med. 1960;56:245–250.
- Mårild K, Ludvigsson J, Sanz Y, et al. Antibiotic exposure in pregnancy and risk of coeliac disease in offspring: a cohort study. BMC Gastroenterol. 2014;14:75.
- Stolte K. Schwere Durchfalle bei neuropathischen Kindern. Jahrb Kinderh. 1917;36:89–127.
- Thompson MW. Heredity, maternal age, and birth order in the etiology of celiac disease. Am J Hum Genet. 1951;3:159–166.
- Zingone F, West J, Crooks CJ, et al. Socioeconomic variation in the incidence of childhood coeliac disease in the UK. Arch Dis Child. 2015;100:466–473.
- Kondrashova A, Mustalahti K, Kaukinen K, et al. Lower economic status and inferior hygienic environment may protect against celiac disease. Ann Med. 2008;40:223–231.
- Olén O, Bihagen E, Rasmussen F, et al. Socioeconomic position and education in patients with coeliac disease. Dig Liver Dis. 2012;44:471–476.
- Whyte LA, Kotecha S, Watkins WJ, et al. Coeliac disease is more common in children with high socio-economic status. Acta Paediatr. 2014;103:289–294.
- Wingren CJ, Björck S, Lynch KF, et al. Coeliac disease in children: a social epidemiological study in Sweden. Acta Paediatr. 2012;101:185–191.
- Ludvigsson JF, ABIS Study Group. Socio-economic characteristics in children with coeliac disease. Acta Paediatr. 2005;94:107–113.
- Snook JA, Duyer L, Lee-Elliott C, et al. Adult coeliac disease and cigarette smoking. Gut. 1996;30:60–62.
- Patel AH, Loftus EV, Jr, Murray JA, et al. Cigarette smoking and celiac sprue: a case-control study. Am J Gastroenterol. 2001;96:2388–2391.
- Vazquez H, Smecuol E, Flores D, et al. Relation between cigarette smoking and celiac disease: evidence from a case-control study. Am J Gastroenterology. 2001;96:798–802.
- Hill AB. The environment and disease: association or causation? Proc R Soc Med. 1965;58:295–300.
- Suman S, Williams EJ, Thomas PW, et al. Is the risk of adult coeliac disease causally related to cigarette exposure? Eur J Gastroenterol Hepatol. 2003;15:995–1000.
- Sugiyama D, Nishimura K, Tamaki K, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2010;69:70–81.
- Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2012;91:142–149.
- Tollerud DJ, Clark JW, Brown LM, et al. The effects of cigarette smoking on T-cell subsets. Am Rev Respir Dis. 1989;139:1446–1451.
- Drago S, El Asmar R, Di Pierro M, et al. Gliadin, zonulin and gut permeability: effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–419.
- Sánchez E, De Palma G, Capilla A, et al. Influence of environmental and genetic factors linked to celiac disease risk on infant gut colonization by Bacteroides species. Appl Environ Microbiol. 2011;77:5316–5323.
- Fasano A. Intestinal permeability and its regulation by zonulin: diagnostic and therapeutic implications. Clin Gastroenterol Hepatol. 2012;10:1096–1100.
- Porras M, Martin MT, Yang PC, et al. Correlation between cyclical epithelial barrier dysfunction and bacterial translocation in the relapses of intestinal inflammation. Inflamm Bowel Dis. 2006;12:843–852.
- Vorobjova T, Raikkerus H, Kadaja L, et al. Circulating zonulin correlates with density of enteroviruses and tolerogenic dendritic cells in the small bowel mucosa of celiac disease patients. Dig Dis Sci. 2017;62:358–371.