
Abstract
Myeloid neoplasms post cytotoxic therapy (MN-pCT) are a category includes AML, MDS, and MDS/MPN arising in patients exposed to cytotoxic (DNA-damaging) therapy for an unrelated condition in 2022 version World Health Organization (WHO) classification. With improved survival of patients with tumors, the incidence of MN-pCT after chemotherapy and/or radiation therapy among patients with tumors has gradually risen. However, the outcome of MN-pCT is poorer than that of primary myeloid neoplasms. This review summarizes the current understanding based on existing research, as a foundation for further research on MN-pCT.
Introduction
With the development of diagnosis and treatment technologies, survival of a variety of cancer patients has been improved, while the incidence of myeloid neoplasms post cytotoxic therapy (MN-pCT) has also increased [Citation1,Citation2]. MN-pCT are defined as myeloid neoplasms that appear after cytotoxic therapy, including chemotherapy and/or radiotherapy [Citation3]. At present, AML-pCT and MDS/MPN-pCT account for approximately 15–20% of AML and MDS/MPN patients [Citation4]. MN-pCT showed a poorer survival compared with patients with primary AML and MDS/MPN [Citation5]. Because of poor response to multiple drugs and therapies, MN-pCT is one of the most feared complications of cytotoxic therapy. Therefore, this paper reviews MN-pCT from the aspects of diagnosis, pathogenesis, prognosis and treatment prospects.
Definition and diagnosis of myeloid neoplasms post cytotoxic therapy
At present, the diagnostic criteria of MN-pCT are based on the diagnostic criteria of AML and MDS/MPN with a previous history of chemotherapy and/or radiotherapy in the 2022 version World Health Organization (WHO) classification. Patients such as NPM1 mutation and corebinding factor leukaemias should still be assigned to this category. Exposure to (poly-ADP-ribose polymerase1, PARP1) inhibitors is added as a qualifying criterion for MN-pCT, and methotrexate has been excluded [Citation3]. For the interval between chemotherapy and/or radiotherapy history and subsequent MN-pCT, there is no clear definition. Radivoyevitch et al. [Citation6] found that prostate cancer patients with radiation therapy showed an increased relative risk to AML and MDS/MPN in 1.5–2.5 years, while patients with lung cancers and breast cancers showed the highest relative risk to AML and MDS/MPN in the next 1–12 years. Wolff et al. [Citation7] found that the occurrence of AML and MDS/MPN after chemotherapy and/or radiotherapy was extremely low, but higher than previously described, and the incidence continued to increase beyond 5 years for early-stage breast cancer patients.
Secondary myeloid neoplasms(s-MDS/s-MPN/s-AML) is another category of myeloid neoplasms, which is a broader, more inclusive diagnosis compared with MN-pCT, it is a newly segregated category encompassing diseases that arise in the setting of certain known predisposing factors, such as MN-pCT, myeloid neoplasms associated with germline predisposition, myeloid neoplasms associated with eosinophilia and tyrosine kinase gene fusions according to the 2022 version WHO classification.
In addition, WHO classification suggested that further classification of MN-pCT may be required based on genetics in the future [Citation3]. Patients with AML-pCT or MDS/MPN-pCT also have different genetic characteristics compared to those with primary AML or MDS/MPN. Some genetic abnormalities were associated with poor prognosis in MN-pCT, such as chromosome 5 abnormalities, chromosome 7 abnormalities, and complex karyotypes. In addition, genetic abnormalities associated with good prognosis or moderate prognosis are less likely to occur in MN-pCT [Citation8]. Genetic abnormalities such as TP53 mutation, RAS mutation, may only be the initial factor, and multiple genetic abnormalities can be acquired subsequently form a secondary strike and induce MN-pCT eventually [Citation9].
Epidemiology and pathogenesis of myeloid neoplasms post cytotoxic therapy
The incidence of MN-pCT is 0.62/100,000 in the general population, furthermore, the incidence of MN-pCT is different in every individual case of cancer. The median age of MN-pCT is 64 years old and chemotherapy treatment has a 4.7-fold increased incidence of MN-pCT, and younger age at the time of exposure showed a higher risk [Citation10]. A large study with more than 700,000 patients and 23 kinds of tumors showed that the 10-year cumulative incidence of MN-pCT in patients with bone and joint malignant tumors was relatively high, about 1.67%. The 10-year cumulative incidence of MN-pCT in breast cancer patients is relatively low, about 0.44%, because of high incidence of breast cancer and large patient base, breast cancer is the tumor with the highest proportion of MN-pCT primary tumors [Citation5].
Chemotherapy and/or radiotherapy not only kill tumor cells, but also damage the deoxyribonucleic acid (DNA) of normal cells, leading to series of genetic changes. These abnormalities of some genetic pathways are major factors to the development of MN-pCT [Citation11]. While the pathogenesis of MN-pCT remains controversial. Some studies have suggested that cytotoxic therapy can directly induce oncogenic gene mutations and genetic instability, thus leading to the occurrence of MN-pCT. For example, alkylating agents may induce mutation of TP53, and tumor suppressor gene TP53 plays a key role in DNA damage repair. Mutation of TP53 affects cell cycle and DNA repair, leading to uncontrolled proliferation and blocked differentiation, resulting in MN-pCT [Citation12]. There are actually emerging data to suggest that cytotoxic therapy in a good subset of patients may not specifically induce the TP53 mutation but rather allow clonal selection among cells already harboring it [Citation11,Citation12]. Studies have also suggested that topoisomerase inhibitors can induce t (15; 17) and promote the development of AML [Citation8]. However, some studies showed that mutations may exist before chemotherapy or radiotherapy. Wong et al. [Citation9]suggested that low-frequency gene mutations may exist before chemotherapy, and hematopoietic stem progenitor cells showed these gene mutations after chemotherapy selectively. Takahashi et al. [Citation13] demonstrated that the presence of clonal hematopoiesis (CH) significantly increases the risk of subsequent treatment of associated myeloid tumors (t-MN) by 14-fold. Similarly, Gillis et al. [Citation14] showed that elderly patients who developed t-MN were more likely to have clonal hematopoiesis of undetermined potential’ (CHIP). CHIP [Citation15] describes the situation in which somatic mutations are present in the blood or bone marrow cells and no other criteria for hematologic neoplasia are met and the identification of factors that promote the transformation from CHIP to t-AML seems useful; it seems that it is not only the matter of CHIP mutations that are useful, but also the germline polymorphism and cell extrinsic factors [Citation15]. During MDS initiation, newly acquired mutations within the previous mutated clone appear, while previous mutations are also passed on. Those acquired mutations may have no consequences as ‘passenger’ mutations or become driver mutations, contributing to clonal evolution [Citation16]. The final stage of the disease progression is an evolution from MDS to s-AML, occurring as a consequence of somatic mutations accumulation. shows the process of clonal evolution schematically. Benjamin et al. [Citation17] revealed a novel mode of t-MN progression that is not reliant on direct mutagenesis or even exposure to chemotherapy. Conversely, for t-MN that evolve under the influence of chemotherapy-induced mutagenesis, distinct chemotherapies not only select preexisting CH but also promote the acquisition of recurrent genomic drivers.
Figure 1. Clonal evolution: from normal haematopoiesis to AML/MDS-pCT.
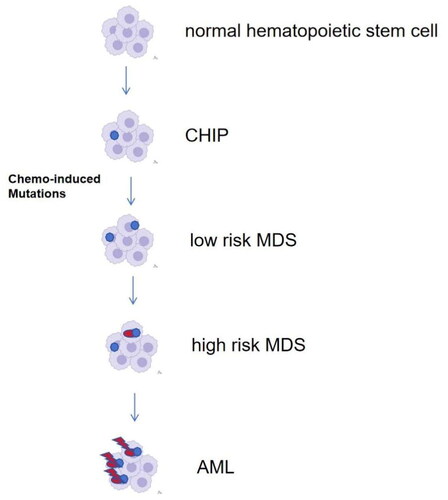
Schwartz et al. [Citation18] found that it takes 405 (118–748) days for most children MN-pCT to develop from mutant clones to MN-pCT by sequencing. An analysis on the risk of using granulocyte colony-stimulating factor (G-CSF) found that tumor patients using G-CSF were more likely to develop MN-pCT than those who did not use G-CSF, the relative risk of MN was 1.92, suggesting that the use of G-CSF increased the risk of MN-pCT [Citation19]. In addition, some studies detected that clonal mutations in cryopreserved stem cells before autologous stem cell transplantation (ASCT) showed a higher risk of MN-pCT, others suggested that preexisting clonal at ASCT scarcely causes MN-pCT directly, while they may contribute to the appearance of new mutations, especially those involving TP53, RUNX1, and RAS, which can facilitate the evolution to MN-pCT [Citation20]. The majority of AML-pCT and MDS-pCT are associated with TP53 mutations. The outcomes of such patients are generally worse with biallelic (multi-hit) TP53 alterations, manifesting as ≥2 TP53 mutations, or with concomitant 17p/TP53 deletion or copy neutral Loss of Heterozygosity (LOH). Less frequent mutations involve genes such as PPM1D and DNA-damage response genes that may require additional work-up for germline predisposition [Citation3].
In approximately 60% of malignant neoplasms in humans, TP53 is mutated or inactivated. TP53 mutations are often detected in MN-pCT patients with chromosome 5 abnormalities and complex karyotypes, and TP53 mutations are associated with poor prognosis [Citation21]. TP53 is located on chromosome17 and encodes a tumor suppression protein p53, widely known as the ‘guardian of the genome’. Activation of p53 takes place in response to DNA damage and replication stress. TP53 activation leads to the elimination or repair of damaged cells, in order to reduce the risk of propagating mutations. It is hypothesized that TP53 promotes a selective growth advantage after exposure to chemo- or radiotherapy, leading to out-competing cells with high p53 activity by those with reduced p53 activity [Citation22]. MN-pCT patients with chromosome 7 abnormality often have RAS pathway activation-related mutations, which are also associated with poor prognosis [Citation23]. At present, no clear gene variation has been found to directly induce MN-pCT.
Latency period was calculated majority as time in months from the date of primary diagnosis of cancer to the date of diagnosis of MN-pCT. Regarding the latency, all those studies in showed the shortest latency from cytotoxic chemotherapy when patients were treated with two leukemogenic agents, suggesting that the more cytotoxicity of leukemogenic agents is intense, the earlier MN-pCT can develop. Hyerim et al. [Citation27] showed the median latency to develop MN-pCT was 5.37 person-years, and patients receiving both chemotherapy and radiotherapy had the highest standard incidence ratio (SIR) (4.64; 95% CI, 4.08–5.20), followed by those receiving CT only (SIR, 3.30; 95% CI, 2.89–3.70). Maung et al. [Citation30] showed that the median latency was 60 months. Latency periods for patients who received chemotherapy alone compared to those who received radiotherapy alone was 56 vs. 13 months (p = 0.45), and patients who developed t-MN following treatment for solid tumour had longer latency period compared to those who had haematological disorders (85 vs. 46 months, p = 0.27).
Table 1. Major studies that investigating the incidence of MN-pCT.
Table 2. Major cytogenetic abnormalities of MN-pCT patients.
In a word, the above-mentioned pathogenesis may play certain roles in the pathogenesis of MN-pCT. Major studies that investigating the incidence and cytogenetic abnormalities were summarized in and Citation2.
Outcomes, prognostic factors of myeloid neoplasms post cytotoxic therapy
Though scarcely occurred, the overall survival (OS) is generally poor for MN-pCT patients, the median OS was 13 months (range 9.1–16.9 months) and 6 months for AML and 15 months for MDS, respectively [Citation34]. Fianchi et al. reported that the median OS was 14.6 months and patients receiving allogeneic stem cell transplantation (allo-SCT) obtained a significantly longer survival based on a study enrolled 277 MN-pCT patients between 1999 and 2013, they also found some factors showing a significant effect on survival by multivariable analysis, such as degree of anemia, age, and unfavorable karyotype. Kuendgen et al. [Citation35] found that cytogenetic features showed an enhanced predictive power by analyzing data of 2087 MDS-pCT patients. Many studies have showed a number of prognostic indicators in AML-pCT, such as unfavorable karyotype, older age, thrombocytopenia, moreover, the outcome was independent of their primary cancer in most studies [Citation36].
For MDS-pCT patients, MD Anderson Cancer Center devised a MDS-pCT prognostic system (TPSS), patients were divided into three prognostic groups: good prognosis (0–2 factors), intermediate prognosis (3–4 factors) and poor prognosis (5–7 factors) according to a number of clinical indicators, including age ≥65 years, ECOG performance status score 2–4 scores, poor cytogenetics (−7 and/or complex), MDS subtype (RARS or RAEB-1/2), hemoglobin (<11 g/dL), platelets (<50 × 109/dL), and transfusion dependency) [Citation37]. Zeidan et al. [Citation38] found that MPSS, TPSS and IPSS-R provide the best predictive power for 370 MDS-pCT patients and identified that MDS-pCT patients showed a significantly higher risk of death compared to primary MDS patients in every prognostic stratification model.
Treatment options for myeloid neoplasms post cytotoxic therapy
Intensive therapy, non- intensive therapy and novel agents
Due to the low incidence, there is no consensus on the treatment of MN-pCT. At present, MN-pCT can be treated with as primary myeloid system neoplasms, including chemotherapy, hematopoietic stem cell transplantation therapy, demethylation drug therapy, immunotherapy [Citation39]. However, it is more difficult to treat MN-pCT patients because of side effects due to previous therapy, including organ dysfunction, chronic immunosuppression, depletion of normal hematopoietic stem cells.
Similar to primary AML/MDS, it is important to determine whether a patient with AML-pCT is able to tolerate intensive induction chemotherapy. Conventional induction regimens showed a median OS of 6 months approximately [Citation36]. Prior exposure to chemotherapy also limits the use of conventional induction regimens in AML-pCT patients. CPX-351 is a liposomal drug of cytarabine and daunorubicin and has been approved by the United States Food and Drug Administration (FDA) for the treatment of newly diagnosed AML-pCT or AML with myelodysplasia-related changes. A randomized, multi-center, open-label, phase 3 trial in the US and Canada showed the median OS was 9.33 months with CPX-351 and 5.95 months with DA regimen, 5-year OS was 18% and 8% respectively. CPX-351 showed a long-term remission and improved OS in AML-pCT patients [Citation40], Geoffrey et al. [Citation41] performed detailed analyses of outcomes after alloHCT following CPX-351 vs 7 1 3 in the phase 3 study, with 3 years OS was 56% vs 23%, respectively.
Lower-intensity therapies can be used for patients who are ineligible for intensive induction therapy. Many drugs have been evaluated in patients with AML-pCT/MDS-pCT, such as hypomethylating agents (azacytidine [Citation42], decitabine [Citation43]), venetoclax alone [Citation44] or in combination with azacytidine [Citation45] or low-dose cytarabine [Citation46], nivolumab [Citation47], dasatinib [Citation48], eprenetapopt [Citation49,Citation50], magrolimab [Citation51], flotetuzumab [Citation52] (). Research data are limited for MDS-pCT. Klimek et al. [Citation53] evaluated response to 5-azacytidine and decitabine in a study enrolled 42 MDS-pCT patients and found that the overall response rate (ORR) was 38%, complete remission (CR) rate (14%), and they also suggested that DNA methyltransferase inhibitors showed an activity in the development of MDS-pCT which is relatively infrequent seen in primary MDS.Menin inhibitors are targeted agents that block the binding of the protein menin to a complex epigenetic entity, which is based on the MLL or KMT2A gene. Multiple clinical trials evaluating oral menin inhibitors in acute leukemias are ongoing [Citation43] (all the above contents are shown in ).
Table 3. Treatment options for MN-pCT patients.
Hematopoietic cell transplantation
Allo-SCT is considered as a consolidative strategy to cure MN-pCT, while outcomes remain poor, with OS estimated at 20–30% at 3 and 5 years in some larger cohort studies [Citation54,Citation55]. Metheny et al. [Citation56] reported a study enrolled 1531 MN-pCT patients from Center for International Blood and Marrow Transplant Research (CIBMT) and showed that the median age was 56 years, 17% of patients had received allo-HSCT, 5 years OS of MDS-pCT and AML-pCT were 27% and 25% respectively, while 5 years disease free survival (DFS) was 19% and 23%. Relapse rates at 5 years of t- MDS and AML-pCT were 46% and 43%, respectively, while non relapse mortality (NRM) was similar at 34% for both MDS-pCT and AML-pCT cohorts.
Summary and future directions
With the development of sequencing technology, significant progress has been made in the pathogenesis, diagnoses and treatment of MN-pCT, early detection of DNA lesions is vital in occurrence of disease, so some authors recommend that primary malignancy may be treated with targeted therapy [Citation17]. In the future, it is urgent to take further studies aiming at the relationship between tumor susceptibility gene mutations and the occurrence of MN-pCT after primary tumor treatment, the risk of developing MN-pCT. If patients are predicted to be at high risk of MN-pCT, it may be necessary to adjust the treatment regimen for the treatment of primary tumors, such as avoiding or reducing the use of alkylating agents and topoisomerase ⅱ inhibitors, reducing the dose, and conducting long-term genetic monitoring in high-risk patients with MN-pCT.
Currently, the diagnosis of MN-pCT depends on a history of exposure to chemotherapy or radiotherapy, but exposure alone is not sufficient to prove causality. In the future, in addition to the existence of chemotherapy and/or radiotherapy treatment history, more specific diagnostic basis should be developed to measure, the onset time interval, genetics and other characteristics of the treatment plan, which are conducive to improving the accuracy of MN-pCT diagnosis. Because of the different pathophysiology to de-novo AML, MN-pCT patient group will often not be appropriate for Clinical trials, which makes the clinical diagnosis and treatment of MN-pCT more difficult, and also limits the clinical research of MN-pCT.
With the improvement of survival of cancer patients, the number of treatment-related tumors will increase, so it is important to do further research in depth. We hope that the pathogenesis of MN-pCT will be more clearly understood in the future, and the occurrence of MN-pCT can be better predicted, thus making the treatment plan more optimized.
Author contributions
Jing Ma and Yafei Wang were involved in the conception of the study. Jing Ma was involved in the analysis. All authors were involved in the interpretation of the findings. All authors were involved in drafting the manuscript. All authors reviewed and approved the final manuscript and agree to be held accountable for all aspects of the work.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Acknowledgements
Yafei Wang reviewed the English language. The authors did not receive support from any organization for the submitted work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Anonymized data are available upon reasonable request.
Additional information
Funding
References
- Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):1–9. doi:10.3322/caac.21149.
- Gurnari C, Fabiani E, Falconi G, et al. From clonal hematopoiesis to therapy-related myeloid neoplasms: the silent way of cancer progression. Biology (Basel). 2021;10(2):128. doi:10.3390/biology10020128.
- Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703–1719. doi:10.1038/s41375-022-01613-1.
- Abou Zahr A, Kavi AM, Mukherjee S, et al. Therapy-related myelodysplastic syndromes, or are they? Blood Rev. 2017;31(3):119–128. doi:10.1016/j.blre.2016.11.002.
- Morton LM, Dores GM, Schonfeld SJ, et al. Association of chemotherapy for solid tumors with development of therapy-related myelodysplastic syndrome or acute myeloid leukemia in the modern era. JAMA Oncol. 2019;5(3):318–325. doi:10.1001/jamaoncol.2018.5625.
- Radivoyevitch T, Sachs RK, Gale RP, et al. Defining AML and MDS second cancer risk dynamics after diagnoses of first cancers treated or not with radiation. Leukemia. 2016;30(2):285–294. doi:10.1038/leu.2015.258.
- Wolff AC, Blackford AL, Visvanathan K, et al. Risk of marrow neoplasms after adjuvant breast cancer therapy: the national comprehensive cancer network experience. J Clin Oncol. 2015;33(4):340–348. doi:10.1200/JCO.2013.54.6119.
- Smith SM, Le Beau MM, Huo D, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102(1):43–52. doi:10.1182/blood-2002-11-3343.
- Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–555. doi:10.1038/nature13968.
- Mcnerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17(9):513–527. doi:10.1038/nrc.2017.60.
- Tiruneh T, Enawgaw B, Shiferaw E. Genetic pathway in the pathogenesis of therapy-related myeloid neoplasms: a literature review. Oncol Ther. 2020;8(1):45–57. doi:10.1007/s40487-020-00111-7.
- Schulz E, Kashofer K, Heitzer E, et al. Preexisting TP53 mutation in therapy-related acute myeloid leukemia. Ann Hematol. 2015;94(3):527–529. doi:10.1007/s00277-014-2191-0.
- Takahashi K, Wang F, Kantarjian H, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. 2017;18(1):100–111. doi:10.1016/S1470-2045(16)30626-X.
- Gillis NK, Ball M, Zhang Q, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017;18(1):112–121. doi:10.1016/S1470-2045(16)30627-1.
- Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoesis of indeterminate potential and its distinction from myelodysplastic synomes. Blood. 2015;126(1):9–16. doi:10.1182/blood-2015-03-631747.
- Higgins A, Shah MV. Genetic and genomic landscape of secondary and therapy-related acute myeloid leukemia. Genes (Basel). 2020;11(7):749. doi:10.3390/genes11070749.
- Diamond B, Ziccheddu B, Maclachlan K, et al. Tracking the evolution of therapy-related myeloid neoplasms using chemotherapy signatures. Blood. 2023;141(19):2359–2371. doi:10.1182/blood.2022018244.
- Schwartz JR, Ma J, Kamens J, et al. The acquisition of molecular drivers in pediatric therapy-related myeloid neoplasms. Nat Commun. 2021;12(1):985. doi:10.1038/s41467-021-21255-8.
- Jabagi MJ, Vey N, Goncalves A, et al. Risk of secondary hematologic malignancies associated with breast cancer chemotherapy and G-CSF support: a nationwide population-based cohort. Int J Cancer. 2021;148(2):375–384. doi:10.1002/ijc.33216.
- Gramegna D, Bertoli D, Cattaneo C, et al. The role of clonal hematopoiesis as driver of therapy-related myeloid neoplasms after autologous stem cell transplantation. Ann Hematol. 2022;101(6):1227–1237. doi:10.1007/s00277-022-04806-x.
- Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol. 2001;19(5):1405–1413. doi:10.1200/JCO.2001.19.5.1405.
- Stefaniuk P, Szymczyk A, Podhorecka M. In pursuit of genetic prognostic factors and treatment approaches in secondary acute myeloid Leukemia-a narrative review of current knowledge. J Clin Med. 2022;11(15):4283. doi:10.3390/jcm11154283.
- Capelli D, Menotti D, Fiorentini A, et al. Secondary acute myeloid leukemia: pathogenesis and treatment. In: Li W, editor. Leukemia [internet]. Brisbane (AU): Exon Publications; 2022. Chapter 7.
- Guru Murthy GS, Hamadani M, Dhakal B, et al. Incidence and survival of therapy related myeloid neoplasm in United States. Leuk Res. 2018;71:95–99. doi:10.1016/j.leukres.2018.07.013.
- Fianchi L, Pagano L, Piciocchi A, et al. Characteristics and outcome of therapy-related myeloid neoplasms: report from the Italian network on secondary leukemias. Am J Hematol. 2015;90(5):E80–5. doi:10.1002/ajh.23966.
- Ong DM, Farrugia H, Wei A. Therapy-related acute myeloid leukaemia and myelodysplastic syndrome in Victoria, Australia 2003–2014. Intern Med J. 2018;48(7):822–829. doi:10.1111/imj.137.
- Ha H, Kim HJ, Park JH, et al. Epidemiologic outlook of therapy-related myeloid neoplasms and selection of high-risk patients: a Korean nationwide study. Cancer. 2022;128(21):3888–3896. doi:10.1002/cncr.34453.
- Eichenauer DA, Thielen I, Haverkamp H, et al. Therapy-related acute myeloid leukemia and myelodysplastic syndromes in patients with Hodgkin lymphoma: a report from the German Hodgkin Study Group. Blood. 2014;123(11):1658–1664. doi:10.1182/blood-2013-07-512657.
- Praga C, Bergh J, Bliss J, et al. Risk of acute myeloid leukemia and myelodysplastic syndrome in trials of adjuvant epirubicin for early breast cancer: correlation with doses of epirubicin and cyclophosphamide. J Clin Oncol. 2005;23(18):4179–4191. doi:10.1200/JCO.2005.05.029.
- Maung SW, Burke C, Hayde J, et al. A review of therapy-related myelodysplastic syndromes and acute myeloid leukaemia (t-MDS/AML) in Irish patients: a single Centre experience. Hematology. 2017;22(6):341–346. Epub 2017 Feb 15. PMID: 28196450. doi:10.1080/10245332.2017.1286539.
- Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–1376. doi:10.1182/blood-2014-11-610543.
- Shih AH, Chung SS, Dolezal EK, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98(6):908–912. doi:10.3324/haematol.2012.076729.
- Bacher U, Haferlach T, Kern W, et al. A comparative study of molecular mutations in 381 patients with myelodysplastic syndrome and in 4130 patients with acute myeloid leukemia. Haematologica. 2007;92(6):744–752. doi:10.3324/haematol.10869.
- Chantadisai M, Kulkarni HR, Baum RP. Therapy-related myeloid neoplasm after peptide receptor radionuclide therapy (PRRT) in 1631 patients from our 20 years of experiences: prognostic parameters and overall survival. Eur J Nucl Med Mol Imaging. 2021;48(5):1390–1398. doi:10.1007/s00259-020-05127-9.
- Kuendgen A, Nomdedeu M, Tuechler H, et al. Therapy-related myelodysplastic syndromes deserve specific diagnostic Sub-classification and risk-stratification-an approach to classification of patients with MDS-pCT. Leukemia. 2021;35(3):835–849. doi:10.1038/s41375-020-0917-7.
- Strickland SA, Vey N. Diagnosis and treatment of therapy-related acute myeloid leukemia. Crit Rev Oncol Hematol. 2022;171:103607. doi:10.1016/j.critrevonc.2022.103607.
- Quintás-Cardama A, Daver N, Kim H, et al. A prognostic model of therapy-related myelodysplastic syndrome for predicting survival and transformation to acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2014;14(5):401–410. doi:10.1016/j.clml.2014.03.001.
- Zeidan AM, Al Ali N, Barnard J, et al. Comparison of clinical outcomes and prognostic utility of risk stratification tools in patients with therapy-related vs de novo myelodysplastic syndromes: a report on behalf of the MDS Clinical Research Consortium. Leukemia. 2017;31(6):1391–1397. doi:10.1038/leu.2017.33.
- Voso MT, Falconi G, Fabiani E. What’s new in the pathogenesis and treatment of therapy-related myeloid neoplasms. Blood. 2021;138(9):749–757. doi:10.1182/blood.2021010764.
- Lancet JE, Uy GL, Newell LF, et al. CPX-351 versus 7 + 3 cytarabine and daunorubicin chemotherapy in older adults with newly diagnosed high-risk or secondary acute myeloid leukaemia: 5-year results of a randomised, open-label, multicentre, phase 3 trial. Lancet Haematol. 2021;8(7):e481–e491. doi:10.1016/S2352-3026(21)00134-4.
- Uy GL, Newell LF, Lin TL, et al. Transplant outcomes after CPX-351 vs 7 + 3 in older adults with newly diagnosed high-risk and/or secondary AML. Blood Adv. 2022;6(17):4989–4993. doi:10.1182/bloodadvances.2023010005.
- Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126(3):291–299. doi:10.1182/blood-2015-01-621664.
- Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30(21):2670–2677. doi:10.1200/JCO.2011.38.9429.
- Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–1117. doi:10.1158/2159-8290.CD-16-0313.
- DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629. doi:10.1056/NEJMoa2012971.
- Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood. 2020;135(24):2137–2145. doi:10.1182/blood.2020004856.
- Daver N, Garcia-Manero G, Basu S, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov. 2019;9(3):370–383. doi:10.1158/2159-8290.CD-18-0774.
- Paschka P, Schlenk RF, Weber D, et al. Adding dasatinib to intensive treatment in core-binding factor acute myeloid leukemia-results of the AMLSG 11-08 trial. Leukemia. 2018;32(7):1621–1630. doi:10.1038/s41375-018-0129-6.
- Sallman DA, DeZern AE, Steensma DP, et al. Phase 1b/2 combination study of APR-246 and azacitidine (AZA) in patients with TP53 mutant myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). Blood. 2018;132(Supplement 1):3091–3091. doi:10.1182/blood-2018-99-119990.
- Cluzeau T, Sebert M, Rahm’e R, et al. APR-246 combined with azacitidine (AZA) in TP53 mutated myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). A phase 2 study by the Groupe Francophone Des Mýelodysplasies (GFM). Blood. 2019;134(Supplement_1):677–677. doi:10.1182/blood-2019-125579.
- Sallman DA, Asch AS, Al Malki MM, et al. The first-in-CD47 class antibody magrolimab combined with azacitidine is well-tolerated and effective in AML patients: phase 1b results. Blood. 2019;134(Supplement_1):569–569. doi:10.1182/blood-2019-126271.
- Aldoss I, Uy GL, Vey N, et al. Flotetuzumab as salvage therapy for primary induction failure and early relapse acute myeloid leukemia. Blood. 2021;136(Supplement 1):16–18. doi:10.1182/blood-2020-134576.
- Klimek VM, Dolezal EK, Tees MT, et al. Efficacy of hypomethylating agents in therapy-related myelodysplastic syndromes. Leuk Res. 2012;36(9):1093–1097. doi:10.1016/j.leukres.2012.04.025.
- Syndax. Syndax announces positive interim data demonstrating robust clinical activity in phase 1 portion of the AUGMENT-101 trial of SNDX-5613 in patients with genetically-defined acute leukemias (Press release). Waltham (MA): Syndax Pharmaceuticals, Inc.; 2021.
- Litzow MR, Tarima S, Pérez WS, et al. Allogeneic transplantation for therapy-related myelodysplastic syndrome and acute myeloid leukemia. Blood. 2010;115(9):1850–1857. doi:10.1182/blood-2009-10-249128.
- Kröger N, Brand R, van Biezen A, et al. Risk factors for therapy-related myelodysplastic syndrome and acute myeloid leukemia treated with allogeneic stem cell transplantation. Haematologica. 2009;94(4):542–549. doi:10.3324/haematol.2008.000927.
- Metheny L, Callander NS, Hall AC, et al. Allogeneic transplantation to treat therapy-related myelodysplastic syndrome and acute myelogenous leukemia in adults. Transplant Cell Ther. 2021;27(11):923.e1–923.e12. doi:10.1016/j.jtct.2021.08.010.