Abstract
Primary ciliary dyskinesia (PCD) is a phenotypically and genetically heterogeneous disorder with an autosomal‐recessive inheritance pattern. Only rarely other modes of inheritance such as X‐linked transmission are observed. The disease phenotype is caused by defects of respiratory cilia, sperm tails and the cilia of the embryonic node. The lack of mucociliary clearance contributes to recurrent respiratory tract infections, that might progress to permanent lung damage (bronchiectasis). The goal of therapy is prevention of bronchiectasis. Male infertility due to sperm tail dysmotility is another frequent finding in PCD. Half of affected individuals have situs inversus (Kartagener's syndrome) due to randomization of left/right body asymmetry. Currently three genes (DNAI1, DNAH5, DNAH11) that encode for dynein proteins have been linked to recessive PCD. Mutations in RPGR located on the X chromosome have been identified in males with retinitis pigmentosa and PCD. As a screening test nasal nitric oxide (NO) measurement is widely used. Establishment of diagnosis currently relies on electron microscopy, direct evaluation of ciliary beat by light microscopy, and/or the novel method of high‐resolution immunofluorecent analysis of respiratory cilia.
Key messages
Primary ciliary dyskinesia is a phenotypically and genetically heterogenous disease.
Early diagnosis of PCD is important and relies so far on a combination of clinical suspicion and confirmatory tests such as ultrastructural (electron microscopy) and/or functional (direct observation by light microscopy) analysis of respiratory cilia.
A novel diagnostic tool – high‐resolution immunofluorescent analysis of respiratory cilia – aids diagnosis.
Primary ciliary dyskinesia
Primary ciliary dyskinesia (PCD) is a rare genetic disorder caused by inherited defects of ciliary function. The genetically, functionally, and ultrastructurally heterogenous disease affects one in 20,000 to one in 60,000 individuals at birth Citation1, Citation2. The pathogenesis of the respiratory disease phenotype reflects defective epithelial ciliary clearance of upper airway compartments such as eustachian tubes and sinuses, and lower airways resulting in chronic inflammatory damage Citation3, Citation4. In approximately half of the PCD patients, situs inversus totalis (complete mirror image of organ positions) is present due to randomization of left/right body asymmetry. The association of PCD and situs inversus is also referred to as Kartagener's syndrome (MM 244400) Citation5.
Pattern of inheritance
PCD is a genetically heterogeneous disorder. Several loci and respective genes have been identified. In the majority of cases PCD is inherited as an autosomal recessive trait, which explains the higher incidence of the disease within inbred populations. In some cases other inheritance patterns have been described. Narayan et al. suggested autosomal dominant or X‐linked inheritance in a family, where an affected mother had five affected children, born to three different fathers Citation6. Furthermore an association between X‐linked retinitis pigmentosa and PCD has been recently reported Citation7.
Clinical presentation
Symptoms of PCD mainly affect the respiratory and reproductive tract. Motile cilia covering epithelial cells lining the upper and lower respiratory tract are responsible for mucociliary clearance of the airways (). Lack of coordinated ciliary movement (i.e. immotility or dysmotility) results in reduced mucociliary clearance and consequent chronic inflammatory damage. The clinical symptoms vary among PCD subjects and depend on age () Citation8, Citation9. In the newborn period unexplained tachypnea, neonatal pneumonia with no risk factors for congenital infections, respiratory distress syndrome, chronic rhinitis, complete mirror image arrangement with structurally normal heart (situs inversus, ), complex heart disease with associated laterality defects, or hydrocephalus may be the first indicators of the disease. Presence of a positive family history aids early diagnosis. In the infant and older child the diagnosis of atypical asthma non‐responsive to treatment, chronic and productive cough, severe gastro‐esophageal reflux, bronchiectasis (), rhino‐sinusitis, chronic and severe secretory otitis media, should alert the physician and promote adequate diagnostic work‐up. In the adult the ciliary defect may become additionally obvious in cases of male and female infertility. A high percentage of males are sterile because of immotility or dysmotility of spermatozoa Citation10–13, but contradictory former assumptions studies have shown that male infertility is not obligatory. Female subfertility is less commonly observed. Lack of ciliary movement in the Fallopian tubes might contribute to subfertility in affected women Citation10, Citation14.
Table I. Clinical findings in patients with PCD.
Figure 1 Photographs of a histology specimen of a murine trachea. Lower(a) and higher (b) magnification photographs depict the respiratory epithelium, which covers the cartilage layer. The ciliated respiratory epithelial cells appear to be brighter and carry hair‐like projections (cilia). Intercalated mucous cells do not carry cilia and are darker in colour.
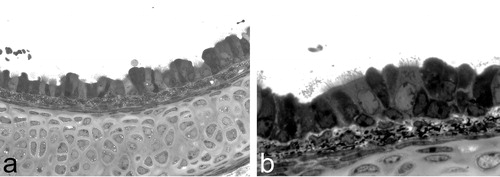
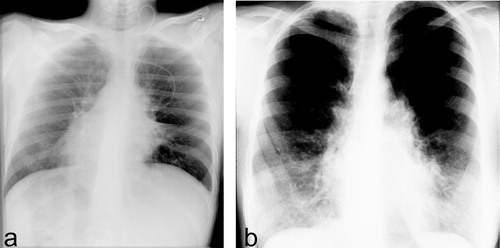
Figure 2 Clinical findings in PCD subjects.a. Radiograph of chest shows dextrocardia in a patient with situs inversus. Note also pulmonary dystelectasis and infiltrates. b. Chest radiograph of an adult PCD patient with chronic airway disease and bronchiectasis.
Diagnosis
There is a large variation in the severity of the clinical phenotype and diagnosis may be difficult. Clinical features of PCD can also mimic other disease, such as cystic fibrosis, allergy or immunologic disorders, Young's syndrome, asplenia/polysplenia Citation4, and there is some risk that the patient's symptoms may be either overestimated or underrated. Therefore, a high index of suspicion for diagnosis of PCD is necessary. The investigation of individuals in whom PCD is suspected should include the exclusion of other differential diagnoses and determination of ciliary function and structure.
The saccharin test has been used to screen mucociliary clearance Citation15. A 1–2 mm particle of saccharin is placed on the inferior nasal turbinate 1 cm from the anterior end. The patient has to sit quietly with the head bent forward and is not allowed to sniff, cough, drink or eat during the investigation. The time is taken for the patient to taste the saccharin and should be less than an hour. If the time is longer than an hour or the patient cannot taste the saccharin mucociliary clearance is impaired. Because the test can only be performed in a cooperative patient, it is only applicable in adolescents and adults. Other disadvantages are the high incidence of false negative and false positive test results and the long test duration. Therefore, the saccharin test is not widely used in clinical practice.
If the diagnosis of PCD is considered, it is much more efficient to perform trans‐nasal brushings and to study the ciliary beat pattern and frequency by direct microscopy Citation16, Citation17. Ciliated epithelial cells are obtained from the inferior or middle turbinate by brushing using a sterile cytology brush. The ciliary beat frequency (CBF) can be measured and the ciliary waveform can be analyzed in detail by digital high speed video imaging differentiating between dyskinetically beating cilia and the normal beat pattern (cilia beat forward and backwards within the same plane without a classical sideway recovery sweep) Citation18. Normal values of CBF depend on the method, on age and vary in different laboratories Citation18, Citation19. CBF of children was found to be significantly greater than in adults Citation18, Citation20. Furthermore, CBF and beat pattern have been correlated with ultrastructural defects (isolated outer arm defects, isolated inner arm or radial spoke defects or transposition defects) and formed three distinct groups of dyskinetic patterns Citation21. However, patients with a transposition defect had cilia with normal CBF, but with an abnormal beat pattern. Therefore, demonstration of normal ciliary function (CBF and beat pattern) excludes diagnosis of PCD and further studies are usually not necessary. Presence of leucocytes should be noted, because inflammatory changes often result in secondary ciliary dysfunction giving rise to false positive test results Citation22–24. Therefore, the investigation should be performed in patients without current infections. Unfortunately this is often difficult to achieve, especially in children with PCD.
In patients with ciliary dysfunction the axonemal structure of respiratory cilia should be investigated by transmission electron microscopy. Typical ultrastructural defects in PCD consist of total or partial absence of dynein arms (∼80%, ), absence or dislocation of central tubules (∼10%), defects of radial spoke (∼6%) and peripheral microtubule anomalies Citation25. Less frequent anomalies include ciliary aplasia, basal apparatus alterations, axoneme‐less cilia, hockey stick cilia and long cilia. In about 3% of subjects with PCD no ultrastructural defects can be detected. In this case, diagnosis of PCD can only be established if dysmotility of cilia or spermatozoa by direct light microscopy is demonstrated. It is also important to know that some axonemal abnormalities like microtubular assembly defects, compound cilia and axonemal membrane blebs can be caused by secondary ciliary alterations due to respiratory infections, smoking or allergic inflammation Citation23. Secondary anomalies are absent after ciliogenesis in respiratory epithelial cell cultures of PCD subjects, while primary ultrastructural abnormalities are still expressed Citation26. Therefore, electron microscopic analysis or light microscopic evaluation of respiratory cilia in cultivated respiratory epithelial cells can help to distinguish between primary and secondary abnormalities. Unfortunately, only a few centers are able to perform these culture techniques and a significant amount of cell material is necessary (i.e. polyps removed after surgery) for such studies. Furthermore, not all primary ultrastructural defects can be easily visualized by electron microscopy. Due to low contrast, inner dynein arms are only difficult to pick up on transmission electron microscopy. Computer‐assisted analysis of conventional electron microscopy using 10 distinct cross‐section photographs improved inner arm visualization Citation27. However, in 14% of cases inner dynein arm morphology was still inconclusive. Another disadvantage of conventional electron microscopy is that many centers perform these studies only on material obtained by clamp biopsies which are more difficult to obtain then transnasal brushings.
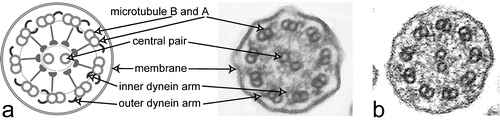
Figure 3 a. Schematic diagram of a cilium cross‐section (left). b. Electron micrograph of a cilium cross‐section from a healthy proband analyzed by transmission electron microscopy. Please note outer dynein arms are visibly attached at almost all nine peripheral doublets. In contrast, inner dynein arms are visible only on a few peripheral doublets (arrow). c. Transmission electron micrograph of a cross‐section from a respiratory cilium originating from a patient carrying DNAH5 mutations shows absence of outer dynein arms.
Nitric oxide (NO) influences various aspects of airway homeostasis and the regulation of ciliary motility and host defense Citation28, Citation29. NO is produced enzymatically from L‐arginine by several isoforms of nitric oxide synthases (NOS), including the inducible form (NOS 2) and constitutive NOS (NOS 1 or neuronal NOS, NOS 3 or endothelial NOS) Citation30. In patients with PCD nasal NO levels are often extremely low compared to controls Citation31–37. NO levels are even lower than in subjects with cystic fibrosis (CF) or other sinus disorders, although there is some overlap with CF Citation37, Citation38. It was also demonstrated that the very low nasal NO levels did not relate to inner or outer arm defects. Interestingly, healthy parents of subjects with PCD (obligate heterozygotes, expected to be carriers for mutations to be associated with PCD) had lower than expected nasal NO levels, suggesting that there is even a relationship between ‘carrier’ status and NO production Citation37. However, because not all PCD individuals show reduced nasal NO levels and other diseases can also result in low nasal NO levels, this method can only be used as a screening test for PCD Citation34, Citation39. Furthermore, the pathogenesis of low nasal NO in PCD is still unknown. It may be secondary to retention of NO caused by airway surface mucus or by impaired enzyme activity of the NOSs. Since retention cannot sufficiently explain reduced upper and lower airway NO Citation33, it seems more likely that the impaired NO production is in some way related to the underlying molecular pathology of the disease and that the production of NO is related to normal ciliary function Citation9.
Unfortunately, to date no easy test is available to establish diagnosis of PCD. Therefore diagnosis should be based on: 1) a typical clinical presentation suggestive for PCD, which includes recurrent infections of the respiratory tract with and without situs inversus, and 2) ciliary dyskinesia or sperm immotility demonstrated by direct visualization of respiratory cilia or sperms, or identification of specific defects of axonemal structures on electron microscopic examination Citation14. Preferably a functional and a morphological cilia defect should be demonstrated in PCD individuals. However, it should be kept in mind that in a subset of patients no ultrastructural defect is present. Direct visualization of ciliary function in PCD individuals is always abnormal, but should be repeated at least once to exclude secondary ciliary dyskinesia.
The difficulties of achieving a correct diagnosis in PCD explain the need for novel diagnostic tools. This prompted us to study respiratory epithelial cells by immunofluorescent microscopy. We recently demonstrated that diagnosis of outer dynein arm defects can be achieved by demonstration of DNAH5 mis‐localization in respiratory epithelial cells obtained by nasal brushing (). We refer to the last chapter of this review (New developments).
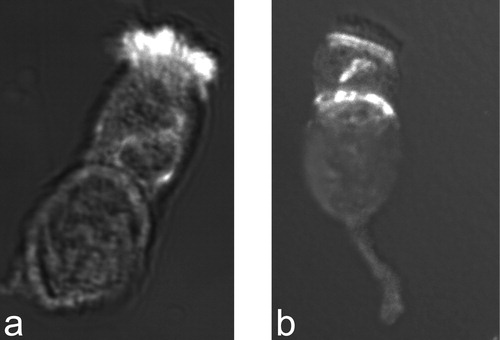
Figure 4 Staining of respiratory epithelial cells with an antibody directed against the axonemal dynein heavy chain DNAH5.a. Respiratory epithelial cell of a healthy proband shows positive staining (white, original red) for the entire length of all cilia. b. In a PCD patient with outer dynein arm defect documented by electron microscopy, no staining of the ciliary compartment is observed. However, there is DNAH5 accumulation in the cytoplasm and at the ciliary base (microtubules organizing centers).
Treatment
Early diagnosis and appropriate therapy might prevent permanent lung damage in individuals with PCD. Regular physiotherapy to maintain clearing of the airways is a very important aspect of the therapy. Bacterial infections need appropriate antibiotic therapy. Interestingly, very often H. influenzae infections are observed. In cases of obstructive lung disease bronchodilator therapy might become necessary. Surgical intervention in cases of chronic sinusitis or chronic otitis media should be considered with care, because of the high rate of spontaneous remissions in adulthood. In addition, insertion of grommets might result in permanent defects of the eardrums. In cases of permanent lung damage with chronic consolidating pneumonia and bronchiectasis, thoracic surgery might be indicated. Chronic respiratory insufficiency rarely develops and in these cases lung transplant might be a therapeutic option. Male infertility due to sperm immotility might be aided by assisted fertilization techniques such as intracytoplasmic sperm injection.
Motile and non‐motile cilia
Cilia are hair‐like extensions (axonemes) of the apical cell surface and contain a highly ordered basic structure of nine peripheral microtubule doublets. Each doublet is composed of an A and a B tubule (of 13 and 11 protofilaments each). In some cilia types (9+2 axoneme) a central pair of single microtubules is present in the center of the ring and extends the length of the axoneme Citation25. In other cilia types the axoneme lacks the central pair apparatus (9+0 axoneme) Citation40. These cilia have been called ‘primary cilia’. Primary cilia were considered to be immotile, but in contrast to this assumption it has been shown that primary cilia of the node rotate clockwise and are critical for left‐right axis determination Citation41. Single 9+0 cilia on a cell are called monocilia. Defects of monocilia are involved in the pathogenesis of various human disorders such as cystic kidney disease, tapeto‐retinal degeneration and liver fibrosis. Occasionally, PCD‐patients also present with cystic kidney disease and tapeto‐retinal degeneration Citation42–45. In this review we focus on the mechanisms that generate and control motility of cilia, because ciliary dysmotility is the key feature of PCD.
Within the axoneme of motile cilia, a number of multiprotein complexes interconnect the different components. Among these are radial spokes, nexin links, central sheath, and dynein arms. Outer and inner dynein arms are attached on the peripheral doublets (). These dynein arms are protein complexes composed of dynein heavy, intermediate and light chains. By means of ATP‐dependent reactions, the dynein arms enable the sliding of adjacent peripheral doublets to each other and generate the ciliary beat. Based on data obtained in Chlamydomonas mutants, outer dynein arms play a major role in generating the ciliary beat frequency, while the inner dynein arms predominantly influence the beat waveform. The other components, such as the central apparatus and radial spokes, provide the structural interface for transmitting regulatory signals to the arms.
Dyneins are large biological motors
Dyneins can be sub‐divided into axonemal and cytoplasmic dyneins. Axonemal dyneins move cilia and flagella, whereas cytoplasmic dyneins are involved in trafficking of vesicles in interphase cells, retrograde axonemal transport, and in organization of the spindle poles during mitosis Citation46. Both dynein protein complexes have a molecular mass greater than a million Dalton. Axonemal dyneins form two structures, inner and outer dynein arms, and are attached to the A microtubule of the nine outer doublets throughout the length of the axoneme, conducting the bending of the cilium or sperm tail.
Both axonemal and cytoplasmic dynein complexes have typically two, and in some cases three, large globular force‐producing heads Citation47. Dynein complexes contain a relatively small number of subunits, corresponding to each of four polypeptide classes: the heavy (HC, 400 to 500 kD), intermediate (IC, 55 to 110 kD), light intermediate (LIC) and light chains (LC, 8 to 45 kD). Heavy chains contain the motor domain responsible for the hydrodynamic coupling of nucleotide hydrolysis with the mechanical movement of dynein along the microtubules. The dynein heavy chain consists of approximately 4600 amino acid residues. The most highly divergent proportion of the heavy chain sequence is the N‐terminal ∼1300 residues. This portion of the protein forms the relatively short tail of dynein. Interactions within the N‐terminal domain are important for heavy chain dimerization Citation48. The dynein motor domain comprises six tandemly linked AAA (ATPases associated diverse cellular activities) modules Citation49. The four centrally located AAA modules include P‐loop motifs that are well conserved in all dynein sequences determined to date. The sequence of the first P‐loop is absolutely conserved and is the site of ATP (adenosine triphosphate) hydrolysis that leads to movement Citation50.
There is a high degree of sequence conservation of dyneins in Chlamydomonas and humans Citation51. In Chlamydomonas, outer dynein arms (ODAs) are spaced along the longitudinal axis of the axoneme with a 24 nm period. The inner dynein arm components (IDA) are arranged along the A tubule with a 96‐nm longitudinal repeat pattern. In Chlamydomonas one ODA complex is known, which contains the three heavy chains (α‐, β‐, and γ‐HC). In humans, at least two distinct ODA types are known. Interestingly, these ODA complexes show distinct localization pattern along the length of the ciliary axoneme. The demonstration that this regional distribution of ODA complexes differs between respiratory cilia and sperm flagella possibly indicates a role in cell‐type specific beat patterns Citation65. The inner dynein arms have a similar basic structure but they appear to be more diverse. In Chlamydomonas, there are several structurally distinct inner arms that contain different dynein isoforms known as I1, I2 and I3. This reflects their complex function in regulating the cilia wave form. The I1 inner dynein arm interacts with the dynein regulatory complex and the radial spokes. IDAs contain probably two HCs in both human and Chlamydomonas. These dimers and trimers of HCs are associated with various ICs and LCs. In humans, four different genes encode ODA‐HCs and seven to eleven genes probably encode IDA‐HCs, respectively Citation52. In addition, there is evidence for different HC isoforms being generated through alternative splicing. The number of genes encoding axonemal dynein ICs and LCs has not been determined precisely yet in mammals.
Several dysmotile strains of Chlamydomonas have been reported, which exhibit ultrastructural defects resembling those of human PCD patients Citation53. By analysis of these strains, mutations have been identified in many different genes encoding axonemal dyneins, including light, intermediate, and heavy chains, which cause PCD in ChlamydomonasCitation54. Mutations affecting the α‐heavy chain of the outer dynein arm cause a reduction in swimming velocity, but have no influence on the assembly of the outer arm. In contrast, mutations of the β‐ and γ‐ heavy chain genes decrease the beat frequency and cause absence of outer dynein arms Citation12. Chlamydomonas mutants lacking some inner arms have nearly normal beat frequency, but the amplitude of the flagella beat appears to be altered Citation55.
Genes involved in primary ciliary dyskinesia
Genome‐wide linkage analyses in European and North American families revealed extensive locus heterogeneity and failed to identify a major gene locus responsible for PCD Citation2, Citation52. Therefore, genes encoding components of the cilium appeared to be interesting candidates. However, there are at least 250 proteins within a single cilium, each encoded by a separate gene. In addition many other genes are involved in the assembly and regulation of the cilium. Mainly studies in Chlamydomonas and Caenorrhabditis elegans have provided significant insights into the identity of components required for cilia/flagella assembly and motility.
To identify responsible loci and genes involved in the pathogenesis of PCD candidate gene and homozygosity mapping strategies were successfully applied Citation56, Citation57. To date several PCD loci on chromosomes 19q13.3‐qter, 9p13‐p21, 7p21 and 5p15‐p14 have been identified Citation9, Citation56–58. However so far, mutations only in three genes encoding dynein chains located on chromosomes 9p13‐21, 5p15‐14 and 7p15.3‐21 have been identified Citation57–59. In addition, an X‐linked variant of PCD with associated retinitis pigmentosa caused by RPGR mutations has been described Citation60, Citation61.
DNAI1 (chromosome 9p)
The DNAI1 gene maps to chromosome 9p13‐p21 and encodes a 699 amino acid residues protein exhibiting high homologies to the Chlamydomonas IC78 dynein chain, which is a component of the outer dynein arm. DNAI1 is composed of 20 exons. Northern blot analysis identified a specific transcript in trachea and testis Citation57, which is compatible with expression in respiratory epithelium and sperm cells. DNAI1 mutations cause PCD with outer dynein arm defects, randomization of left/right body asymmetry, and male infertility Citation57, Citation62. Rarely germline mutations of the DNAI1 gene have been reported Citation63. Interestingly, so far three groups independently reported the 219+3insT mutation in intron 1, which might be explained either by a mutational hot‐spot or a founder effect.
DNAH5 (chromosome 5p)
The PCD locus on the short arm of chromosome 5 was identified by a homozygosity mapping strategy in one large Arab family with PCD and absence of outer dynein arms Citation56. Subsequently the responsible gene DNAH5 was identified. The DNAH5 genomic region comprises 79 exons and one alternative first exon, and spans 250kb. The DNAH5 gene is highly orthologous to the Clamydomonas reinhardtii gene encoding the γ‐axonemal heavy chain of the outer dynein arm. The proteins show a high degree of sequence conservation (40% identity, 59% similarity). Moreover, mutations in the Chlamydomonas orthologue are suspected to result in slow swimming algae with a phenotype highly similar on the ultrastructural level Citation53. Olbrich et al. reported four homozygous mutations and six heterozygous mutations within the coding region of the DNAH5 gene, which were identified by sequencing all 80 DNAH5 exons Citation59. Seven nucleotide changes introduced a premature stop codon, resulting in peptides lacking the motor domains and microtubule binding domains. In addition two missense mutations in conserved protein domains and one splice‐site mutation were found. These mutations were associated with partial or total loss of ODA and with left‐right asymmetry anomalies.
The analysis of respiratory cilia in three families carrying homozygous DNAH5 mutations indicated a genotype‐phenotype correlation Citation64. Mutations causing premature translational termination of DNAH5 (1855NfsX5, 2814fsX1) resulted in a complete absence of all outer dynein arms (ODA) in respiratory cilia, whereas a splice site mutation did not cause total absence of ODA. Semiquantitatively assessed cilia of affected subjects revealed shortened stubby ODA compared to controls. Furthermore 54% of the ODA were less than half the average length of ODA in normal subjects, indicating a partial ODA deficiency.
In summary, DNAI1 and DNAH5 are important PCD genes, that if mutated cause ultrastructural defects resulting in partial or total loss of the outer dynein arm. These defects can now also be demonstrated by immunofluorescent studies ().
DNAH11 (chromosome 7p)
DNAH11, encoding the axonemal heavy chain dynein 11, has also been proposed as a candidate gene for PCD Citation58. The gene comprises 82 exons, extending over 353kb of genomic sequence on chromosome 7p15.3‐21. The orthologous mouse gene Dnah11 (lrd) has already been described in the iv/iv (inversus viscerum) mouse model, where a missense mutation causes situs inversus and immotile cilia of the embryonic node. Dnah11 is expressed in the node at day 7.5 dpc and disruption of the mouse Dnah11 (lrd) results in randomization of the left‐right axis. Mutational analysis by direct sequencing revealed in one patient with uniparental isodisomy and cystic fibrosis a homozygous non‐sense mutation (c.8554C‐>T; R2852X) in exon 52. The mutant protein is predicted not to be able to apply force to the adjacent microtubule doublets, because of the absence of the microtobule binding domain. The patient exhibited situs inversus and a severe respiratory phenotype. Electron microscopy revealed normal respiratory cilia axonemes without any dynein arm defects. Thus the respiratory phenotype in this patient might be related to the CFTR mutation (cystic fibrosis) and situs inversus due to the DNAH11 mutation. Sequence analysis in additional PCD sibships revealed several sequence variants but no other DNAH11 mutations.
RPGR (X chromosome)
RPGR localizes to the X chromosome. 15%–20% of retinitis pigmentosa cases are caused by RPGR mutations. Affected individuals are usually males, who inherit RPGR mutations by their mothers. Rarely typical symptoms of PCD were reported in males with retinitis pigmentosa carrying RPGR mutations Citation60, Citation61, Citation65, Citation66. Diagnosis of PCD could be confirmed by detection of ultrastructural defects of respiratory cilia. Some affected individuals had additional sensory hearing deficits.
Other candidate genes
Several other candidate genes involved in cilia function have been studied. The inner dynein arm heavy chain 7 (DNAH7) shares high homologies with the Drosophila inner arm chain Dhc36c and inner arm chains from ChlamydomonasCitation67. DNAH7 is not expressed in undifferentiated human bronchial epithelial cells, but is strongly induced in differentiated ciliated cells. In cilia from a studied PCD patient DNAH7 was undetectable, whereas intracellular DNAH7 was present. Zhang et al. concluded that DNAH7 is synthesized in respiratory cells but not assembled in cilia of the studied PCD patient Citation67. Therefore, they considered DNAH7 to be an interesting candidate gene for inner arm defects. However, they failed to identify mutations in DNAH7.
Other genes encoding for axonemal dyneins, such as the heavy chain DNAH9, the intermediate chain DNAI2, and the light chains LC8 and TCTE3, were so far also excluded as a major cause for PCD by candidate gene analyses Citation68–70.
Besides dynein defects, the absence of central complexes of cilia have been described. Therefore, hpF20, has been considered as a candidate gene, as defects of the Chlamydomonas pf20 protein induce paralyzed flagella lacking the central complex. Hpf20 is located on chromosome 2q34 and comprises 16 exons. Until now, no disease‐causing mutations were identified in five PCD subjects with central complex alterations Citation71.
Other candidate genes for PCD include transcription factors that regulate dynein gene expression. FOXJ1 encodes such a factor, but has also been excluded as a candidate gene for PCD Citation72.
Models for PCD
To date only a few mouse models with known genetic defects and a phenotype affecting respiratory cilia function have been generated. Axonemal dynein heavy chain 5 (Mdnah5)‐deficient mice exhibit respiratory cilia immotility, random left‐right axis determination, chronic airway disease and hydrocephalus formation. Affected mice develop early chronic rhinitis. Outer dynein arms of respiratory cilia are completely absent in mutant mice Citation73. In situ hybridization experiments identified cell‐specific expression in ciliated cells of the upper and lower airways, and the ventral node from 7 days post coitum (dpc) to 8.5 dpc during embryonic development Citation59, Citation64. A detailed analysis of wild‐type mice showed that ependymal cells lining the brain ventricles and the aqueduct carry motile cilia, which produce an ‘ependymal flow’ of cerebro‐spinal fluid. This ependymal flow is apparently critical to maintain patency of the aqueductal region during late brain development. In Mdnah5‐deficient mice ependymal cilia show ODA alterations and severe ependymal cilia dysfunction, which results in absence of ependymal flow. As a consequence Mdnah5‐deficient mice develop aqueduct closure with triventricular hydrocephalus formation during late (postnatal) brain development. Interestingly, the risk for triventricular hydrocephalus due to aqueduct closure is also increased in PCD patients, which indicates that this disease mechanism also plays a role for human hydrocephalus formation Citation73.
In contrast, targeted mutations of Mdhc7, which encodes for an inner dynein arm heavy chain result in reduced sperm motility with male infertility and only a mild respiratory phenotype. Mdhc7 deficient mice show a 50% reduction of respiratory ciliary beat frequency. However, this does not cause any apparent airway disease in affected mice Citation12.
Because Mdnah5 deficient mice die early (∼4–5 weeks) due to hydrocephalus formation and Mdhc7 deficient mice do not exhibit a respiratory PCD phenotype, to date no good animal model to study primary ciliary dyskinesia in mice is available.
New developments
Identification of genes involved in the pathogenesis of PCD facilitates development of novel diagnostic tools. High‐resolution immunofluorescent studies demonstrated in PCD patients with outer dynein arm defects and known DNAH5 and DNAI1 mutations mis‐localization of the axonemal chain DNAH5 Citation74. The protein accumulated in the microtubules organizing centers (MTOC), but was absent throughout the whole or distal parts of the cilia axonemes (). The advantage of this tool is that respiratory cells obtained by transnasal brushings, a non‐invasive method, can be stained and also used for functional analysis of ciliary beat frequency and pattern. In a large cohort of subjects including healthy controls, CF‐patients and PCD affected, blinded investigation of transnasal brushing material could reliably detect outer dynein arm defects in all individuals. Healthy controls and cystic fibrosis patients showed normal staining pattern. These findings indicate that this novel tool based on immunofluorescent microscopy can be applied in diagnosis of PCD in order to detect all outer dynein arm defects (regardless of mutational status), which represent the most common ultrastructural defects. Current research focuses on generation of additional antibodies, which should enable diagnosis of additional ultrastructural defects. An antibody for an inner dynein arm component will be soon available (personal data).
Linkage analyses in informative families (consanguineous parents or several affected family members) have largely facilitated PCD research. Therefore, recruitment of informative PCD families for future genetic analyses is very important (please contact corresponding author). Identification of additional genes involved in cilia function will provide new insights into the molecular mechanisms and help to develop novel techniques to diagnose subjects with PCD with ultrastructural defects other than outer arm defects. Until now, diagnosis of PCD has relied on a combination of clinical history evaluation, analysis of ciliary structure/function, and in some cases genotyping. New and easy‐to‐perform techniques may aid early diagnosis and start of treatment, which will hopefully result in reduction of chronic lung injury such as bronchiectasis in affected patients.
Acknowledgements
We are grateful to the German patient support group ‘Kartagener Syndrom und Primäre Ciliäre Dyskinesie e.V.’. H.O. is supported from the German Research Foundation DFG Om 6/2 and the SFB 592, Michael‐Wagner‐Stiftung ‘Kinderlachen’, and by the Zentrum Klinische Forschung, Freiburg. We very much appreciate the help of Heike Olbrich for the preparation of figures. In addition we would like to thank all members of the scientific adivisory board of the German PCD support group in particular Peter Ahrens, Stephan Illing, Thomas Nüsslein, and Klaus Seithe for valuable discussions.
References
- Afzelius B. A. A human syndrome caused by immotile cilia. Science 1976; 193: 317–9
- Blouin J. L., Meeks M., Radhakrishna U., Sainsbury A., Gehring C., Sail G. D. Primary ciliary dyskinesia: a genome‐wide linkage analysis reveals extensive locus heterogeneity. Eur J Hum Genet 2000; 8: 109–18
- Afzelius B. A. Genetics and pulmonary medicine. 6. Immotile cilia syndrome: past, present, and prospects for the future. Thorax 1998; 53: 894–7
- Schidlow D. V. Primary ciliary dyskinesia (the immotile cilia syndrome). Ann Allergy 1994; 73: 457–68, quiz 468–70
- Kartagener M., Stucki P. Bronchiectasis with situs inversus. Arch Pediatr 1962; 79: 193–207
- Narayan D., Krishnan S. N., Upender M., Ravikumar T. S., Mahoney M. J., Dolan T. F Jr. Unusual inheritance of primary ciliary dyskinesia (Kartagener's syndrome). J Med Genet 1994; 31: 493–6
- Krawczynski M. R., Witt M. PCD and RP: X‐linked inheritance of both disorders?. Pediatr Pulmonol 2004; 38: 88–9
- Bush A., Cole P., Hariri M., Mackay I., Phillips G., O'Callaghan C. Primary ciliary dyskinesia: diagnosis and standards of care. Eur Respir J 1998; 12: 982–8
- Meeks M., Bush A. Primary ciliary dyskinesia (PCD). Pediatr Pulmonol 2000; 29: 307–16
- Afzelius B. A., Eliasson R. Male and female infertility problems in the immotile‐cilia syndrome. Eur J Respir Dis Suppl 1983; 127: 144–7
- Munro N. C., Currie D. C., Lindsay K. S., Ryder T. A., Rutman A., Dewar A. Fertility in men with primary ciliary dyskinesia presenting with respiratory infection. Thorax 1994; 49: 684–7
- Neesen J., Kirschner R., Ochs M., Schmiedl A., Habermann B., Mueller C. Disruption of an inner arm dynein heavy chain gene results in asthenozoospermia and reduced ciliary beat frequency. Hum Mol Genet 2001; 10: 1117–28
- Sapiro R., Kostetskii I., Olds‐Clarke P., Gerton G. L., Radice G. L., Strauss I. J. Male infertility, impaired sperm motility, and hydrocephalus in mice deficient in sperm‐associated antigen 6. Mol Cell Biol 2002; 22: 6298–305
- Afzelius B. A. Immotile Cilia Syndrome (Primary Ciliary Dyskinesia) Including Kartagener Syndrome. The Metabolic and molecular bases of inherited disease, C. R Scriver, W. S Sly. McGraw‐Hill, Inc, New York 1995
- Canciani M., Barlocco E. G., Mastella G., de Santi M. M., Gardi C., Lungarella G. The saccharin method for testing mucociliary function in patients suspected of having primary ciliary dyskinesia. Pediatr Pulmonol 1988; 5: 210–4
- Rutland J., Griffin W., Cole P. Nasal brushing and measurement of ciliary beat frequency. An in vitro method for evaluating pharmacologic effects on human cilia. Chest 1981; 80((6 Suppl))865–7
- MacCormick J., Robb I., Kovesi T., Carpenter B. Optimal biopsy techniques in the diagnosis of primary ciliary dyskinesia. J Otolaryngol 2002; 31: 13–7
- Chilvers M. A., Rutman A., O'Callaghan C. Functional analysis of cilia and ciliated epithelial ultrastructure in healthy children and young adults. Thorax 2003; 58: 333–8
- Jorissen M., Willems T., Van der Schueren B. Nasal ciliary beat frequency is age independent. Laryngoscope 1998; 108: 1042–7
- O'Callaghan C., Smith K., Wilkinson M., Morgan D., Priftis K. Ciliary beat frequency in newborn infants. Arch Dis Child 1991; 66: 443–4
- Chilvers M. A., Rutman A., O'Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J Allergy Clin Immunol 2003; 112: 518–24
- Jorissen M., Willems T., Van der Schueren B., Verbeken E. Secondary ciliary dyskinesia is absent after ciliogenesis in culture. Acta Otorhinolaryngol Belg 2000; 54: 333–42
- Afzelius B. A. Genetical and ultrastructural aspects of the immotile‐cilia syndrome. Am J Hum Genet 1981; 33: 852–64
- Rutland J., Cole P. J. Nasal mucociliary clearance and ciliary beat frequency in cystic fibrosis compared with sinusitis and bronchiectasis. Thorax 1981; 36: 654–8
- Ibanez‐Tallon I., Heintz N., Omran H. To beat or not to beat: roles of cilia in development and disease. Hum Mol Genet 2003; 12 Spec No 1: R27–35
- Jorissen M., Willems T., Van der Schueren B., Verbeken E., De Boeck K. Ultrastructural expression of primary ciliary dyskinesia after ciliogenesis in culture. Acta Otorhinolaryngol Belg 2000; 54: 343–56
- Escudier E., Couprie M., Duriez B., Roudot‐Thoraval F., Millepied M. C., Pruliere‐Escabasse V. Computer‐assisted analysis helps detect inner dynein arm abnormalities. Am J Respir Crit Care Med 2002; 166: 1257–62
- Hibbs J. B, Jr. Synthesis of nitric oxide from L‐arginine: a recently discovered pathway induced by cytokines with antitumour and antimicrobial activity. Res Immunol 1991; 142: 565–9, discussion 596–8
- Jain B., Rubinstein I., Robbins R. A., Leise K. L., Sisson J. H. Modulation of airway epithelial cell ciliary beat frequency by nitric oxide. Biochem Biophys Res Commun 1993; 191: 83–8
- Zapol W. M., Rimar S., Gillis N., Marletta M., Bosken C. H. Nitric oxide and the lung. Am J Respir Crit Care Med 1994; 149: 1375–80
- Lundberg J. O., Rinder J., Weitzberg E., Lundberg J. M., Alving K. Nasally exhaled nitric oxide in humans originates mainly in the paranasal sinuses. Acta Physiol Scand 1994; 152: 431–2
- Loukides S., Kharitonov S., Wodehouse T., Cole P. J., Barnes P. J. Effect of arginine on mucociliary function in primary ciliary dyskinesia. Lancet 1998; 352: 371–2
- Grasemann H., Gartig S. S., Wiesemann H. G., Teschler H., Konietzko N., Ratjen F. Effect of L‐arginine infusion on airway NO in cystic fibrosis and primary ciliary dyskinesia syndrome. Eur Respir J 1999; 13: 114–8
- Karadag B., James A. J., Gultekin E., Wilson N. M., Bush A. Nasal and lower airway level of nitric oxide in children with primary ciliary dyskinesia. Eur Respir J 1999; 13: 1402–5
- Wodehouse T., Kharitonov S. A., Mackay I. S., Barnes P. J., Wilson R., Cole P. J. Nasal nitric oxide measurements for the screening of primary ciliary dyskinesia. Eur Respir J 2003; 21: 43–7
- Baraldi E., Pasquale M. F., Cangiotti A. M., Zanconato S., Zacchello F. Nasal nitric oxide is low early in life: case study of two infants with primary ciliary dyskinesia. Eur Respir J 2004; 24: 881–3
- Noone P. G., Leigh M. W., Sannuti A., Minnix S. L., Carson J. L., Hazucha M. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med 2004; 169: 459–67
- Deja M., Busch T., Bachmann S., Riskowski K., Campean V., Wiedmann B. Reduced nitric oxide in sinus epithelium of patients with radiologic maxillary sinusitis and sepsis. Am J Respir Crit Care Med 2003; 168: 281–6
- Corbelli R., Bringolf‐Isler B., Amacher A., Sasse B., Spycher M., Hammer J. Nasal nitric oxide measurements to screen children for primary ciliary dyskinesia. Chest 2004; 126: 1054–9
- Wheatley D. N., Wang A. M., Strugnell G. E. Expression of primary cilia in mammalian cells. Cell Biol Int 1996; 20: 73–81
- Nonaka S., Tanaka Y., Okada Y., Takeda S., Harada A., Kanai Y. Randomization of left‐right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell 1998; 95: 829–37
- Svedbergh B., Jonsson V., Afzelius B. Immotile‐cilia syndrome and the cilia of the eye. Albrecht Von Graefes Arch Klin Exp Ophthalmol 1981; 215: 265–72
- Bonneau D., Raymond F., Kremer C., Klossek J. M., Kaplan J., Patte F. Usher syndrome type I associated with bronchiectasis and immotile nasal cilia in two brothers. J Med Genet 1993; 30: 253–4
- Ohga H., Suzuki T., Fujiwara H., Furutani A., Koga H. [A case of immotile cilia syndrome accompanied by retinitis pigmentosa]. Nippon Ganka Gakkai Zasshi 1991; 95: 795–801
- Segal P., Kikiela M., Mrzyglod S., Zeromska‐Zbierska I. Kartagener's syndrome with familial eye changes. Am J Ophthalmol 1963; 55: 1043–9
- Holzbaur E. L., Vallee R. B. DYNEINS: molecular structure and cellular function. Annu Rev Cell Biol 1994; 10: 339–72
- Vallee R. B., Gee M. A. Make room for dynein. Trends Cell Biol 1998; 8: 490–4
- Habura A., Tikhonenko I., Chisholm R. L., Koonce M. P. Interaction mapping of a dynein heavy chain. Identification of dimerization and intermediate‐chain binding domains. J Biol Chem 1999; 274: 15447–53
- Asai D. J., Koonce M. P. The dynein heavy chain: structure, mechanics and evolution. Trends Cell Biol 2001; 11: 196–202
- Gibbons I. R., Lee‐Eiford A., Mocz G., Phillipson C. A., Tang W. J., Gibbons B. H. Photosensitized cleavage of dynein heavy chains. Cleavage at the “V1 site” by irradiation at 365 nm in the presence of ATP and vanadate. J Biol Chem 1987; 262: 2780–6
- Silflow C. D., Lefebvre P. A. Assembly and motility of eukaryotic cilia and flagella. Lessons from Chlamydomonas reinhardtii. Plant Physiol 2001; 127: 1500–7
- Maiti A. K., Mattei M. G., Jorissen M., Volz A., Zeigler A., Bouvagnet P. Identification, tissue specific expression, and chromosomal localisation of several human dynein heavy chain genes. Eur J Hum Genet 2000; 8: 923–32
- Wilkerson C. G., King S. M., Witman G. B. Molecular analysis of the gamma heavy chain of Chlamydomonas flagellar outer‐arm dynein. J Cell Sci 1994; 107: 497–506
- Kamiya R. Exploring the function of inner and outer dynein arms with Chlamydomonas mutants. Cell Motil Cytoskeleton 1995; 32: 98–102
- Brokaw C. J., Kamiya R. Bending patterns of Chlamydomonas flagella: IV. Mutants with defects in inner and outer dynein arms indicate differences in dynein arm function. Cell Motil Cytoskeleton 1987; 8: 68–75
- Omran H., Haffner K., Volkel A., Kuehr J., Ketelsen U. P., Ross U. H. Homozygosity mapping of a gene locus for primary ciliary dyskinesia on chromosome 5p and identification of the heavy dynein chain DNAH5 as a candidate gene. Am J Respir Cell Mol Biol 2000; 23: 696–702
- Pennarun G., Escudier E., Chapelin C., Bridoux A. M., Cacheux V., Roger G. Loss‐of‐function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am J Hum Genet 1999; 65: 1508–19
- Bartoloni L., Blouin J. L., Pan Y., Gehrig C., Maiti A. K., Scamuffa N. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci U S A 2002; 99: 10282–6
- Olbrich H., Haffner K., Kispert A., Volkel A., Volz A., Sasmaz G. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left‐right asymmetry. Nat Genet 2002; 30: 143–4
- van Dorp D. B., Wright A. F., Carothers A. D., Bleeker‐Wagemakers E. M. A family with RP3 type of X‐linked retinitis pigmentosa: an association with ciliary abnormalities. Hum Genet 1992; 88: 331–4
- Dry K. L., Manson F. D., Lennon A., Bergen A. A., Van Dorp D. B., Wright A. F. Identification of a 5' splice site mutation in the RPGR gene in a family with X‐linked retinitis pigmentosa (RP3). Hum Mutat 1999; 13: 141–5
- Guichard C., Harricane M. C., Lafitte J. J., Godard P., Zaegel M., Tack V. Axonemal dynein intermediate‐chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (Kartagener syndrome). Am J Hum Genet 2001; 68: 1030–5
- Zariwala M., Noone P. G., Sannuti A., Minnix S., Zhou Z., Leigh M. W. Germline mutations in an intermediate chain dynein cause primary ciliary dyskinesia. Am J Respir Cell Mol Biol 2001; 25: 577–83
- Kispert A., Petry M., Olbrich H., Volz A., Ketelsen U. P., Horvath J. Genotype‐phenotype correlations in PCD patients carrying DNAH5 mutations. Thorax 2003; 58: 552–4
- Iannaccone A., Breuer D. K., Wang X. F., Kuo S. F., Normando E. M., Filippova E. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J Med Genet 2003; 40: e118
- Zito I., Downes S. M., Patel R. J., Cheetham M. E., Ebenezer N. D., Jenkins S. A. RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J Med Genet 2003; 40: 609–15
- Zhang Y. J., O'Neal W. K., Randell S. H., Blackburn K., Moyer M. B., Boucher R. C. Identification of dynein heavy chain 7 as an inner arm component of human cilia that is synthesized but not assembled in a case of primary ciliary dyskinesia. J Biol Chem 2002; 277: 17906–15
- Bartoloni L., Blouin J. L., Maiti A. K., Sainsbury A., Rossier C., Gehrig C. Axonemal beta heavy chain dynein DNAH9: cDNA sequence, genomic structure, and investigation of its role in primary ciliary dyskinesia. Genomics 2001; 72: 21–33
- Pennarun G., Chapelin C., Escudier E., Bridoux A. M., Dastot F., Cacheux V. The human dynein intermediate chain 2 gene (DNAI2): cloning, mapping, expression pattern, and evaluation as a candidate for primary ciliary dyskinesia. Hum Genet 2000; 107: 642–9
- Neesen J., Drenckhahn J. D., Tiede S., Burfeind P., Grzmil M., Konietzko J. Identification of the human ortholog of the t‐complex‐encoded protein TCTE3 and evaluation as a candidate gene for primary ciliary dyskinesia. Cytogenet Genome Res 2002; 98: 38–44
- Pennarun G., Bridoux A. M., Escudier E., Dastot‐Le Moal F., Cacheux V., Amselem S. Isolation and expression of the human hPF20 gene orthologous to Chlamydomonas PF20: evaluation as a candidate for axonemal defects of respiratory cilia and sperm flagella. Am J Respir Cell Mol Biol 2002; 26: 362–70
- Maiti A. K., Bartoloni L., Mitchison H. M., Meeks M., Chung E., Spiden S. No deleterious mutations in the FOXJ1 (alias HFH‐4) gene in patients with primary ciliary dyskinesia (PCD). Cytogenet Cell Genet 2000; 90: 119–22
- Ibanez‐Tallon I., Gorokhova S., Heintz N. Loss of function of axonemal dynein Mdnah5 causes primary ciliary dyskinesia and hydrocephalus. Hum Mol Genet 2002; 11: 715–21
- Fliegauf M., Olbrich H., Horvath J., Wildhaber J. H., Zariwala M. A., Kennedy M. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am J Respir Crit Care Med 2005; 171: 1343–9