
Abstract
Purpose
It has long been thought that the carcinogenic effect of radiation resulted from the induction of oncogenic mutations which then led to an increase in the proportion of cancer-bearing individuals. However, even as early as the 1960s, there were indications that the carcinogenic effect of radiation might result from the induction of an earlier onset of cancer. Recently, the former notion was challenged by its inability to explain time-dependent decline of the relative risk following an exposure to radiation, and a parallel shift of mouse survival curves toward younger ages following an exposure to radiation. The two observations are clearly understood if it is assumed only that a radiation exposure causes an earlier onset of spontaneously occurring cancers.
Method
In the present study, a critical review was conducted which examined papers that showed dose responses which apparently supported the mutation induction theory of radiation carcinogenesis.
Results
It was found that there were two types of misleading experimental designs: one consisted of studies in which observations were prematurely terminated, and which consequently hid a complete story of radiation carcinogenesis. The other set of papers used age adjustments which were derived from the idea that the life shortening effect of radiation needs to be compensated for since tumor mortality becomes higher among older subjects. This type of adjustment appeared reasonable but was found actually to be a different form of description on an earlier onset of cancer following radiation exposures.
Conclusion
In mouse experiments, radiation exposures did not lead to the induction of a large increase in the proportion of tumor deaths when life-long observations were made. Human epidemiologic data are also in line with the earlier onset hypothesis of radiation action. It should be cautioned, however, that the earlier onset model applies only to malignancies whose mortality increases rapidly with the increase of age and does not apply to diseases of short latency such as childhood leukemia and thyroid cancers.
Introduction
It happened once for us how to select cancer cases for studying radiation effects on the TP53 tumor suppressor gene in cancers which occurred in atomic-bomb (A-bomb) survivors. While it was obvious to prioritize cancers which occurred among the survivors of the highest dose group, the next question was how to deal with the latency period. This is because relative risk (RR) for cancer decreases with the increase of years since radiation exposure. Should early onset cases be selected as their RR is higher? Now the question is when we see a general statement that the RR is elevated, one can easily take it to mean that excess cases had occurred, but is this correct?
In fact, this is not always correct because, although it is true that the mortality or incidence rate has increased compared to that of sex- and age-matched controls (the induction theory) at one time point, this observation may also occur if the age of onset of spontaneous cancers (which were destined to occur) is shifted toward younger ages (the etiologic theory) because the background cancer mortality increases rapidly with advancement of the age. Thus, an increased risk for cancer does not necessarily mean that an excess of cancer cases has occurred and the RR of two, for example, would not necessarily mean that one sample out of two is radiation affected and the other is spontaneous in origin.
To describe the risk of radiation-induced cancer mortality, either interpretation (an upward or leftward shift of the mortality curve) can be used. However, if we want to understand the underlying molecular mechanisms involved in cancer induction by radiation, only one interpretation will be correct. Since the 1960s, entire lifespan observations of irradiated mice showed, rather surprisingly, that there is little evidence for a large increase in the number of tumor deaths other than a dose-dependent life shortening. Instead, there were publications which raised the possibility of an earlier onset of cancer. It is noted that cancers that we are concerned consist of those which show an increase of background mortality or incidence with a power function of the age, and do not include those with short latency periods (time from exposure to disease onset) like leukemia or thyroid cancers.
For example, Upton et al. (Citation1960) wrote that ‘If a somatic mutation is involved, radiation may merely create conditions favorable for the selection of spontaneous carcinogenic mutants, possibly by excessive growth stimulation through homeostatic mechanisms’.
Lindop and Rotblat (Citation1962) concluded that ‘all causes are accelerated by radiation but not at the same rate, and leukemia has been brought forward the most, and pulmonary tumors the least’.
Kohn and Guttman (Citation1963) presented data which showed that in CAF1 mice, radiation exposures did not increase the proportion of animals bearing tumors (pulmonary tumors consisted of 60 to 90% of all tumors in this strain of mice) but led to a shortening of the lifespan. The number of tumors per tumor-bearing mouse did not increase either.
Fry et al. (Citation1978) stated that in B6CF1 mice, ‘in the case of lethal lung tumors, the slope of the age-specific rates for tumors after irradiation is similar to the slope for control mice but is displaced to the left (toward younger ages)’.
Storer (Citation1986) also mentioned that ‘For those cancers that show a continuing increase in incidence or mortality rate with increasing age, it is impossible to distinguish between a higher total incidence and simply an earlier onset in an irradiated population. Albert and Altshuler (Citation1976) suggested that the principal effect of a carcinogen exposure may be due to a decrease in the latent period required for the expression of a tumor in the subpopulation that would have developed the tumor later in the absence of an exposure. On the other hand, for cancers that do not show a progressive increase with age, such as some forms of leukemia, this reasoning does not apply’.
Takahashi et al. (Citation1992) mentioned in their mouse study that ‘the liver tumors observed in the present study are histologically quite similar to spontaneous tumors, but they are clearly different from the tumors induced by chemical carcinogens’, which can be interpreted to mean that radiation-related tumors result from accelerated manifestation of spontaneous tumors caused by other factors.
The aim of the present study is to review the mutation induction theory of radiation carcinogenesis, and to revisit the general thoughts that radiation exposures induce oncogenic mutations which lead to carcinogenesis, and that once oncogenic mutations were induced, an intervention in this process would no longer be possible.
Pitfalls in studies which indicated an increased cancer incidence
Premature termination of the experiments provides only a one-time cross-sectional picture of total carcinogenesis processes which take a whole life
It was not rare in the past to find studies which dealt with radiation exposure and cancer, but which were terminated prematurely at the time when the tumor deaths just started in the control group. The underlying motivations were two-fold; first, it can save time and costs, and second, to avoid a gradual increase in the proportion of cancer deaths in the control group, which decreases the impact of contrasting results between the irradiated and control group (). Under such conditions, one can only see the tip of an iceberg, which hid the inconvenient fact that the results gradually changed if the mice were allowed to live longer.
Figure 1. (A) Tumor mortality starts to increase earlier in the exposed group. Thus, premature termination of the follow up shows nothing except an increase in the frequency which is a false interpretation. (B) Liver tumor incidence in male B6C3F1 mice sacrificed at 13 months of age (Takahashi et al. Citation1992). (C) Incidence of mammary neoplasms in rats at 11 months of age (Bond et al. Citation1960).
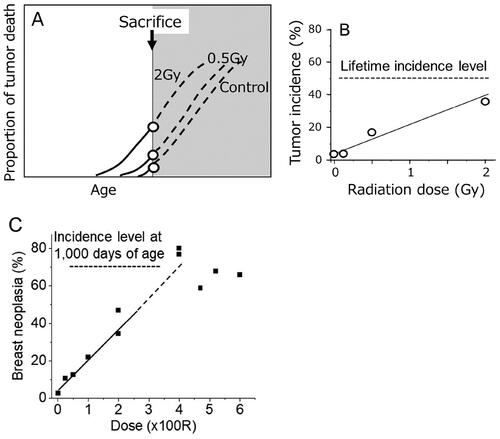
One example is irradiation of male B6C3F1 mice where observations were terminated at 13 months of age when over 90% of the animals were still alive (). The results may appear to show a clear dose-related increase in the incidence of liver tumors (Takahashi et al. Citation1992). However, the control frequency of liver tumors in male mice of this strain is about 50% if they were allowed to live their whole lifespan (Tanaka et al. Citation2007). In other words, if a life-long observation were made, no such dose response would have been observed. Because life-long observations showed a linear relationship between life shortening and radiation dose (e.g. Storer,Citation1965 see ), the observation shown in merely indicates a cross-sectional picture during gradual tumor development which takes place over an entire life. It should be stressed that such an experimental design hid the actual whole course of events.
shows another example of a prematurely terminated study on mammary neoplasms in rats which consisted of adenocarcinomas (AC), adenofibromas, fibroadenomas (FA), and fibrosarcomas which was terminated when the animals were 11 months of age (Bond et al. Citation1960). Here too, although a clear dose response is shown, such a dose response merely represents a one-time snapshot out of continuously developing events since about 70% of the unirradiated rats had AC and/or FA at 1000 days of age (Shellabarger et al. Citation1980).
Age-adjusted tumor mortality or incidence merely calculated the radiation effect on earlier onset
Age adjustment is a standard technique used in epidemiology when the mortality or incidence of age-related diseases such as cancer is to be compared between two populations in which the age distributions are different. For this purpose, one first adjusts the number of individuals in the study population to fit with the number in the reference group for each age stratum. Second, one multiplies the corrected number of individuals by the mortality rate or incidence observed in the population in concern. Then, finally the data is summed in all age strata to determine the age-adjusted mortality or incidence in the population.
There is a series of studies which used this technique to describe the carcinogenic effects of radiation. The reason why age adjustments were thought necessary was to avoid underestimates of radiation effects because the lifespan is shortened among the irradiated animals, and tumors are a disease of the elderly (Ullrich et al. Citation1977). Here, the animals were followed until their natural deaths. One example is shown in which describes the dose response seen with the incidence of lung adenocarcinomas in female BALB/c mice. The incidence of tumors is noted as being age adjusted (Ullrich and Storer Citation1979). Let us examine what that means.
Figure 2. (A) Age-adjusted incidence of lung adenocarcinomas in female BALB/c mice (Ullrich and Storer Citation1979). (B) the first step of age adjustment is to increase the number of individuals at age A (upward arrow). (C) the second step is to multiply the adjusted number by the observed mortality rate, which is equal to the mortality of the control group, but the age was advanced by X days here.
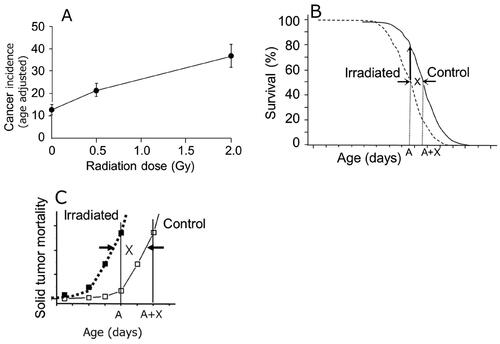
The first step is to correct the number of individuals in each age stratum or interval (the upward arrow in ). The second step is to multiply the adjusted number by the observed mortality rate (). Although the authors of the papers were not aware of it, this observed mortality in the exposed group is in fact equal to the mortality of the control group, but the age was advanced by X days which is the amount of time the mortality curve was shifted to the left or toward younger ages from the control mortality curve. Therefore, the mean age-adjusted mortality (or incidence) of the exposed group expressed the mortality (or incidence) of the control group, but the age was advanced by X days. Thus, the age adjustment performed here is a different way of estimating the effect of an earlier onset. Having said this, the age adjustment performed under such circumstances does not seem adequate because life shortening does not happen by chance but as a natural consequence in the processes of radiation carcinogenesis and thus should not be manipulated.
In order to evaluate how much of an impact such age adjustments can produce, let us assume there are two mouse groups consisting of 130 individuals each (), and the incidence of tumor death is advanced by 100 days in the exposed group. Here, it is assumed that all individuals die by cancer. The first step of calculating the age adjustment is to correct the number of individuals in each age stratum, and then multiply the number by the tumor mortality which was observed. Summing over all age strata gave rise to 216.7 out of 520 (42%) in the exposed group (last line in ) and 130 tumor deaths out of 520 (25%) in the control group. This means that when the mean lifespan was shortened by 100 days, the age adjustment had caused an increase in the proportion of tumor deaths by 1.7 times (42%/25%).
Table 1. Age-adjusted tumor mortality in the exposed group when deaths occurred 100 days earlier than control.
Because the latency period between a radiation exposure and tumor detection may vary among different tissues, the age adjustment does not imply that every tissue behaves in the same fashion. For example, mouse immature oocytes are extremely sensitive to the killing effect of radiation (Dobson and Kwan Citation1977), which causes an earlier onset of menopause and hormonal perturbations. Such a condition can elevate sensitivities toward the development of ovarian and hormone-related tumors. Thus, the proportion of mice bearing ovarian tumors increases very rapidly to reach about 40% at doses up to 0.5 Gy and then levels off (Ullrich and Storer Citation1979) probably because an exposure to 0.5 Gy kills over 90% of immature oocytes, and consequently a further increase in the dose would not lead to a further strengthening of the pro-tumorigenic microenvironment.
In contrast, instead of thinking that the tumor mortality might be higher and calculating a hypothetical age-adjusted mortality, if the epidemiologic concept of incidence, which is expressed per Person⋅Years (PY) or Mouse⋅Days (MD), were used, the results could be described more easily because mortality can be expressed as the number of deaths divided by PY (or MD), and an earlier onset makes the denominator smaller even though the actual number of tumor deaths is the same. Nevertheless, whether or not an excessive number of tumor deaths has occurred cannot be determined until an entire lifespan observation has been made.
No large increase in the proportion of tumor deaths is observed in other lifespan studies
The case of BDF1 mice
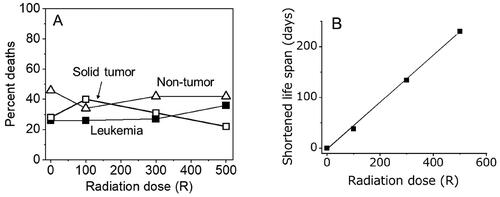
About 55% of female BDF1 mice (a hybrid of the C57BL and DBA strains) die naturally from malignancies (leukemia and solid tumors), and about 45% die from non-tumor causes (Storer Citation1965). Radiation exposures delivered to such mice with doses of 100 R, 300 R or 500 R (observations were made until over 80% of the animals had died naturally) did not increase the proportion of tumor death () but shortened the mean lifespan by 38, 134, and 230 days, respectively, which was linearly related to the dose () (Storer Citation1965). The results clearly showed that radiation exposure shortened the latency of cancer development but did not increase the proportion of tumor deaths. Similar results for no increased proportion of tumor death by radiation exposure are reported for BALB/c and C57BL/6 mice (Maisin et al. Citation1991).
Figure 3. (A) Neither an increased proportion in tumor deaths nor a decreased proportion of non-tumor deaths were observed following radiation exposures of up to 500 R (Storer Citation1965). (B) in the same study, life span was reduced in a dose dependent manner.
The case of B6C3F1 mice also indicates no large increase in the proportion of tumor deaths
is a summary of a rather recent mouse study consisting of 500 males and 500 females each in control and exposed groups. Mice were allowed to live their entire life and were autopsied. A total dose of 8 Gy was given at a very low dose rate (about 20 mGy/day for 400 days) (Tanaka et al. Citation2007). The mice used were the B6C3F1 strain (a hybrid of C57BL and C3H), and about 90% of them died as a result of tumors, which is quite different from humans. Nevertheless, it is impressive to see that the results did not indicate a large increase in the proportion of tumor deaths either as a whole or by organs (). Because as much as 90% of the animals died as a result of malignancies, this mouse strain might not be best suited for detecting an increase in the proportion of tumor deaths since a tumor death can happen only once in a lifetime. Nevertheless, the results may look somewhat unexpected by the eyes which support the mutation theory of radiation carcinogenesis, especially since the proportion of tumor deaths did not increase and reach 100% but remained at the same 90% value following an exposure to as high as 8 Gy of chronic radiation. However, there is some evidence for the induction of tumors. Specifically, the mean tumor multiplicity per mouse which includes both fatal and incidental tumors increased from 1.97 to 2.33 (a 1.2-times increase) in males and 1.75 to 2.54 (a 1.5-times increase) in females even though the irradiated animals died earlier than the control animals by 100 days. The slightly larger increase in females was due to an increased proportion of hormone-related tumors, which needs caution in its interpretation (see below). Admitting that an about 1.2-times increase occurred in tumor multiplicity with an exposure to 8 Gy, this is remarkably small. The authors mentioned that ‘the life shortening … is due to death from a variety of neoplasms and not from increased incidence of specific neoplasms’. In this context, it is also described that in CAF1 mice, the tumor multiplicity per tumor-bearing mouse did not increase after exposure to various doses of radiation (Kohn and Guttman Citation1963).
Figure 4. Proportion of fatal tumors (A) and incidence of tumors (B) in B6C3F1 mice that were exposed chronically to 8 Gy and observed until natural deaths (Tanaka et al. Citation2007). The two symbols connected by each line are the control group (left) and the exposed group (right). Open symbols indicate statistically significant cases when compared to the control values. In this study, the mean life span was shortened by about 100 days (Tanaka et al. Citation2003).
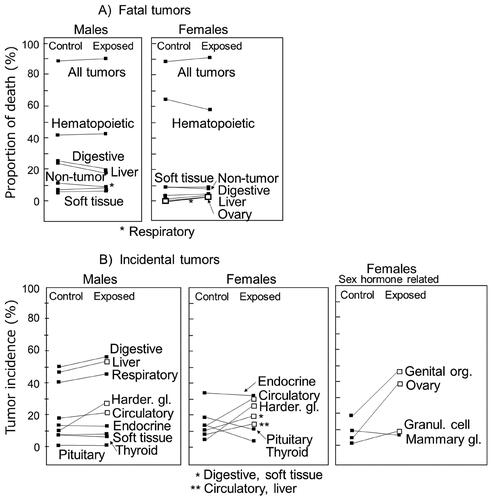
On the other hand, the proportion of incidental tumors (those that were found at autopsy) was found to be increased in some organs (), but tumors in genital organs, ovaries, and granulosa cells in female mice are related to an unusually high vulnerability of resting oocytes to radiation (e.g. about half of them would die with an exposure to 0.2 Gy) (Dobson and Kwan Citation1977). As a consequence, a radiation exposure can easily cause an early menopause and hormonal derangements which increases sensitivity toward the development of hormone-receptor positive tumors. However, human resting oocytes are not so sensitive to radiation, and hence mice are not considered to be a suitable animal model for human females. On the other hand, the proportion of Harderian gland tumors was also increased in both sexes, but this organ does not seem to exist in humans (Albert et al. Citation1986) and the reason for its high sensitivity in developing tumors is not understood.
Paths toward understanding radiation carcinogenesis
Tumor mortality begin to increase earlier following an exposure to radiation but not in proportion to the background rate
The mutation induction theory of radiation carcinogenesis hypothesizes that only a fraction of the irradiated individuals will be affected, and that the remainder will not be affected. This means that among radiation-exposed individuals, tumor deaths would begin to occur earlier than usual due to the presence of affected individuals but would end with normal ages due to the presence of non-affected individuals. In other words, as it is assumed that there exist two subpopulations with different mean lifespan in the irradiated population, the distribution of total individual lifespan should become wider which may cause a shallower mean slope in the survival curve or mortality curve, but this is clearly not the case as the slopes are nearly the same in both irradiated and control groups ( for example). Indeed, the tumor mortality data fit closely to the shifted control curve toward younger ages (). Thus, it is highly likely that the earlier occurrence of tumor deaths following an exposure to radiation is caused by a horizontal shift of the age-related mortality curve and that all the tumors in the exposed group are affected.
Figure 5. (A) Temporal changes in solid tumor mortality in mice after acute exposures to 1.9 Gy (Sasaki and Fukuda Citation2005; Nakamura Citation2023). B) Temporal changes in cancer mortality in A-bomb survivors who were exposed to 1 Gy at age 10 (Nakamura Citation2023). C) RR for solid tumor mortality in mice (Nakamura Citation2023).
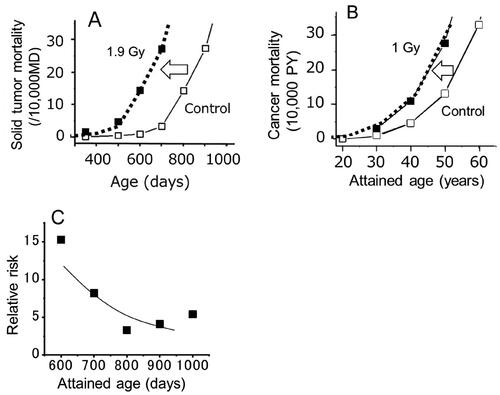
In both mice and humans, a close inspection of cancer mortality data at the time when the cancer mortality starts to increase reveals that the mortality in the exposed group does not increase in proportion to the mortality of the control group, but started to increase when the baseline mortality was still nearly zero ( is for mice and for A-bomb survivors) (Nakamura Citation2023). Thus, if we assume that the curve of the exposed group had shifted upwardly (the induction model), the ratio of the two mortality values at the same age (which represents the RR) changes with the attained age. Specifically, at a young age, even though the number of the deaths in the exposed group is small, the RR becomes a large value because the corresponding mortality of the control group is still close to zero. In contrast, in middle to old ages, although the number of affected individuals in the exposed group may become larger, the mortality of the control group has also increased, and thus the RR becomes smaller ().
With the mutation theory, the temporal changes of the RR cannot be explained by simple numbers, and thus in recent epidemiologic studies of A-bomb survivors, the cancer risk is expressed as the RR of survivors who were 30 years of age at the time of exposure and reached age 70 along with instructions for calculation of the RR for those who were exposed at different ages and reached different ages (Brenner et al. Citation2022). However, this RR value at age 70 does not express by itself the fact that individuals with a young age at exposure were at a higher risk of developing cancers at young ages, which does not happen in those who were exposed at older ages.
On the other hand, if we think that the mortality curve of the exposed group has shifted horizontally toward younger ages (with the shape remaining unchanged), a life-time risk value can be shown as weeks, months or years of life lost if one dies from cancer (life shortening is another form of health risk in life). The thick dotted lines in are not fitted curves of the exposed group but are shifted curves of the control response. It is quite impressive to find that simply shifting the control curve horizontally can lead to an exact fit of the observed points in both mice and humans.
Increased cancer mortality does not necessarily mean an increased number of cancer deaths
Epidemiologic studies of A-bomb survivors indicated that the proportion of people dying from cancer following an exposure to 0.1 Sv was estimated to be about a 2% net increase from a background risk of 30% (males: M) or 20% (females: F) exposed at age 10; about a 1% net increase from 25% (M) or 19% (F) exposed at age 30; and a 0.3% or 0.4% net increase from 20% (M) or 16% (F) exposed at age 50 (Preston et al. Citation2003). However, it should be noted that the estimates were made under the condition that both, the control and exposed groups reached the same age. In contrast, if the carcinogenic effect of radiation is to stimulate spontaneously arising tumors to appear earlier than usual, the accelerated tumors would mostly be derived from a spontaneous origin, and the excess number of individuals dying from cancer would remain nearly zero, but of course the lifespan would be shortened (). Why is this nearly zero but not zero? This is because some cancers such as leukemia and thyroid cancer may develop with short latency periods, and at least some of them might arise from the mutagenic effects of radiation (Albert and Altshuler Citation1976; Nakamura Citation2005).
Figure 6. Cancer mortality in A-bomb survivors who were exposed to radiation at the age of 10 (Nakamura Citation2023). The thick dotted line represents a control curve shifted toward younger ages which agreed closely with the epidemiological estimates (■). Because the two curves are essentially parallel, there would be no excess number of cancers in the exposed group if the risks are compared over the entire life span.
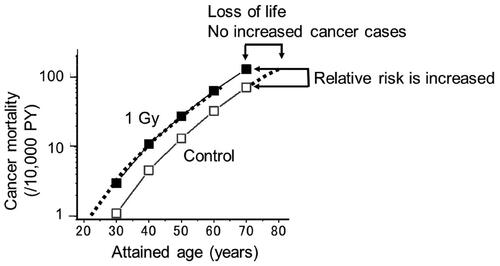
When calculating RR, the exposed to control ratio of cancer mortality or incidence is calculated and it is attempted to show an upward shift in RR at some attained age. However, laymen will ask if the proportion of people dying from cancer may or may not increase during one’s life. In the latter case, since the area below the mortality curves in represents the total number of individuals who died from cancer, and the two curves are essentially parallel, the life-time risk for the number of people dying from cancer would not be increased in the exposed group.
In support of this notion, a recent mouse study using serial sacrifices showed that malignant lymphoma cells were detected at the same age in both exposed (with a total dose of 8 Gy from a chronic exposure) and control animals (Tanaka et al. Citation2017), but a previous study revealed that deaths from malignant lymphoma occurred about 100 days earlier in the exposed group (Tanaka et al. Citation2003; Tanaka et al. Citation2007). This is new and clear evidence that a radiation exposure does not affect the age of tumor cell emergence, but rather accelerates tumor formation and kills the host earlier than usual.
Radiation-induced tissue inflammation and natural aging of the stroma contribute to this effect
The next question to ask is what are the possible biological events which are associated to radiation exposure and affect most natural cancers to appear earlier than usual. To my knowledge, tissue inflammation is the most likely event. For example, a radiation exposure to a tissue appears to create conditions which mimic the feeder layer effect in primary cell cultures in vitro, and there are multiple pathways leading toward the development of tissue inflammation (Nakamura Citation2021). Nevertheless, it is certain that a radiation risk for cancer does not appear immediately after an exposure but takes many months in mice and years in humans until exposed individuals reach their so-called cancer age. Therefore, it seems likely that radiation-induced tissue damage which leads to inflammation could likely persist for a long period until it becomes able to interact with aged stroma in elderly individuals (Fane and Weeraratna Citation2020). Unfortunately, the detailed mechanisms of long-lasting tissue damage are only poorly understood.
Possible reasons why some types of malignancies do not exhibit earlier onset
Lastly, if radiation exposure causes earlier onset of spontaneously arising malignancies, it may be worth asking why some types of cancers do not exhibit earlier onset following exposure to radiation. In this regard, mouse studies are not informative because individual ages at death with specific tumors are not usually presented. Even among the Life Span Study (LSS) population of A-bomb survivors, substantial number of organs show the lack of significantly increased radiogenic risk due to small number of cases (insufficient statistical power). Even among major types of cancers, several of them do not show elevated risks by an exposure; these included cancers of cervix, kidney, gallbladder, rectum, prostate, and pancreas (Preston et al. Citation2007), and each of these seems to manifest a specific reason for its behavior.
First, the RR for prostate cancer was not significantly elevated in the 2007 incidence report (Preston et al. Citation2007) but it became significant in a subsequent study during which time the number of the cases doubled and more than half of them were diagnosed among those who were less than 20 years of age at the time of the bombing (Mabuchi et al. Citation2021). Likewise, although the trend is not yet certain, the RR for pancreatic cancer might be in a process of revealing the radiation effect as it is now significantly elevated in women (excess RR per Gy = 0.7; n = 417) but not in men (excess RR per Gy = 0.07; n = 306). The number of cases increased by 40% during the 11 years of additional follow up since 2007 (Sadakane et al. Citation2019).
As for cervical cancer, it is well known to be caused mostly by infection with human papilloma virus (HPV). The background incidence rate did not show an age-related rapid increase but was approximately linear after the age of 20 years until the 1980s, but then the incidence at age 40 and onward rapidly declined to give to a plateau value while the incidence at ages below 40 remained nearly the same (Supplemental Figure S1). Thus, even if an earlier shift of the onset age could be caused by irradiation, it appears difficult to detect an increased risk from the data.
Gallbladder cancer is known to be associated with gallstone and bacterial infection. It is hypothesized that epithelial damage caused by gallstone and/or bacterial infection leads to cell-death-and-regeneration cycles (chronic inflammation) (Espinoza et al. Citation2016), which leads to cancer development. Thus, the effect of radiation might be masked by the natural promotive effects.
Renal cancer consists of only 218 cases among the LSS population (Grant et al. Citation2021). While the increased risk for renal cancer is reported among ankylosing spondylitis patients (Weiss et al. Citation1994) and cervical cancer patients (Boice et al. Citation1988; Kleinerman et al. Citation1995) who underwent radiotherapy, those patients received fractionated multiple exposures. On the other hand, a single exposure to 10 Gy to the kidneys of rhesus monkeys did not induce any sign of inflammation during 6 months of post-exposure observations (Cohen et al. Citation2020). Similar lack of glomerular inflammation was observed in CAF1 and BALB/c mice following an exposure to about 5 Gy (Guttman and Kohn Citation1960). These data indicate that the lack of evidence for the increased risk among A-bomb survivors might be due to the kidney’s refractory nature against the induction of inflammation from a single exposure to radiation.
For rectum cancer, it seems most difficult to understand the lack of evidence for an elevated risk. After whole genome analysis of cancers, no difference in the mutation spectrum was detected between the colon cancer (for which radiation exposure can increase the risk) and rectal cancer (radiation exposure does not seem to increase the risk) (Cancer Genome Atlas Network Citation2012). Although it has been reported that the risk may increase following radiotherapy for prostate cancer (e.g. Rombouts et al. Citation2018) or cervical cancer (Chaturvedi et al. Citation2007), the total dose is quite high (e.g. 30–60 Gy: Kleinerman et al. Citation1995). In view of these observations, it seems reasonable to accept the notion that there are probably ‘effective’ thresholds (Boice et al. Citation1988).
Conclusion
There are four major observations.
In the present earlier onset hypothesis, it is those common cancers which develop late in one’s life that we are concerned, while childhood leukemia and thyroid cancer are probably exceptions since their age response is quite different from common old-age related malignancies.
A unit of radiation risk for cancer as a whole (except for some short latency types as mentioned above) may be described by the time lost in life, which can be defined by the age at the time of exposure, sex, and radiation dose. It seems likely that the value is constant throughout one’s life and can be easily understood by individuals when compared with the relative risk which is a measure for a population under study.
Since somatic mutations in tissue stem cells do not seem likely to be the cause of radiation carcinogenesis but a tissue response is, the carcinogenic effect of radiation is better to be classified as a deterministic effect, except for some childhood malignancies as mentioned above.
Because inflammation is likely to be involved in radiation carcinogenesis, there should be pathways which can be used to intervene in the process of radiation-related carcinogenesis to reduce its effects. For example, non-steroid anti-inflammatory drugs (NSAIDs) are reported to mitigate, although not completely, the development of cancers induced by a potent chemical carcinogen (e.g. Harris et al. Citation2000). Also, calorie restriction has been recognized as a potent approach to reduce the tumor incidence in both irradiated and non-irradiated mice, and to extend life spans (Yoshida et al. Citation1997; Shang et al. Citation2014). Calorie restrictions affect various biological functions, and the effect is not yet fully understood, but anti-inflammatory responses appear to be involved (e.g. Chung et al. Citation2002). Thus, here may be a possible link between calorie restriction and a reduction of tumor development, and if so, studies aimed at preventing development of radiation-related solid cancers may also lead to prevention of spontaneous cancers.
Supplemental Material
Download MS Word (23.3 KB)Acknowledgement
The author is grateful to Dr. Leon Kapp for his careful reading of the manuscript, Dr. Koji Ono (THCU) for his continued encouragement, and Dr. Hiromi Sugiyama (RERF) for her advice.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes on contributors
Nori Nakamura
Nori Nakamura, PhD, is a radiation biologist working at the Radiation Effects Research Foundation for more than 30 years and interested in the mechanisms of radiation carcinogenesis.
References
- Albert RE, Altshuler B. 1976. Assessment of environmental carcinogen risks in terms of life shortening. Environ Health Perspect. 13:91–94. doi:10.2307/3428242
- Albert DM, Frayer WC, Black HE, Massicotte SJ, Sang DN, Soque J. 1986. The harderian gland: its tumors and its relevance to humans. Trans Am Ophthalmol Soc. 84:321–341.
- Boice JDJr, Engholm G, Kleinerman RA, Blettner M, Stovall M, Lisco H, Moloney WC, Austin DF, Bosch A, Cookfair DL, et al. 1988. Radiation dose and second cancer risk in patients treated for cancer of the cervix. Radiat Res. 116(1):3–55. doi:10.2307/3577477
- Bond VP, Cronkite EP, Lippincott SW, Shellabarger CJ. 1960. Studies on radiation-induced mammary gland neoplasia in the rat. III. Relation of the neoplastic response to dose of total-body radiation. Radiat Res. 12(3):276–285. doi:10.2307/3570916
- Brenner AV, Preston DL, Sakata R, Cologne J, Sugiyama H, Utada M, Cahoon EK, Grant E, Mabuchi K, Ozasa K. 2022. Comparison of all solid cancer mortality and incidence dose-response in the Life Span Study of Atomic bomb survivors, 1958-2009. Radiat Res. 197(5):491–508. doi:10.1667/RADE-21-00059.1
- Cancer Genome Atlas Network. 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 487(7407):330–337. doi:10.1038/nature11252
- Chaturvedi AK, Engels EA, Gilbert ES, Chen BE, Storm H, Lynch CF, Hall P, Langmark F, Pukkala E, Kaijser M, et al. 2007. Second cancers among 104,760 survivors of cervical cancer: evaluation of long-term risk. J Natl Cancer Inst. 99(21):1634–1643. doi:10.1093/jnci/djm201
- Chung HY, Kim HJ, Kim KW, Choi JS, Yu BP. 2002. Molecular inflammation hypothesis of aging based on the anti-aging mechanism of calorie restriction. Microsc Res Tech. 59(4):264–272. doi:10.1002/jemt.10203
- Cohen EP, Farese AM, Parker GA, Kane MA, MacVittie TJ. 2020. Lack of cellular inflammation in a non-human primate model of radiation nephropathy. Health Phys. 119(5):588–593. doi:10.1097/HP.0000000000001329
- Dobson RL, Kwan TC. 1977. The tritium RBE at low-level exposure—variation with dose, dose rate, and exposure duration. Curr Top Radiat Res Q. 12:44–62.
- Espinoza JA, Bizama C, García P, Ferreccio C, Javle M, Miquel JF, Koshiol J, Roa JC. 2016. The inflammatory inception of gallbladder cancer. Biochim Biophys Acta. 1865(2):245–254. doi:10.1016/j.bbcan.2016.03.004
- Fane M, Weeraratna AT. 2020. How the ageing microenvironment influences tumour progression. Nat Rev Cancer. 20(2):89–106. doi:10.1038/s41568-019-0222-9
- Fry RJM, Staffeldt E, Tyler SA. 1978. Some problems arising in analysis of large-scale animal irradiation experiments. Environ Int. 1(6):361–366. doi:10.1016/0160-4120(78)90015-6
- Grant EJ, Yamamura M, Brenner AV, Preston DL, Utada M, Sugiyama H, Sakata R, Mabuchi K, Ozasa K. 2021. Radiation risks for the incidence of kidney, bladder and other urinary tract cancers: 1958–2009. Radiat Res. 195(2):140–148. doi:10.1667/RADE-20-00158.1
- Guttman PH, Kohn HI. 1960. Progressive intercapillary glomerulosclerosis in the mouse, rat, and Chinese hamster, associated with aging and x-ray exposure. Am J Pathol. 37(3):293–307.
- Harris RE, Alshafie GA, Abou-Issa H, Seibert K. 2000. Chemoprevention of breast cancer in rats by celecoxib, a cyclooxygenase 2 inhibitor. Cancer Res. 60(8):2101–2103.
- Kleinerman RA, Boice JDJr, Storm HH, Sparen P, Andersen A, Pukkala E, Lynch CF, Hankey BF, Flannery JT. 1995. Second primary cancer after treatment for cervical cancer. An international cancer registries study. Cancer. 76(3):442–452. doi:10.1002/1097-0142(19950801)76:3<442::AID-CNCR2820760315>3.0.CO;2-L
- Kohn HI, Guttman PH. 1963. Age at exposure and the late effects of x-rays. Survival and tumor incidence in CAF mice irradiated at 1 to 2 years of age. Radiat Res. 18(3):348–373. doi:10.2307/3571501
- Lindop PJ, Rotblat J. 1962. The age factor in the susceptibility of man and animals to radiation. I. The age factor in radiation sensitivity in mice. Br J Radiol. 35(409):23–31. doi:10.1259/0007-1285-35-409-23
- Mabuchi K, Preston DL, Brenner AV, Sugiyama H, Utada M, Sakata R, Sadakane A, Grant EJ, French B, Cahoon EK, et al. 2021. Risk of prostate cancer incidence among atomic bomb survivors: 1958–2009. Radiat Res. 195(1):66–76. doi:10.1667/RR15481.1
- Maisin JR, Wambersie A, Gerber GB, Mattelin G, Lambiet-Collier M, De Coster B, Gueulette J. 1991. Life-shortening and disease incidence in mice after exposure to gamma rays or high-energy neutrons. Radiat Res. 128(1 Suppl):S117–S123.
- Nakamura N. 2005. A hypothesis: radiation-related leukemia is mainly attributable to the small number of people who carry pre-existing clonally expanded preleukemic cells. Radiat Res. 163(3):258–265. doi:10.1667/rr3311
- Nakamura N. 2021. Reexamining the role of tissue inflammation in radiation carcinogenesis: a hypothesis to explain an earlier onset of cancer. Int J Radiat Biol. 97(10):1341–1351. doi:10.1080/09553002.2021.1955998
- Nakamura N. 2023. Radiation-induced increases in cancer mortality result from an earlier onset of the disease in mice and atomic bomb survivors. Int J Radiat Biol. 99(8):1139–1147. doi:10.1080/09553002.2023.2158246
- Preston DL, Ron E, Tokuoka S, Funamoto S, Nishi N, Soda M, Mabuchi K, Kodama K. 2007. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat Res. 168(1):1–64. doi:10.1667/RR0763.1
- Preston DL, Shimizu Y, Pierce DA, Suyama A, Mabuchi K. 2003. Studies of mortality of atomic bomb survivors. Report 13: Solid cancer and noncancer disease mortality: 1950–1997. Radiat Res. 160(4):381–407.
- Rombouts AJM, Hugen N, van Beek JJP, Poortmans PMP, de Wilt JHW, Nagtegaal ID. 2018. Does pelvic radiation increase rectal cancer incidence? - A systematic review and meta-analysis. Cancer Treat Rev. 68:136–144. doi:10.1016/j.ctrv.2018.05.008
- Sadakane A, French B, Brenner AV, Preston DL, Sugiyama H, Grant EJ, Sakata R, Utada M, Cahoon EK, Mabuchi K, et al. 2019. Radiation and risk of liver, biliary tract, and pancreatic cancers among atomic bomb survivors in Hiroshima and Nagasaki: 1958–2009. Radiat Res. 192(3):299–310. doi:10.1667/RR15341.1
- Sasaki S, Fukuda N. 2005. Temporal variation of excess mortality rate from solid tumors in mice irradiated at various ages with gamma rays. J Radiat Res. 46(1):1–19. doi:10.1269/jrr.46.1
- Shang Y, Kakinuma S, Yamauchi K, Morioka T, Kokubo T, Tani S, Takabatake T, Kataoka Y, Shimada Y. 2014. Cancer prevention by adult-onset calorie restriction after infant exposure to ionizing radiation in B6C3F1 male mice. Int J Cancer. 135(5):1038–1047. doi:10.1002/ijc.28751
- Shellabarger CJ, Chmelevsky D, Kellerer AM. 1980. Induction of mammary neoplasms in the Sprague-Dawley rat by 430keV neutrons and X-rays. J Natl Cancer Inst. 64(4):821–833.
- Storer JB. 1965. Radiation resistance with age in normal and irradiated population of mice. Radiat Res. 25(3):435–459. doi:10.2307/3571760
- Storer JB. 1986. Carcinogenic effects: an overview. In: AC Upton, RE Albert, FJ Burns, RE Shore, editors. Radiation carcinogenesis. New York: Elsevier; p. 11–22.
- Takahashi T, Watanabe H, Dohi K, Ito A. 1992. 252Cf relative biological effectiveness and inheritable effect of fission neutrons in mouse liver tumorigenesis. Cancer Res. 52(7):1948–1953.
- Tanaka S, Tanaka IB, 3rd, Sasagawa S, Ichinohe K, Takabatake T, Matsushita S, Matsumoto T, Otsu H, Sato F. 2003. No lengthening of life span in mice continuously exposed to gamma rays at very low dose rates. Radiat Res. 160(3):376–379. doi:10.1667/rr3042
- Tanaka IB, 3rd, Tanaka S, Ichinohe K, Matsushita S, Matsumoto T, Otsu H, Oghiso Y, Sato F. 2007. Cause of death and neoplasia in mice continuously exposed to very low dose rates of gamma rays. Radiat Res. 167(4):417–437. doi:10.1667/RR0728.1
- Tanaka IB, 3rd, Komura J, Tanaka S. 2017. Pathology of serially sacrificed female B6C3F1 mice continuously exposed to very low-dose-rate gamma rays. Radiat Res. 187(3):346–360. doi:10.1667/RR14617.1
- Ullrich RL, Jernigan MC, Storer JB. 1977. Neutron carcinogenesis. Dose and dose-rate effects in BALB/c mice. Radiat Res. 72(3):487–498.
- Ullrich RL, Storer JB. 1979. Influence of γ irradiation on the development of neoplastic disease in mice. III Dose-rate effects. Radiat Res. 80(2):325–342.
- Upton AC, Kimball AW, Furth J, Christenberry KW, Benedict WH. 1960. Some delayed effects of atom-bomb radiations in mice. Cancer Res. 20(2):1–60.
- Weiss HA, Darby SC, Doll R. 1994. Cancer mortality following X-ray treatment for ankylosing spondylitis. Int J Cancer. 59(3):327–338. doi:10.1002/ijc.2910590307
- Yoshida K, Inoue T, Nojima K, Hirabayashi Y, Sado T. 1997. Calorie restriction reduces the incidence of myeloid leukemia induced by a single whole-body radiation in C3H/He mice. Proc Natl Acad Sci U S A. 94(6):2615–2619. doi:10.1073/pnas.94.6.2615