Abstract
Familial combined hyperlipidemia (FCHL) constitutes a substantial risk factor for atherosclerosis since it is observed in about 20% of coronary heart disease (CHD) patients under 60 years. FCHL, characterized by elevated levels of total cholesterol (TC) and triglycerides (TGs), or both, is also one of the most common familial hyperlipidemias with a prevalence of 1%–6% in Western populations. Numerous studies have been performed to identify genes contributing to FCHL. The recent linkage and association studies and their replications are beginning to elucidate the genetic variations underlying the susceptibility to FCHL. Three chromosomal regions on 1q21–23, 11p and 16q22–24.1 have been replicated in different study samples, offering targets for gene hunting. In addition, several candidate gene studies have replicated the influence of the lipoprotein lipase (LPL) gene and apolipoprotein A1/C3/A4/A5 (APOA1/C3/A4/A5) gene cluster in FCHL. Recently, the linked region on chromosome 1q21 was successfully fine‐mapped and the upstream transcription factor 1 (USF1) gene identified as the underlying gene for FCHL. This finding has now been replicated in independent FCHL samples. However, the total number of variants, the risk related to each variant and their relative contributions to the disease susceptibility are not known yet.
| Abbreviations | ||
| APOA1 | = | apolipoprotein A1 |
| APOA2 | = | apolipoprotein A2 |
| APOA5 | = | apolipoprotein A5 |
| APOB | = | apolipoprotein B |
| APOC3 | = | apolipoprotein C3 |
| APOE | = | apolipoprotein E |
| APOA1/C3/A4/A5 | = | apolipoprotein A1/C3/A4/A5 |
| CHD | = | coronary heart disease |
| FCHL | = | familial combined hyperlipidemia |
| HDL‐C | = | high‐density lipoprotein cholesterol |
| HNF4A | = | hepatic nuclear factor 4 alpha |
| LD | = | linkage disequilibrium |
| LDL‐C | = | low‐density lipoprotein cholesterol |
| LEPR | = | leptin receptor |
| LIPC | = | hepatic lipase |
| LPL | = | lipoprotein lipase |
| SNP | = | single nucleotide polymorphism |
| TC | = | total cholesterol |
| TG | = | triglycerides |
| TNFRSF1B | = | tumor necrosis factor receptor superfamily, member 1B |
| TXNIP | = | thioredoxin interacting protein |
| T2DM | = | type 2 diabetes mellitus |
| USF1 | = | upstream transcription factor 1 |
Introduction
Familial combined hyperlipidemia (FCHL), first described among young survivors of myocardial infarction in 1973 Citation1–3, is the most common familial dyslipidemia predisposing to coronary heart disease (CHD). FCHL is a common disease with an estimated population prevalence of 1%–6% Citation3,4. Elevated levels of serum total cholesterol (TC), triglycerides (TGs), or both characterize the FCHL disorder. FCHL also features low levels of high‐density lipoprotein cholesterol (HDL‐C), elevated levels of serum apolipoprotein B (apoB), and glucose intolerance as component traits Citation2,3,Citation5.
It has been evident for 30 years that FCHL has a strong genetic component Citation2,3,Citation6. The genetic component in FCHL has been suggested by familial aggregation of dyslipidemia Citation2 and in fact, FCHL was originally suggested to be inherited as an autosomal dominant disorder due to the vertical transmission pattern Citation1. A more complex polygenic background is, however, likely, as suggested by metabolic Citation7 and segregation studies Citation8,9. Furthermore, whole‐genome searches and candidate gene studies performed in FCHL families originating from different populations have identified several putative loci for FCHL Citation10–16. The recent findings and replications of the linkage and association studies are beginning to identify the DNA sequence variations contributing to FCHL. Fine‐mapping of one of the linked regions recently resulted in the characterization of the first positionally cloned gene for FCHL, upstream transcription factor 1 (USF1) Citation17. Since USF1 is a transcription factor known to regulate the expression of a number of genes participating in glucose and lipid metabolism, it provides an excellent candidate for FCHL. A recent study also indicates that common variants and haplotypes in the hepatic nuclear factor 4 alpha (HNF4A) gene are associated with high serum lipid levels and the metabolic syndrome in FCHL families Citation18. Interestingly, cooperative effects of USF1 and HNF4A have been suggested to control the regulation of multiple genes, including apolipoprotein A2 (APOA2) and apolipoprotein C3 (APOC3) Citation19–21. Interactions between DNA sequence variants are likely to contribute to the complex pathogenic mechanism(s) of common cardiovascular traits, raising the possibility that variants in USF1, HNF4A and apolipoproteins may interactively confer susceptibility to FCHL. To summarize, these accumulating data indicate that multiple variants contribute to the susceptibility to FCHL.
Both rare and common variants have been suggested to contribute for example to low plasma levels of HDL‐C in the general population Citation22,23. However, the extent to which each group influences this susceptibility is not known. It is thus likely that multiple DNA sequence variants, both common and rare, underlie the genetic susceptibility to FCHL. In addition to the human DNA sequence and variation data produced by the Human Genome Project and the HapMap Project, recent advances in genotyping technologies and statistical approaches should enable an accelerated investigation of the sequence variations at the genomic level to identify all the variants involved in the disease susceptibility of FCHL.
Key messages
Familial combined hyperlipidemia (FCHL) is a common complex disorder with both genetic and environmental factors affecting the disease susceptibility.
Most likely the genetic susceptibility to FCHL is determined by multiple DNA sequence variants and their interactions.
This review focuses on the recent findings that are beginning to elucidate the FCHL susceptibility genes, including the upstream transcription factor 1 (USF1) and lipoprotein lipase (LPL) genes, as well as the apolipoprotein A1/C3/A4/A5 (APOA1/C3/A4/A5) gene cluster.
Genes conferring the susceptibility to FCHL
USF1 identified as the gene underlying the linkage signal on chromosome 1q21
Previously a locus for FCHL was identified on human chromosome 1q21–q23 in FCHL families originating from the genetically relatively isolated population of Finland Citation10. Since then, this finding has been replicated in several FCHL samples, originating from other, more heterogeneous populations Citation15,Citation24–26. Linkage has been observed to FCHL, as well as to several FCHL component traits, including TG, TC and apoB levels Citation10,Citation15,Citation25,26. On 1q21 no significant evidence of genetic heterogeneity was observed in the Finns Citation17. However, in Mexican, German and Chinese samples the proportion of families contributing to linkage ranged from 22% to 71% Citation24,Citation26. Interestingly, the same markers in the 1q21 region have also been linked to type 2 diabetes mellitus (T2DM) in numerous studies Citation27–33. Most recently, 1q23–31 was also shown to be linked to the metabolic syndrome Citation34. The evidence for linkage obtained for 1q21 has varied in these FCHL and T2DM studies, most likely reflecting the underlying genetic heterogeneity, as well as population‐based and diagnostic differences. Interestingly, many of the critical metabolic features of FCHL, e.g. hypertriglyceridemia and insulin resistance, also represent trait components of T2DM. Taken together, these linkage findings suggest that one or more genes in this particular chromosomal region predispose to both FCHL and T2DM, two clinical phenotypes with overlapping component traits and shared diagnostic features.
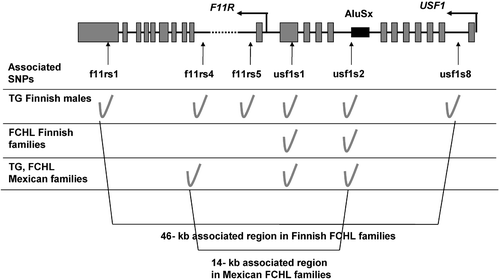
The upstream transcription factor 1 (USF1) was recently linked and associated with FCHL in 60 extended Finnish FCHL families with 721 genotyped individuals (P = 0.00002) Citation17. The evidence for association was strongest among males for high serum TGs (P = 0.0000009) and extended to a ∼46‐kb region, containing also the adjacent F11 receptor (F11R) gene Citation17 (). An association was also observed for apoB, TC and the LDL peak particle size Citation17. The known functions of F11R, related to regulation of tight junction assembly in epithelia, pathogenesis of viral infections, and transendothelial migration of certain T cell types Citation35–37, make it a less likely candidate gene for FCHL than USF1. USF1 is a ubiquitously expressed transcription factor of the basic helix‐loop‐helix leucine zipper family. It forms homo‐ and heterodimers (with USF2) and recognizes a CACGTG motif termed E box, resulting in activation of the gene transcription and enhanced expression in response to various stimuli such as glucose and dietary carbohydrates Citation38. USF1 regulates the expression of several genes participating in glucose and lipid metabolism, such as apolipoprotein C3 (APOC3), apolipoprotein A2 (APOA2), apolipoprotein A5 (APOA5), apolipoprotein E (APOE), hormone sensitive lipase (LIPE), hepatic lipase (LIPC), glucokinase (GCK), islet‐specific glucose‐6‐phosphatase catalytic‐subunit‐related protein (IGRP), insulin (INS), glucagon receptor (GCGR), ATP‐binding cassette transporter A1 (ABCA1), fatty acid synthase (FAS), acetyl‐CoA carboxylase alpha (ACACA) and plasminogen activator inhibitor‐1 (PAI1) Citation20,21,Citation38–51. A more complete list of the USF1 target genes is available in a recent paper Citation52.
Figure 1. Overview of the associatedUSF1 region in Finnish and Mexican familial combined hyperlipidemia (FCHL) families. The associated region was restricted by ∼70% (from 46 kb to 14 kb) using two different populations, the Finns and Mexicans Citation17,Citation26. The rs numbers of the single nucleotide polymorphisms (SNPs) are as follows: f11rs1 (rs836), f11rs4 (hCV1459766), f11rs5 (rs4339888), usf1s1 (rs3737787), usf1s2 (rs2073658), usf1s8 (rs2516838).
In the original study, genetic data were supported by preliminary functional data, as the USF1 risk haplotype had an effect on the expression profiles of fat biopsies Citation17. Expression profiles of fat biopsies of FCHL cases seemed to differ depending on their carrier status for the associated USF1 haplotype Citation17. A total of 25 genes were significantly upregulated and 73 genes downregulated in the susceptibility haplotype carriers Citation17. The upregulated genes belonged to functional classes mainly related to fat metabolism, and the downregulated genes included several functional classes related to immune response. Interestingly, the upregulated genes included the stearoyl‐CoA desaturase (SCD) gene Citation17. The SCD activity has been shown to be associated with plasma TG levels Citation53. The downregulated genes also included several known important atherosclerosis‐related genes, such as APOE, phospholipid transfer protein (PLTP), macrophage scavenger receptor 1 (MSP1), arachidonate 5‐lipoxygenase (ALOX5), and complement component 3a receptor 1 (C3AR1).
A recent study further demonstrated differential expression of known USF1 target genes, APOE, ABCA1 and angiotensinogen (AGT), between USF1 risk allele carriers and non‐carriers using a larger number of fat biopsies of FCHL and low HDL‐C‐affected patients (n = 19) Citation52. No differences in the characteristically low USF1 transcript levels were observed between carriers of the USF1 risk versus non‐risk alleles in the original Citation17 or in this subsequent study Citation52. However, it is possible that even a subtle difference may have significant effects on the expression of USF1 target genes.
Hoffstedt et al. studied the effect of USF1 variants on lipolysis in adipocytes Citation54. Lipolysis is a critical aspect of adipocyte function and manifests large differences between individuals potentially due to genetic differences. In this study including fat biopsies from 196 normolipidemic obese women, the usf1s2 (rs2073658) variant was associated with the maximum lipolytic action in response to stimulation by noradrenaline, as well as by β1‐, β2‐ and β3‐ adrenergic receptor agonists (dobutamine, terbutaline, CGP12177 and forskolin). Importantly, the carriers of the protective allele of the usf1s2 variant had a significantly increased maximum lipolytic activity in response to these drugs. This association between the USF1 variant and the maximum lipolytic activity was observed in normolipidemic women without any disease such as FCHL, CHD or T2DM. The result may imply that the defect caused by USF1 is present already before the clinical manifestation of the disease. Defect in USF1 may ultimately lead to disease when other genetic or environmental factors accumulate in the same individual.
Since the original finding in Finnish FCHL families, the association between the DNA sequence variations in USF1 and FCHL has been replicated in several study samples Citation26,Citation55–57 (). The first replication study including 24 extended multigenerational Mexican FCHL families, replicated the association, and moreover the associated region was restricted to 14 kb Citation26 versus more than 46 kb in the Finns Citation17 (). These data provides a shorter region with fewer variants available for functional analyses. No gender differences were observed in Mexican FCHL families Citation26. The most significantly associated single nucleotide polymorphisms (SNPs), usf1s1 (rs3737787) and usf1s2 (rs2073658), or SNPs in linkage disequilibrium (LD) with usf1s1/s2, were investigated in three other study samples ascertained for CHD or for family history of CHD Citation55–57. In extended Utah pedigrees with family members suffering early death due to CHD, early strokes, or early onset hypertension, the USF1 SNPs and haplotype were associated with FCHL, TG and low‐density lipoprotein cholesterol (LDL‐C) levels Citation57. In the European Atherosclerosis Research Study II, Putt et al. examined the lipid levels before and after meal and after oral glucose tolerance test Citation56. They found differences in the correlation between body mass index (BMI), fasting LDL‐C and glucose levels according to the USF1 genotypes. It is worth noting that as in Finns Citation17 the common allele of usf1s1 (rs3737787), usf1s2 (rs2073658) or SNPs in LD with usf1s1/s2 represents the associated allele in all of these studies, replicating the original study. It can be concluded that these SNPs seem to capture the disease‐associated signal, although their direct relationship to the functional defect contributing to FCHL pathogenesis is not known yet.
Table I. Summary of the genetic studies on the USF1 gene, the first gene identified for familial combined hyperlipidemia (FCHL) using positional cloning approach.
Most recently, Komulainen et al. investigated the role of USF1 variants and haplotypes as a risk factor for cardiovascular disease events at the population level Citation55 (). Importantly, they observed that female carriers of a USF1 risk allele had a two‐fold risk of a cardiovascular event and an increased risk of all‐cause mortality during the follow‐up period in a Finnish prospective population cohort consisting of 14,000 individuals, followed up for cardiovascular events in a period of 7–10 years Citation55.
Two different studies have focused on the contribution of USF1 SNPs to T2DM Citation58,59 (). In the first study by Ng et al., a significant association of USF1 polymorphisms with T2DM and the metabolic syndrome‐related traits was observed in Chinese T2DM families, although no single SNP explained the previous linkage to the 1q21 Citation58. In the Chinese case‐control sample, the population‐based hospital cases of T2DM were, however, not associated with usf1s1 Citation58. In the second study, Gibson et al. did not find differences in allele frequencies between French diabetics and healthy controls Citation59. To summarize, the results for USF1 with T2DM and the metabolic syndrome have included both positive and negative associations Citation58,59. These data imply that there are also other regional genes on 1q21 that contribute to the linkage signals of these two disorders.
Interestingly, a gene for combined hyperlipidemia (Hyplip1) in mouse was mapped to a region on chromosome 3 in the HcB‐19/Dem mouse that was orthologous to human chromosome 1q21 Citation60. The underlying gene, thioredoxin interacting protein (TXNIP), was recently identified as a gene for combined hyperlipidemia in mouse Citation61. TXNIP provided thus a strong positional candidate for human FCHL Citation62. However, several recent studies show that variations in the TXNIP gene do not confer the susceptibility to FCHL Citation17,Citation63,64. Regarding the possibility whether the identified USF1 risk haplotype would have a long‐range effect on the expression of TXNIP, the TXNIP expression profiles of fat biopsies from affected Finnish FCHL family members having the USF1 risk haplotype were compared with affected FCHL family members, homozygous for the putative protective haplotype. No haplotype‐dependent difference in TXNIP expression was detected Citation17. To summarize, it seems unlikely that TXNIP accounts for the observed evidence of linkage between FCHL and the 1q21 region.
Variants in the apolipoprotein gene cluster APOA1/C3/A4/A5 are implicated in FCHL
Multiple studies predict the importance of the APOA1/C3/A4 gene cluster as a modifier gene complex in the development of FCHL Citation65–73 (), although not all studies have shown the connection Citation74–76. The most investigated polymorphisms of this gene cluster are three restriction enzyme polymorphisms, XmnI and MspI residing upstream to the apolipoprotein A1 (APOA1) gene and the SstI site in the 3′ untranslated region of exon 4 of the APOC3 gene Citation77. The positive studies include a Dutch study, in which the minor alleles of these polymorphisms were associated with elevated plasma TC, TG, LDL‐C, apoB, and apoC3 levels in Dutch FCHL families Citation67. Furthermore, a suggestive evidence for linkage between the MspI minor allele and plasma LDL‐C levels was detected Citation67. Based on the results, Dallinga‐Thie et al. suggested that the APOAI/C3/A4 gene cluster is not the primary cause of FCHL, but rather it has a specific modifying effect on plasma TG and LDL‐C levels in this lipid disorder Citation67. The negative studies include a Finnish study in which the MspI polymorphism was associated with serum TC and apoB levels in spouses, but no evidence of direct involvement of the APOAI/C3/A4 loci or haplotypes in the expression of FCHL in the Finnish FCHL families was found Citation76.
Table II. Genes associated with familial combined hyperlipidemia (FCHL) in previous studies.
Recently, APOA5 gene was added as part of the APOA1/C3/A4 gene cluster Citation78. Variants of this gene cluster have been linked to high TGs in both the general population Citation78–87 and in FCHL Citation88–90. In the Dutch FCHL families, APOA1/C3/A4/A5 showed an association with TG levels and LDL particle size, and the strongest evidence of association was obtained with SNPs in APOA1 and APOA5Citation89. Ribalta and colleagues also suggested a potential implication of APOA5 in the hypertriglyceridemia present in FCHL, because in hyperlipidemic patients with FCHL, the carriers of the minor allele of a SNP in APOA5 had significantly increased plasma TG levels when compared with the carriers of the common allele Citation90. In British FCHL families, alleles of the APOA1/C3/A4/A5 gene cluster were overtransmitted to subjects with FCHL, and the transmission of the common APOA1/C3/A4/A5 haplotype to the affected subjects was reduced Citation88. Several studies also imply that APOA5 is a potential risk factor for cardiovascular disease Citation82,Citation91,92.
APOA5 is suggested to reduce plasma TGs by inhibiting lipidation of apoB and thus reducing the hepatic very low‐density lipoprotein (VLDL) production rate, as well as by stimulating LPL‐mediated clearance of TG‐rich lipoproteins Citation93. Overexpression of APOA5 has been shown to lower TGs in mice and ApoA5 knockout mice have severe hypertriglyceridemia Citation78,Citation94. Recently, a study by Nowak et al. implied that APOA5 is regulated by insulin Citation46. Interestingly, insulin induces a dose‐dependent downregulation of APOA5 expression by reducing the binding of upstream stimulatory factors USF1 and USF2 to E‐Box (5′‐CACGTG‐3′) of the APOA5 promoter. It was also suggested, that the inhibitory effect of insulin on the APOA5 transcription involves a phosphorylation mechanism of USF that modulates their binding to the APOA5 promoter and results in APOA5 transrepression Citation46. The downregulation of APOA5 by insulin could explain the association between hypertriglyceridemia and hyperinsulinemia. Interestingly, APOA5 is also a highly responsive target gene of the peroxisome proliferator‐activated receptor alpha and may act as a major mediator for fibrates in reduction of plasma TGs Citation95.
Lipoprotein lipase and hepatic lipase genes in FCHL
The variants in the LPL gene have been of interest in FCHL because LPL catalyzes the hydrolysis of TGs of VLDL and chylomicrons and thus delivers fatty acids to tissues (). Nevin et al. identified variations in the coding region of the LPL gene in 6 of 20 FCHL patients, the variations including Asp9Asn, Val108Val and Ser447stop Citation96. Reymer et al. found a common Asn291Ser variant of the LPL gene in FCHL patients and observed an association with FCHL and HDL‐C levels, as well as with catalytic activity of LPLCitation97. Since these studies, the Asn291Ser and Asp9Asn variants have been associated with reduced HDL‐C, elevated TG, apoB and/or insulin levels in FCHL patients in several different studies Citation98–103. In addition to coding variants in LPL, variations in the promoter region have been identified in FCHL patients Citation101,102,Citation104,105. Yang et al. reported a heterozygous carrier of a promoter variant (−39C/T) in LPL when screening 20 FCHL probands for mutations in the putative LPL promoter region Citation105. The variant resulted in diminished transcriptional activity of the LPL gene, potentially by abolishing the transcription factor Oct‐1 binding site. The −93G/T and −53C/G variants were also reported to affect promoter activity Citation104. However, not all studies have supported the role of LPL in FCHL Citation74,Citation106,107. In addition to FCHL, variations in LPL have been associated with CHD Citation108. Evidence for linkage to the hepatic lipase (LIPC) locus (P<0.003) and to the lecithin:cholesterol acyltransferase (LCAT) locus (P<0.0006) has also been observed Citation13 and a coding variant in LIPC was associated with elevated TC and apoB levels in the Dutch FCHL families Citation109. However, the linkage to LIPC locus was not confirmed in the Finnish FCHL families Citation106.
Variants in HNF4A gene associated with lipid levels in FCHL
A region on chromosome 20q12–q13 has been linked to T2DM and obesity in numerous studies Citation110–114. Recently, several independent groups have identified associations between SNPs in the HNF4A gene residing in this 20q12–q13.1 region and T2DM Citation115–119. Previously mutations in HNF4A have been demonstrated to cause maturity onset diabetes of the young type I (MODY1) Citation120. Interestingly, the same chromosomal region on 20q12–q13 has also been linked to TGs and low HDL‐C in FCHL families Citation11,Citation121,122. Subsequently, evidence for linkage has been found with high TGs in other populations Citation123,124. There is a clear phenotypic overlap between FCHL and T2DM, patients with T2DM exhibiting often hypertriglyceridemia and patients with FCHL glucose intolerance and/or insulin resistance Citation5,Citation125. Both diseases also predispose to CHD.
Considering this clear phenotypic overlap between T2DM and FCHL, and the fact that both disorders have been linked to the same chromosomal region on 20q Citation11,Citation110,111,Citation113,114,Citation121,122, it is possible that HNF4A contributes to linkage signals in both diseases. Recently common HNF4A variants and their haplotypes were investigated for association in Finnish and Mexican FCHL families, comprising 1020 subjects Citation18 (). The common HNF4A variants and haplotypes were associated with elevated serum lipid levels, the metabolic syndrome as well as with elevated glucose parameters in the Finnish FCHL families Citation18. Importantly, both the Finnish and Mexican FCHL families shared two common lipid‐associated HNF4A haplotypes Citation18. This is the first study demonstrating that common HNF4A variants and their haplotypes are associated with high plasma lipid levels and the metabolic syndrome.
HNF4A is a transcription factor, regulating several genes in lipid and glucose metabolism. Interestingly, it has been shown that cooperative binding of USF1 and HNF4A drives the transcription of the human APOA2 gene Citation20. In addition, sequences from sites bound by HNF4A and USF1 were demonstrated to show significant overlap in HepG2 cells Citation19. As interactions between DNA sequence variants are suggested to be critical in the etiology of complex traits such as FCHL, it is possible that DNA sequence variants in USF1 and HNF4A interactively influence the susceptibility to FCHL.
TNFRSF1B and LEPR as candidate genes for QTL affecting apoB levels on chromosome 1
A genome‐wide scan in 18 Dutch FCHL families revealed a quantitative trait locus (QTL) affecting apoB levels on chromosome 1 short arm Citation15. The candidate genes associated with FCHL in this region include tumor necrosis factor receptor superfamily, member 1B (TNFRSF1B) Citation126 and leptin receptor (LEPR)Citation127. TNFRSF1B, located on 1p36.2, was associated with susceptibility to FCHL in the Dutch family sample Citation126. For further support, the levels of soluble extracellular domain of TNF‐R p75 were lower in the hyperlipidemic than in the normolipidemic relatives of FCHL patients Citation128. A polymorphism in the coding region of the LEPR gene (Gln223Arg), located on 1p31 was associated with an increased risk of FCHL and a difference in HDL‐C levels Citation127. To determine whether one or both of these genes explain the linkage signal, or if there are additional regional genes involved, requires further investigation.
Other candidate genes investigated for FCHL include manganese superoxide dismutase locus on chromosome 6, which showed suggestive evidence of linkage in Dutch FCHL families with FCHL, as well as with related traits such as TC, apoB and apoC3 levels (P<0.02) Citation13. Biochemical and genetic associations of plasma apoA2 levels with FCHL have also been suggested Citation129. The role of APOA2 in FCHL has been discussed recently Citation130,131.
Gene expression profiling provides novel candidate genes for FCHL
Gene expression is the major determinant of the phenotype and function of a living organism. The profile of expressed genes in a particular cell is highly dynamic and changes rapidly in response to cellular events and external stimuli. For long, methods have been available to measure the gene expression of a limited number of genes at a time. Recently, DNA microarray technology has provided a tool to monitor the expression pattern of a whole genome simultaneously on a single chip Citation132. This technology provides a useful tool to profile gene expression at the genomic level, and to determine sets of genes expressed or turned off together. Consequently, these techniques allow for identification of the genes and metabolic pathways that are differentially expressed in healthy and diseased individuals and, thus, provide an alternative approach to identify novel genes and pathways involved in FCHL.
Three studies have been performed to identify the differential gene expression between FCHL patients and healthy controls Citation133–135. One of the studies investigated lymphoblastic cell lines Citation133, and two others focused on adipose tissue Citation134,135. The first study by Erlings et al. detected 25 differentially expressed genes out of 588 genes in subcutaneous adipose tissue obtained from 5 unrelated FCHL and 4 control individuals Citation134. Many of the differentially expressed genes had been implicated in the activation of the adipocyte cell cycle. Expression of tumor necrosis factor, alpha (TNFA) was upregulated in FCHL patients, which is of particular interest since TNFRSF1B variants and decreased levels of soluble extracellular domain of TNF‐R p75 have been observed to be associated with FCHL previously Citation126,Citation128.
Morello et al. studied the gene expression in immortalized lymphoblastic cells obtained from FCHL cases and their normolipidemic spouses and relatives Citation133. Of the 7647 genes expressed in these samples, 166 genes were differentially expressed between cases and controls. Categorizing the differentially expressed genes according to biological processes that the genes were involved in revealed that almost half of the genes were taking part in metabolism. Interestingly, the early growth response 1 gene (EGR‐1) encoding a transcription factor was upregulated in lymphoblasts Citation133, as well as in the adipocytes derived from FCHL patients in the previous study Citation134. Of the 166 genes differentially expressed in the lymphoblastic cell lines, surrounding sequences of 16 genes contained the EGR‐1 consensus sequence (5′‐CGCCCCCGC‐3′) Citation133.
Meex et al. Citation135 measured the expression levels of 640 genes in 5 unrelated FCHL patients and 10 control individuals. Initially, 27 genes were identified differentially expressed between FCHL cases and controls. After validation CD36/FAT was shown to be upregulated in the original sample of FCHL cases using quantitative reverse transcription polymerase chain reaction (RT‐PCR), as well as in five additional pairs of FCHL cases and controls. CD36/FAT is a multiligand class B scavenger receptor that has been implicated in the transmembrane transport of long‐chain fatty acids and in the regulation of angiogenesis. Interestingly, Meex et al. found a significant correlation between the CD36/FAT expression and esterification of fatty acids into phospholipids and TGs, suggesting that this gene has a functional role in lipid metabolism Citation135.
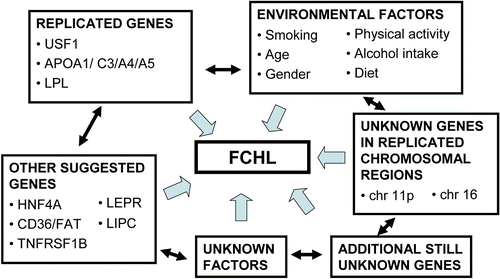
To summarize the candidate gene studies and the evidence obtained in replication studies (), it seems evident that DNA sequence variants at least in USF1, LPL and the APOA1/C3/A4/A5 gene cluster confer the susceptibility to FCHL. However, none of the candidate genes investigated so far account solely for a major genetic component in FCHL, further confirming the current understanding that the complex FCHL phenotype is an end result of genetic and environmental interplay of multiple factors ().
Figure 2. Multiple genetic and environmental factors confer the susceptibility to familial combined hyperlipidemia(FCHL).
Chromosomal loci identified for FCHL
To identify novel loci for FCHL, genome‐wide scans have been performed in the Finnish Citation11,Citation122, Dutch Citation14,15 and British families with FCHL Citation16. Loci on chromosomes 1q21, 2p, 2q31, 8q, 10p11.2, 10q11.2–10qter, 16q, 20q and 21q21 were identified in the Finnish FCHL families Citation10,11,Citation122, and loci on chromosomes 1p, 2p, 11p, 16q and 19q in the Dutch FCHL families Citation14,15. A combined genome scan of the Dutch and Finnish families with FCHL identified three loci for HDL‐C on 2p, 9p and 16q Citation12. In addition, a recent genome scan in the British FCHL families replicated the 11p locus and identified two novel candidate loci on 6q and 8p Citation16.
Three of these chromosomal regions, 1q21–23, 11p and 16q22–24.1, have been replicated in several FCHL study samples. The 1q21 locus has been observed in the Finnish and Dutch genome scans, as well as in independent studies of Chinese, German, US Caucasian and Mexican families with FCHL Citation10,11,Citation15,Citation24–26. Another replicated locus is the 11p region, which has been detected in the Dutch and British FCHL families Citation14,Citation16. This region showed evidence of linkage to FCHL, as well as to the TC and TG traits in the British study sample Citation16. No evidence of linkage for apoB levels was observed for this 11p region Citation14, implying that this locus may not directly contribute to the observed elevation of apoB‐containing particles in FCHL. Genetic heterogeneity was demonstrated for the 11p region, as 49% of the British FCHL families were linked to this region Citation16.
Given the known difficulties in replicating and verifying the results of complex traits, international collaboration to replicate findings in independent and combined study samples is of utmost importance to accelerate the gene identification process. This strategy is based on increased statistical power to verify those of the identified regions that have the highest statistical likelihood to harbor causative genes. The power of this strategy has recently been demonstrated with other complex diseases, such as inflammatory bowel disease and asthma Citation136–138, as well as in a combined data analysis of Dutch and Finnish genome‐wide scans for FCHL Citation12. In that study, three regions, 16q24.1, 2p25.1, and 9p23, were identified where the evidence for linkage emerged from the combined study sample Citation12. The region on 16q24.1 resulted in most significant evidence for linkage, a lod score of 3.4, for HDL‐C, 50% of the Finnish and Dutch families being linked to this region. This very same 16q24.1 region was also recently implicated for HDL‐C in Mexican Americans Citation139, as well as in several other independent studies Citation12,Citation26,Citation122,Citation139–141. Different study populations could thus be utilized first to replicate the loci that harbor susceptibility genes for FCHL, and second, to fine‐map these verified regions.
It has not been directly evaluated whether the three replicated regions on 1q21, 11p and 16q explain the dyslipidemia in some or all FCHL families. These types of studies are clearly warranted to estimate the risk related to each variant, their contribution to FCHL and its component traits, as well as to investigate the likely gene‐gene interactions in FCHL.
Future directions and concluding comments
FCHL is a typical complex trait with several genes, environmental factors and their interactions contributing to the disease phenotype (). The possible high genetic complexity and the fact that the current strategies are based on functional characterization of mutations of monogenic diseases make the dissection of the wide allelic spectrum of variants conferring the susceptibility to complex diseases very challenging. These variants can result in minor changes in protein product or reside in promoters or other regulatory regions; or they can cause small changes in the binding affinity of a transcription factor or slightly alter gene expression levels, timing and tissue specificity. In all of these cases, it may be difficult to demonstrate not only the functional consequences of a single variant but also of the allelic combinations of multiple variants. Nutrients or other environmental factors may potentially also modulate differences introduced by genetic variation. It is thus possible that each individual variant causes subtle changes in the disease phenotype. Accumulation of multiple deleterious gene variants and environmental factors in a particular individual will then ultimately lead to the expression of the disease phenotype.
Candidate gene studies, as well as positional cloning have provided novel candidate genes for FCHL of which variants in USF1, the APOA1/C3/A4/A5 gene cluster and LPL have been replicated in several study samples from distinct populations. Many of the variants currently associated with FCHL are common and the risk involved with them at the population level is not known at the moment. Extensive population‐based studies, such as recently conducted for USF1Citation55 and multiple T2DM candidate genes Citation142, estimating the risk related to each variant and their relative contribution to the disease susceptibility, are warranted.
Now in the post‐genomic era, over a hundred different genomes including the human genome have been sequenced, and the HapMap project has described the human sequence variation throughout the human genome Citation143. This vast amount of information together with advancing genotyping technologies have enabled genome‐wide LD and association studies using hundreds of thousands of SNPs. These novel genomic approaches are expected to facilitate the dissection of the molecular basis of human complex diseases and provide novel insights into disease pathogenesis in near future. Utilization of the HapMap data offers short‐cuts to gene identification providing that common variants contribute significantly to common diseases Citation144–146. Moreover, strategies using tag SNPs, derived from the HapMap data, have most power to detect these common causative variants Citation147. Consequently, the novel genome‐wide association approaches are more likely to identify common than rare variants influencing the FCHL susceptibility. Based on the recent candidate gene studies of DNA sequence variants contributing to levels of plasma HDL‐cholesterol in different populations, it is, however, likely that rare DNA sequence variants also contribute to disease susceptibility Citation22,23. As large‐scale sequencing of candidate genes in affected individuals is currently the only approach to identify new rare variants, more efficient and cost‐effective sequencing approaches are warranted.
The Human Genome Project and HapMap Project have provided the basic tools for unraveling the complex genetics of FCHL and other complex traits. However, this challenging task can be accomplished only by combining the accumulating biological information with interdisciplinary knowledge and international collaboration. Skillful combining of molecular medicine and systems biology is needed to overcome the greatest challenge of any complex disease, i.e. the meaningful analysis of the enormous amount of data using tools reflecting accurately the underlying phenotypic and genetic complexity.
Acknowledgements
Many students and collaborators contributed to the work and ideas presented in this review and we would like to thank them. Work in the laboratory of PP is supported by the National Institutes of Health grants HL‐28481, HL‐82762 and HL‐70150, as well as the American Heart Association grant 0430180N.
References
- Rose H. G., Kranz P., Weinstock M., Juliano J., Haft J. I. Inheritance of combined hyperlipoproteinemia: evidence for a new lipoprotein phenotype. Am J Med 1973; 54: 148–60
- Nikkila E. A., Aro A. Family study of serum lipids and lipoproteins in coronary heart‐disease. Lancet 1973; 1: 954–9
- Goldstein J. L., Schrott H. G., Hazzard W. R., Bierman E. L., Motulsky A. G. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest 1973; 52: 1544–68
- Hopkins P. N., Heiss G., Ellison R. C., Province M. A., Pankow J. S., Eckfeldt J. H., et al. Coronary artery disease risk in familial combined hyperlipidemia and familial hypertriglyceridemia: a case‐control comparison from the National Heart, Lung, and Blood Institute Family Heart Study. Circulation 2003; 108: 519–23
- Vakkilainen J., Porkka K. V., Nuotio I., Pajukanta P., Suurinkeroinen L., Ylitalo K., et al. Glucose intolerance in familial combined hyperlipidaemia. EUFAM study group. Eur J Clin Invest 1998; 28: 24–32
- Aro A. Serum lipids and lipoproteins in first degree relatives of young survivors of myocardial infarction. Acta Med Scand Suppl 1973; 553: 1–103
- de Graaf J., Stalenhoef A. F. Defects of lipoprotein metabolism in familial combined hyperlipidaemia. Curr Opin Lipidol 1998; 9: 189–96
- Cullen P., Farren B., Scott J., Farrall M. Complex segregation analysis provides evidence for a major gene acting on serum triglyceride levels in 55 British families with familial combined hyperlipidemia. Arterioscler Thromb 1994; 14: 1233–49
- Jarvik G. P., Brunzell J. D., Austin M. A., Krauss R. M., Motulsky A. G., Wijsman E. Genetic predictors of FCHL in four large pedigrees. Influence of ApoB level major locus predicted genotype and LDL subclass phenotype. Arterioscler Thromb 1994; 14: 1687–94
- Pajukanta P., Nuotio I., Terwilliger J. D., Porkka K. V., Ylitalo K., Pihlajamaki J., et al. Linkage of familial combined hyperlipidaemia to chromosome 1q21–q23. Nat Genet 1998; 18: 369–73
- Pajukanta P., Terwilliger J. D., Perola M., Hiekkalinna T., Nuotio I., Ellonen P., et al. Genomewide scan for familial combined hyperlipidemia genes in finnish families, suggesting multiple susceptibility loci influencing triglyceride, cholesterol, and apolipoprotein B levels. Am J Hum Genet 1999; 64: 1453–63
- Pajukanta P., Allayee H., Krass K. L., Kuraishy A., Soro A., Lilja H. E., et al. Combined analysis of genome scans of Dutch and Finnish families reveals a susceptibility locus for high‐density lipoprotein cholesterol on chromosome 16q. Am J Hum Genet 2003; 72: 903–17
- Aouizerat B. E., Allayee H., Cantor R. M., Dallinga‐Thie G. M., Lanning C. D., de Bruin T. W., et al. Linkage of a candidate gene locus to familial combined hyperlipidemia: lecithin:cholesterol acyltransferase on 16q. Arterioscler Thromb Vasc Biol 1999; 19: 2730–6
- Aouizerat B. E., Allayee H., Cantor R. M., Davis R. C., Lanning C. D., Wen P. Z., et al. A genome scan for familial combined hyperlipidemia reveals evidence of linkage with a locus on chromosome 11. Am J Hum Genet 1999; 65: 397–412
- Allayee H., Krass K. L., Pajukanta P., Cantor R. M., van der Kallen C. J., Mar R., et al. Locus for elevated apolipoprotein B levels on chromosome 1p31 in families with familial combined hyperlipidemia. Circ Res 2002; 90: 926–31
- Naoumova R. P., Bonney S. A., Eichenbaum‐Voline S., Patel H. N., Jones B., Jones E. L., et al. Confirmed locus on chromosome 11p and candidate loci on 6q and 8p for the triglyceride and cholesterol traits of combined hyperlipidemia. Arterioscler Thromb Vasc Biol 2003; 23: 2070–7
- Pajukanta P., Lilja H. E., Sinsheimer J. S., Cantor R. M., Lusis A. J., Gentile M., et al. Familial combined hyperlipidemia is associated with upstream transcription factor 1 (USF1). Nat Genet 2004; 36: 371–6
- Weissglas‐Volkov D., Huertas‐Vazquez A., Suviolahti E., Lee J. C., Plaisier C., Canizales‐Quinteros S., Tusie‐Luna T., et al. Common hepatic nuclear factor 4 alpha variants are associated with high serum lipid levels and the metabolic syndrome. Diabetes 2006; 55: 1970–7
- Rada‐Iglesias A., Wallerman O., Koch C., Ameur A., Enroth S., Clelland G., et al. Binding sites for metabolic disease related transcription factors inferred at base pair resolution by chromatin immunoprecipitation and genomic microarrays. Hum Mol Genet 2005; 14: 3435–47
- Ribeiro A., Pastier D., Kardassis D., Chambaz J., Cardot P. Cooperative binding of upstream stimulatory factor and hepatic nuclear factor 4 drives the transcription of the human apolipoprotein A‐II gene. J Biol Chem 1999; 274: 1216–25
- Pastier D., Lacorte J. M., Chambaz J., Cardot P., Ribeiro A. Two initiator‐like elements are required for the combined activation of the human apolipoprotein C‐III promoter by upstream stimulatory factor and hepatic nuclear factor‐4. J Biol Chem 2002; 277: 15199–206
- Frikke‐Schmidt R., Nordestgaard B. G., Jensen G. B., Tybjaerg‐Hansen A. Genetic variation in ABC transporter A1 contributes to HDL cholesterol in the general population. J Clin Invest 2004; 114: 1343–53
- Cohen J. C., Kiss R. S., Pertsemlidis A., Marcel Y. L., McPherson R., Hobbs H. H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 2004; 305: 869–72
- Pei W., Baron H., Muller‐Myhsok B., Knoblauch H., Al‐Yahyaee S. A., Hui R., et al. Support for linkage of familial combined hyperlipidemia to chromosome 1q21–q23 in Chinese and German families. Clin Genet 2000; 57: 29–34
- Coon H., Myers R. H., Borecki I. B., Arnett D. K., Hunt S. C., Province M. A., et al. Replication of linkage of familial combined hyperlipidemia to chromosome 1q with additional heterogeneous effect of apolipoprotein A‐I/C‐III/A‐IV locus. The NHLBI Family Heart Study. Arterioscler Thromb Vasc Biol 2000; 20: 2275–80
- Huertas‐Vazquez A., Aguilar‐Salinas C., Lusis A. J., Cantor R. M., Canizales‐Quinteros S., Lee J. C., et al. Familial combined hyperlipidemia in Mexicans: association with upstream transcription factor 1 and linkage on chromosome 16q24.1. Arterioscler Thromb Vasc Biol 2005; 25: 1985–91
- Hanson R. L., Ehm M. G., Pettitt D. J., Prochazka M., Thompson D. B., Timberlake D., et al. An autosomal genomic scan for loci linked to type II diabetes mellitus and body‐mass index in Pima Indians. Am J Hum Genet 1998; 63: 1130–8
- Elbein S. C., Hoffman M. D., Teng K., Leppert M. F., Hasstedt S. J. A genome‐wide search for type 2 diabetes susceptibility genes in Utah Caucasians. Diabetes 1999; 48: 1175–82
- Vionnet N., Hani El H., Dupont S., Gallina S., Francke S., Dotte S., et al. Genomewide search for type 2 diabetes‐susceptibility genes in French whites: evidence for a novel susceptibility locus for early‐onset diabetes on chromosome 3q27‐qter and independent replication of a type 2‐diabetes locus on chromosome 1q21–q24. Am J Hum Genet 2000; 67: 1470–80
- Wiltshire S., Hattersley A. T., Hitman G. A., Walker M., Levy J. C., Sampson M., et al. A genomewide scan for loci predisposing to type 2 diabetes in a U.K. population (the Diabetes UK Warren 2 Repository): analysis of 573 pedigrees provides independent replication of a susceptibility locus on chromosome 1q. Am J Hum Genet 2001; 69: 553–69
- Hsueh W. C., St Jean P. L., Mitchell B. D., Pollin T. I., Knowler W. C., Ehm M. G., et al. Genome‐wide and fine‐mapping linkage studies of type 2 diabetes and glucose traits in the Old Order Amish: evidence for a new diabetes locus on chromosome 14q11 and confirmation of a locus on chromosome 1q21–q24. Diabetes 2003; 52: 550–7
- Watanabe R. M., Ghosh S., Langefeld C. D., Valle T. T., Hauser E. R., Magnuson V. L., et al. The Finland‐United States investigation of non‐insulin‐dependent diabetes mellitus genetics (FUSION) study. II. An autosomal genome scan for diabetes‐related quantitative‐trait loci. Am J Hum Genet 2000; 67: 1186–200
- Xiang K., Wang Y., Zheng T., Jia W., Li J., Chen L., et al. Genome‐wide search for type 2 diabetes/impaired glucose homeostasis susceptibility genes in the Chinese: significant linkage to chromosome 6q21–q23 and chromosome 1q21–q24. Diabetes 2004; 53: 228–34
- Langefeld C. D., Wagenknecht L. E., Rotter J. I., Williams A. H., Hokanson J. E., Saad M. F., et al. Linkage of the metabolic syndrome to 1q23–q31 in Hispanic families: the Insulin Resistance Atherosclerosis Study Family Study. Diabetes 2004; 53: 1170–4
- Liu Y., Nusrat A., Schnell F. J., Reaves T. A., Walsh S., Pochet M., et al. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci 2000; 113((Pt 13))2363–74
- Barton E. S., Forrest J. C., Connolly J. L., Chappell J. D., Liu Y., Schnell F. J., et al. Junction adhesion molecule is a receptor for reovirus. Cell 2001; 104: 441–51
- Ostermann G., Weber K. S., Zernecke A., Schroder A., Weber C. JAM‐1 is a ligand of the beta(2) integrin LFA‐1 involved in transendothelial migration of leukocytes. Nat Immunol 2002; 3: 151–8
- Vallet V. S., Casado M., Henrion A. A., Bucchini D., Raymondjean M., Kahn A., et al. Differential roles of upstream stimulatory factors 1 and 2 in the transcriptional response of liver genes to glucose. J Biol Chem 1998; 273: 20175–9
- Casado M., Vallet V. S., Kahn A., Vaulont S. Essential role in vivo of upstream stimulatory factors for a normal dietary response of the fatty acid synthase gene in the liver. J Biol Chem 1999; 274: 2009–13
- Iynedjian P. B. Identification of upstream stimulatory factor as transcriptional activator of the liver promoter of the glucokinase gene. Biochem J 1998; 333((Pt 3))705–12
- Kutz S. M., Higgins C. E., Samarakoon R., Higgins S. P., Allen R. R., Qi L., et al. TGF‐beta‐induced PAI‐1 expression is E box/USF‐dependent and requires EGFR signaling. Exp Cell Res 2006; 312: 1093–105
- Yang X. P., Freeman L. A., Knapper C. L., Amar M. J., Remaley A., Brewer H. B., Jr., et al. The E‐box motif in the proximal ABCA1 promoter mediates transcriptional repression of the ABCA1 gene. J Lipid Res 2002; 43: 297–306
- Wang D., Sul H. S. Upstream stimulatory factor binding to the E‐box at ‐65 is required for insulin regulation of the fatty acid synthase promoter. J Biol Chem 1997; 272: 26367–74
- Botma G. J., Verhoeven A. J., Jansen H. Hepatic lipase promoter activity is reduced by the C‐480T and G‐216A substitutions present in the common LIPC gene variant, and is increased by Upstream Stimulatory Factor. Atherosclerosis 2001; 154: 625–32
- Salero E., Gimenez C., Zafra F. Identification of a non‐canonical E‐box motif as a regulatory element in the proximal promoter region of the apolipoprotein E gene. Biochem J 2003; 370((Pt 3))979–86
- Nowak M., Helleboid‐Chapman A., Jakel H., Martin G., Duran‐Sandoval D., Staels B., et al. Insulin‐mediated down‐regulation of apolipoprotein A5 gene expression through the phosphatidylinositol 3‐kinase pathway: role of upstream stimulatory factor. Mol Cell Biol 2005; 25: 1537–48
- Portois L., Tastenoy M., Viollet B., Svoboda M. Functional analysis of the glucose response element of the rat glucagon receptor gene in insulin‐producing INS‐1 cells. Biochim Biophys Acta 2002; 1574: 175–86
- Read M. L., Clark A. R., Docherty K. The helix‐loop‐helix transcription factor USF (upstream stimulating factor) binds to a regulatory sequence of the human insulin gene enhancer. Biochem J 1993; 295((Pt 1))233–7
- Martin C. C., Svitek C. A., Oeser J. K., Henderson E., Stein R., O'Brien R. M. Upstream stimulatory factor (USF) and neurogenic differentiation/beta‐cell E box transactivator 2 (NeuroD/BETA2) contribute to islet‐specific glucose‐6‐phosphatase catalytic‐subunit‐related protein (IGRP) gene expression. Biochem J 2003; 371((Pt 3))675–86
- Travers M. T., Vallance A. J., Gourlay H. T., Gill C. A., Klein I., Bottema C. B., et al. Promoter I of the ovine acetyl‐CoA carboxylase‐alpha gene: an E‐box motif at ‐114 in the proximal promoter binds upstream stimulatory factor (USF)‐1 and USF‐2 and acts as an insulin‐response sequence in differentiating adipocytes. Biochem J 2001; 359((Pt 2))273–84
- Smih F., Rouet P., Lucas S., Mairal A., Sengenes C., Lafontan M., et al. Transcriptional regulation of adipocyte hormone‐sensitive lipase by glucose. Diabetes 2002; 51: 293–300
- Naukkarinen J., Gentile M., Soro‐Paavonen A., Saarela J., Koistinen H. A., Pajukanta P., et al. USF1 and dyslipidemias: converging evidence for a functional intronic variant. Hum Mol Genet 2005; 14: 2595–605
- Attie A. D., Krauss R. M., Gray‐Keller M. P., Brownlie A., Miyazaki M., Kastelein J. J., et al. Relationship between stearoyl‐CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. J Lipid Res 2002; 43: 1899–907
- Hoffstedt J., Ryden M., Wahrenberg H., van Harmelen V., Arner P. Upstream transcription factor‐1 gene polymorphism is associated with increased adipocyte lipolysis. J Clin Endocrinol Metab 2005; 90: 5356–60
- Komulainen K., Alanne M., Auro K., Kilpikari R., Pajukanta P., Saarela J., et al. Risk Alleles of USF1‐Gene Predict Cardiovascular Disease of Women in Two Prospective Studies. PLoS Genetics 2006; 2: e69
- Putt W., Palmen J., Nicaud V., Tregouet D. A., Tahri‐Daizadeh N., Flavell D. M., et al. Variation in USF1 shows haplotype effects, gene:gene and gene:environment associations with glucose and lipid parameters in the European Atherosclerosis Research Study II. Hum Mol Genet 2004; 13: 1587–97
- Coon H., Xin Y., Hopkins P. N., Cawthon R. M., Hasstedt S. J., Hunt S. C. Upstream stimulatory factor 1 associated with familial combined hyperlipidemia, LDL cholesterol, and triglycerides. Hum Genet 2005; 117: 444–51
- Ng M. C., Miyake K., So W. Y., Poon E. W., Lam V. K., Li J. K., et al. The linkage and association of the gene encoding upstream stimulatory factor 1 with type 2 diabetes and metabolic syndrome in the Chinese population. Diabetologia 2005; 48: 2018–24
- Gibson F., Hercberg S., Froguel P. Common polymorphisms in the USF1 gene are not associated with type 2 diabetes in French Caucasians. Diabetes 2005; 54: 3040–2
- Castellani L. W., Weinreb A., Bodnar J., Goto A. M., Doolittle M., Mehrabian M., et al. Mapping a gene for combined hyperlipidaemia in a mutant mouse strain. Nat Genet 1998; 18: 374–7
- Bodnar J. S., Chatterjee A., Castellani L. W., Ross D. A., Ohmen J., Cavalcoli J., et al. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat Genet 2002; 30: 110–6
- Pajukanta P., Bodnar J. S., Sallinen R., Chu M., Airaksinen T., Xiao Q., et al. Fine mapping of Hyplip1 and the human homolog, a potential locus for FCHL. Mamm Genome 2001; 12: 238–45
- Coon H., Singh N., Dunn D., Eckfeldt J. H., Province M. A., Hopkins P. N., et al. TXNIP gene not associated with familial combined hyperlipidemia in the NHLBI Family Heart Study. Atherosclerosis 2004; 174: 357–62
- van der Vleuten G. M., Hijmans A., Heil S., Blom H. J., Stalenhoef A. F., de Graaf J. Can we exclude the TXNIP gene as a candidate gene for familial combined hyperlipidemia?. Am J Med Genet A 2006; 140: 1010–2
- Wojciechowski A. P., Farrall M., Cullen P., Wilson T. M., Bayliss J. D., Farren B., et al. Familial combined hyperlipidaemia linked to the apolipoprotein AI‐CII‐AIV gene cluster on chromosome 11q23–q24. Nature 1991; 349: 161–4
- Hayden M. R., Kirk H., Clark C., Frohlich J., Rabkin S., McLeod R., et al. DNA polymorphisms in and around the Apo‐A1‐CIII genes and genetic hyperlipidemias. Am J Hum Genet 1987; 40: 421–30
- Dallinga‐Thie G. M., Bu X. D., van Linde‐Sibenius Trip M., Rotter J. I., Lusis A. J., de Bruin T. W. Apolipoprotein A‐I/C‐III/A‐IV gene cluster in familial combined hyperlipidemia: effects on LDL‐cholesterol and apolipoproteins B and C‐III. J Lipid Res 1996; 37: 136–47
- Dallinga‐Thie G. M., van Linde‐Sibenius Trip M., Rotter J. I., Cantor R. M., Bu X., Lusis A. J., et al. Complex genetic contribution of the Apo AI‐CIII‐AIV gene cluster to familial combined hyperlipidemia. Identification of different susceptibility haplotypes. J Clin Invest 1997; 99: 953–61
- Xu C. F., Talmud P., Schuster H., Houlston R., Miller G., Humphries S. Association between genetic variation at the APO AI‐CIII‐AIV gene cluster and familial combined hyperlipidaemia. Clin Genet 1994; 46: 385–97
- Groenendijk M., Cantor R. M., Blom N. H., Rotter J. I., de Bruin T. W., Dallinga‐Thie G. M. Association of plasma lipids and apolipoproteins with the insulin response element in the apoC‐III promoter region in familial combined hyperlipidemia. J Lipid Res 1999; 40: 1036–44
- Ribalta J., La Ville A. E., Vallve J. C., Humphries S., Turner P. R., Masana L. A variation in the apolipoprotein C‐III gene is associated with an increased number of circulating VLDL and IDL particles in familial combined hyperlipidemia. J Lipid Res 1997; 38: 1061–9
- Groenendijk M., Cantor R. M., De Bruin T. W., Dallinga‐Thie G. M. New genetic variants in the apoA‐I and apoC‐III genes and familial combined hyperlipidemia. J Lipid Res 2001; 42: 188–94
- Deeb S. S., Nevin D. N., Iwasaki L., Brunzell J. D. Two novel apolipoprotein A‐IV variants in individuals with familial combined hyperlipidemia and diminished levels of lipoprotein lipase activity. Hum Mutat 1996; 8: 319–25
- Marcil M., Boucher B., Gagne E., Davignon J., Hayden M., Genest J., Jr. Lack of association of the apolipoprotein A‐I‐C‐III‐A‐IV gene XmnI and SstI polymorphisms and of the lipoprotein lipase gene mutations in familial combined hyperlipoproteinemia in French Canadian subjects. J Lipid Res 1996; 37: 309–19
- Wijsman E. M., Brunzell J. D., Jarvik G. P., Austin M. A., Motulsky A. G., Deeb S. S. Evidence against linkage of familial combined hyperlipidemia to the apolipoprotein AI‐CIII‐AIV gene complex. Arterioscler Thromb Vasc Biol 1998; 18: 215–26
- Tahvanainen E., Pajukanta P., Porkka K., Nieminen S., Ikavalko L., Nuotio I., et al. Haplotypes of the ApoA‐I/C‐III/A‐IV gene cluster and familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol 1998; 18: 1810–7
- Groenendijk M., Cantor R. M., de Bruin T. W., Dallinga‐Thie G. M. The apoAI‐CIII‐AIV gene cluster. Atherosclerosis 2001; 157: 1–11
- Pennacchio L. A., Olivier M., Hubacek J. A., Cohen J. C., Cox D. R., Fruchart J. C., et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science 2001; 294: 169–73
- Pennacchio L. A., Olivier M., Hubacek J. A., Krauss R. M., Rubin E. M., Cohen J. C. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum Mol Genet 2002; 11: 3031–8
- Endo K., Yanagi H., Araki J., Hirano C., Yamakawa‐Kobayashi K., Tomura S. Association found between the promoter region polymorphism in the apolipoprotein A‐V gene and the serum triglyceride level in Japanese schoolchildren. Hum Genet 2002; 111: 570–2
- Baum L., Tomlinson B., Thomas G. N. APOA5‐1131T>C polymorphism is associated with triglyceride levels in Chinese men. Clin Genet 2003; 63: 377–9
- Pennacchio L. A., Rubin E. M. Apolipoprotein A5, a newly identified gene that affects plasma triglyceride levels in humans and mice. Arterioscler Thromb Vasc Biol 2003; 23: 529–34
- Nabika T., Nasreen S., Kobayashi S., Masuda J. The genetic effect of the apoprotein AV gene on the serum triglyceride level in Japanese. Atherosclerosis 2002; 165: 201–4
- Talmud P. J., Hawe E., Martin S., Olivier M., Miller G. J., Rubin E. M., et al. Relative contribution of variation within the APOC3/A4/A5 gene cluster in determining plasma triglycerides. Hum Mol Genet 2002; 11: 3039–46
- Kao J. T., Wen H. C., Chien K. L., Hsu H. C., Lin S. W. A novel genetic variant in the apolipoprotein A5 gene is associated with hypertriglyceridemia. Hum Mol Genet 2003; 12: 2533–9
- Martin S., Nicaud V., Humphries S. E., Talmud P. J. Contribution of APOA5 gene variants to plasma triglyceride determination and to the response to both fat and glucose tolerance challenges. Biochim Biophys Acta 2003; 1637: 217–25
- Wright W. T., Young I. S., Nicholls D. P., Patterson C., Lyttle K., Graham C. A. SNPs at the APOA5 gene account for the strong association with hypertriglyceridaemia at the APOA5/A4/C3/A1 locus on chromosome 11q23 in the Northern Irish population. Atherosclerosis 2006; 185: 353–60
- Eichenbaum‐Voline S., Olivier M., Jones E. L., Naoumova R. P., Jones B., Gau B., et al. Linkage and association between distinct variants of the APOA1/C3/A4/A5 gene cluster and familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol 2004; 24: 167–74
- Mar R., Pajukanta P., Allayee H., Groenendijk M., Dallinga‐Thie G., Krauss R. M., et al. Association of the APOLIPOPROTEIN A1/C3/A4/A5 gene cluster with triglyceride levels and LDL particle size in familial combined hyperlipidemia. Circ Res 2004; 94: 993–9
- Ribalta J., Figuera L., Fernandez‐Ballart J., Vilella E., Castro Cabezas M., Masana L., et al. Newly identified apolipoprotein AV gene predisposes to high plasma triglycerides in familial combined hyperlipidemia. Clin Chem 2002; 48: 1597–600
- Aouizerat B. E., Kulkarni M., Heilbron D., Drown D., Raskin S., Pullinger C. R., et al. Genetic analysis of a polymorphism in the human apoA‐V gene: effect on plasma lipids. J Lipid Res 2003; 44: 1167–73
- Hubacek J. A. Apolipoprotein A5 and triglyceridemia. Focus on the effects of the common variants. Clin Chem Lab Med 2005; 43: 897–902
- Schaap F. G., Rensen P. C., Voshol P. J., Vrins C., Van Der Vliet H. N., Chamuleau R. A., et al. ApoAV reduces plasma triglycerides by inhibiting very low density lipoprotein‐triglyceride (VLDL‐TG) production and stimulating lipoprotein lipase‐mediated VLDL‐TG hydrolysis. J Biol Chem 2004; 279: 27941–7
- van der Vliet H. N., Schaap F. G., Levels J. H., Ottenhoff R., Looije N., Wesseling J. G., et al. Adenoviral overexpression of apolipoprotein A‐V reduces serum levels of triglycerides and cholesterol in mice. Biochem Biophys Res Commun 2002; 295: 1156–9
- Vu‐Dac N., Gervois P., Jakel H., Nowak M., Bauge E., Dehondt H., et al. Apolipoprotein A5, a crucial determinant of plasma triglyceride levels, is highly responsive to peroxisome proliferator‐activated receptor alpha activators. J Biol Chem 2003; 278: 17982–5
- Nevin D. N., Brunzell J. D., Deeb S. S. The LPL gene in individuals with familial combined hyperlipidemia and decreased LPL activity. Arterioscler Thromb 1994; 14: 869–73
- Reymer P. W., Gagne E., Groenemeyer B. E., Zhang H., Forsyth I., Jansen H., et al. A lipoprotein lipase mutation (Asn291Ser) is associated with reduced HDL cholesterol levels in premature atherosclerosis. Nat Genet 1995; 10: 28–34
- de Bruin T. W., Mailly F., van Barlingen H. H., Fisher R., Castro Cabezas M., Talmud P., et al. Lipoprotein lipase gene mutations D9N and N291S in four pedigrees with familial combined hyperlipidaemia. Eur J Clin Invest 1996; 26: 631–9
- Reymer P. W., Groenemeyer B. E., Gagne E., Miao L., Appelman E. E., Seidel J. C., et al. A frequently occurring mutation in the lipoprotein lipase gene (Asn291Ser) contributes to the expression of familial combined hyperlipidemia. Hum Mol Genet 1995; 4: 1543–9
- Campagna F., Montali A., Baroni M. G., Maria A. T., Ricci G., Antonini R., et al. Common variants in the lipoprotein lipase gene, but not those in the insulin receptor substrate‐1, the beta3‐adrenergic receptor, and the intestinal fatty acid binding protein‐2 genes, influence the lipid phenotypic expression in familial combined hyperlipidemia. Metabolism 2002; 51: 1298–305
- Samuels M. E., Forbey K. C., Reid J. E., Abkevich V., Bulka K., Wardell B. R., et al. Identification of a common variant in the lipoprotein lipase gene in a large Utah kindred ascertained for coronary heart disease: the ‐93G/D9N variant predisposes to low HDL‐C/high triglycerides. Clin Genet 2001; 59: 88–98
- Hoffer M. J., Bredie S. J., Snieder H., Reymer P. W., Demacker P. N., Havekes L. M., et al. Gender‐related association between the ‐93T–>G/D9N haplotype of the lipoprotein lipase gene and elevated lipid levels in familial combined hyperlipidemia. Atherosclerosis 1998; 138: 91–9
- Hoffer M. J., Bredie S. J., Boomsma D. I., Reymer P. W., Kastelein J. J., de Knijff P., et al. The lipoprotein lipase (Asn291–>Ser) mutation is associated with elevated lipid levels in families with familial combined hyperlipidaemia. Atherosclerosis 1996; 119: 159–67
- Yang W. S., Nevin D. N., Iwasaki L., Peng R., Brown B. G., Brunzell J. D., et al. Regulatory mutations in the human lipoprotein lipase gene in patients with familial combined hyperlipidemia and coronary artery disease. J Lipid Res 1996; 37: 2627–37
- Yang W. S., Nevin D. N., Peng R., Brunzell J. D., Deeb S. S. A mutation in the promoter of the lipoprotein lipase (LPL) gene in a patient with familial combined hyperlipidemia and low LPL activity. Proc Natl Acad Sci U S A 1995; 92: 4462–6
- Pajukanta P., Porkka K. V., Antikainen M., Taskinen M. R., Perola M., Murtomaki‐Repo S., et al. No evidence of linkage between familial combined hyperlipidemia and genes encoding lipolytic enzymes in Finnish families. Arterioscler Thromb Vasc Biol 1997; 17: 841–50
- Gagne E., Genest J., Jr., Zhang H., Clarke L. A., Hayden M. R. Analysis of DNA changes in the LPL gene in patients with familial combined hyperlipidemia. Arterioscler Thromb 1994; 14: 1250–7
- Stein Y., Stein O. Lipoprotein lipase and atherosclerosis. Atherosclerosis 2003; 170: 1–9
- Hoffer M. J., Snieder H., Bredie S. J., Demacker P. N., Kastelein J. J., Frants R. R., et al. The V73M mutation in the hepatic lipase gene is associated with elevated cholesterol levels in four Dutch pedigrees with familial combined hyperlipidemia. Atherosclerosis 2000; 151: 443–50
- Bowden D. W., Sale M., Howard T. D., Qadri A., Spray B. J., Rothschild C. B., et al. Linkage of genetic markers on human chromosomes 20 and 12 to NIDDM in Caucasian sib pairs with a history of diabetic nephropathy. Diabetes 1997; 46: 882–6
- Ji L., Malecki M., Warram J. H., Yang Y., Rich S. S., Krolewski A. S. New susceptibility locus for NIDDM is localized to human chromosome 20q. Diabetes 1997; 46: 876–81
- Lembertas A. V., Perusse L., Chagnon Y. C., Fisler J. S., Warden C. H., Purcell‐Huynh D. A., et al. Identification of an obesity quantitative trait locus on mouse chromosome 2 and evidence of linkage to body fat and insulin on the human homologous region 20q. J Clin Invest 1997; 100: 1240–7
- Zouali H., Hani E. H., Philippi A., Vionnet N., Beckmann J. S., Demenais F., et al. A susceptibility locus for early‐onset non‐insulin dependent (type 2) diabetes mellitus maps to chromosome 20q, proximal to the phosphoenolpyruvate carboxykinase gene. Hum Mol Genet 1997; 6: 1401–8
- Ghosh S., Watanabe R. M., Hauser E. R., Valle T., Magnuson V. L., Erdos M. R., et al. Type 2 diabetes: evidence for linkage on chromosome 20 in 716 Finnish affected sib pairs. Proc Natl Acad Sci U S A 1999; 96: 2198–203
- Damcott C. M., Hoppman N., Ott S. H., Reinhart L. J., Wang J., Pollin T. I., et al. Polymorphisms in both promoters of hepatocyte nuclear factor 4‐alpha are associated with type 2 diabetes in the Amish. Diabetes 2004; 53: 3337–41
- Silander K., Mohlke K. L., Scott L. J., Peck E. C., Hollstein P., Skol A. D., et al. Genetic variation near the hepatocyte nuclear factor‐4 alpha gene predicts susceptibility to type 2 diabetes. Diabetes 2004; 53: 1141–9
- Hansen S. K., Rose C. S., Glumer C., Drivsholm T., Borch‐Johnsen K., Jorgensen T., et al. Variation near the hepatocyte nuclear factor (HNF)‐4alpha gene associates with type 2 diabetes in the Danish population. Diabetologia 2005; 48: 452–8
- Love‐Gregory L. D., Wasson J., Ma J., Jin C. H., Glaser B., Suarez B. K., et al. A common polymorphism in the upstream promoter region of the hepatocyte nuclear factor‐4 alpha gene on chromosome 20q is associated with type 2 diabetes and appears to contribute to the evidence for linkage in an ashkenazi jewish population. Diabetes 2004; 53: 1134–40
- Winckler W., Graham R. R., de Bakker P. I., Sun M., Almgren P., Tuomi T., et al. Association testing of variants in the hepatocyte nuclear factor 4alpha gene with risk of type 2 diabetes in 7,883 people. Diabetes 2005; 54: 886–92
- Yamagata K., Furuta H., Oda N., Kaisaki P. J., Menzel S., Cox N. J., et al. Mutations in the hepatocyte nuclear factor‐4alpha gene in maturity‐onset diabetes of the young (MODY1). Nature 1996; 384: 458–60
- Lilja H. E., Suviolahti E., Soro‐Paavonen A., Hiekkalinna T., Day A., Lange K., et al. Locus for quantitative HDL‐cholesterol on chromosome 10q in Finnish families with dyslipidemia. J Lipid Res 2004; 45: 1876–84
- Soro A., Pajukanta P., Lilja H. E., Ylitalo K., Hiekkalinna T., Perola M., et al. Genome scans provide evidence for low‐HDL‐C loci on chromosomes 8q23, 16q24.1‐24.2, and 20q13.11 in Finnish families. Am J Hum Genet 2002; 70: 1333–40
- Yu Y., Wyszynski D. F., Waterworth D. M., Wilton S. D., Barter P. J., Kesaniemi Y. A., et al. Multiple QTLs influencing triglyceride and HDL and total cholesterol levels identified in families with atherogenic dyslipidemia. J Lipid Res 2005; 46: 2202–13
- Li W. D., Dong C., Li D., Garrigan C., Price R. A. A genome scan for serum triglyceride in obese nuclear families. J Lipid Res 2005; 46: 432–8
- Olefsky J. M., Farquhar J. W., Reaven G. M. Reappraisal of the role of insulin in hypertriglyceridemia. Am J Med 1974; 57: 551–60
- Geurts J. M., Janssen R. G., van Greevenbroek M. M., van der Kallen C. J., Cantor R. M., Bu X., et al. Identification of TNFRSF1B as a novel modifier gene in familial combined hyperlipidemia. Hum Mol Genet 2000; 9: 2067–74
- van der Vleuten G. M., Kluijtmans L. A., Hijmans A., Blom H. J., Stalenhoef A. F., de Graaf J. The Gln223Arg polymorphism in the leptin receptor is associated with familial combined hyperlipidemia. Int J Obes (Lond) 2006; 30: 892–8
- van Greevenbroek M. M., van der Kallen C. J., Geurts J. M., Janssen R. G., Buurman W. A., de Bruin T. W. Soluble receptors for tumor necrosis factor‐alpha (TNF‐R p55 and TNF‐R p75) in familial combined hyperlipidemia. Atherosclerosis 2000; 153: 1–8
- Allayee H., Castellani L. W., Cantor R. M., de Bruin T. W., Lusis A. J. Biochemical and genetic association of plasma apolipoprotein A‐II levels with familial combined hyperlipidemia. Circ Res 2003; 92: 1262–7
- Aouizerat B. E., Kane J. P. Apolipoprotein A‐II: active or passive role in familial combined hyperlipidemia. Circ Res 2003; 92: 1179–81
- Martin‐Campos J. M., Escola‐Gil J. C., Ribas V., Blanco‐Vaca F. Apolipoprotein A‐II, genetic variation on chromosome 1q21–q24, and disease susceptibility. Curr Opin Lipidol 2004; 15: 247–53
- Lockhart D. J., Winzeler E. A. Genomics, gene expression and DNA arrays. Nature 2000; 405: 827–36
- Morello F., de Bruin T. W., Rotter J. I., Pratt R. E., van der Kallen C. J., Hladik G. A., et al. Differential gene expression of blood‐derived cell lines in familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol 2004; 24: 2149–54
- Eurlings P. M., Van Der Kallen C. J., Geurts J. M., Kouwenberg P., Boeckx W. D., De Bruin T. W. Identification of differentially expressed genes in subcutaneous adipose tissue from subjects with familial combined hyperlipidemia. J Lipid Res 2002; 43: 930–5
- Meex S. J., van der Kallen C. J., van Greevenbroek M. M., Eurlings P. M., El Hasnaoui M., Evelo C. T., et al. Up‐regulation of CD36/FAT in preadipocytes in familial combined hyperlipidemia. FASEB J 2005; 19: 2063–5
- Van Eerdewegh P., Little R. D., Dupuis J., Del Mastro R. G., Falls K., Simon J., et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 2002; 418: 426–30
- Hugot J. P., Chamaillard M., Zouali H., Lesage S., Cezard J. P., Belaiche J., et al. Association of NOD2 leucine‐rich repeat variants with susceptibility to Crohn's disease. Nature 2001; 411: 599–603
- Ogura Y., Bonen D. K., Inohara N., Nicolae D. L., Chen F. F., Ramos R., et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001; 411: 603–6
- Mahaney M. C., Almasy L., Rainwater D. L., VandeBerg J. L., Cole S. A., Hixson J. E., et al. A quantitative trait locus on chromosome 16q influences variation in plasma HDL‐C levels in Mexican Americans. Arterioscler Thromb Vasc Biol 2003; 23: 339–45
- Shearman A. M., Ordovas J. M., Cupples L. A., Schaefer E. J., Harmon M. D., Shao Y., et al. Evidence for a gene influencing the TG/HDL‐C ratio on chromosome 7q32.3‐qter: a genome‐wide scan in the Framingham study. Hum Mol Genet 2000; 9: 1315–20
- Dastani Z., Quiogue L., Plaisier C., Engert J. C., Marcil M., Genest J., et al. Evidence for a gene influencing high‐density lipoprotein cholesterol on chromosome 4q31.21. Arterioscler Thromb Vasc Biol 2006; 26: 392–7
- Lyssenko V., Almgren P., Anevski D., Orho‐Melander M., Sjögren M., Saloranta C., Tuomi T., Groop L., the Botnia Study Group. Genetic Prediction of Future Type 2 Diabetes. PLoS Med 2005; 2: 1299–308
- Altshuler D., Brooks L. D., Chakravarti A., Collins F. S., Daly M. J., Donnelly P. A haplotype map of the human genome. Nature 2005; 437: 1299–320
- Collins F. S., Guyer M. S., Charkravarti A. Variations on a theme: cataloging human DNA sequence variation. Science 1997; 278: 1580–1
- Risch N., Merikangas K. The future of genetic studies of complex human diseases. Science 1996; 273: 1516–7
- Lander E. S. The new genomics: global views of biology. Science 1996; 274: 536–9
- Jorgenson E., Witte J. Coverage and Power in Association Studies. Am J Hum Genet 2006; 78: 884–8