Abstract
Molecular phylogenetics has become a prominent aspect of algal systematics. The field of phylogenetic reconstruction is fast-evolving and novel techniques take time to penetrate taxonomic research. We highlight a selection of advances in phylogenetic inference and evolutionary analysis methods that could, in our opinion, benefit algal systematic studies. The focus of the paper is on model-based techniques. Following a brief introduction to maximum likelihood and Bayesian phylogenetic inference methods, we address model selection and partitioning strategies, and illustrate some issues concerning systematic error (phylogenetic bias), data saturation and tree rooting. We discuss the importance of experimental design (taxon and character sampling) and explore methods to test the reliability of phylogenetic results. Finally, we address methods for estimating ancestral states of discrete and continuous characters and techniques for dating phylogenetic trees. For each of these topics, we provide a brief circumscription, refer to the more specialized literature, and list a selection of software to carry out the analyses.
Introduction
From the early 1990s onwards, molecular phylogenetic techniques have been playing an increasingly important role in algal taxonomic studies (Brodie & Lewis, Citation2007). Several methods are available for inferring phylogenies from molecular data, of which maximum parsimony (MP), maximum likelihood (ML) and Bayesian inference (BI) are the most commonly used (Maggs et al., Citation2007; Mann & Evans, Citation2007). Phylogenetic analysis techniques evolve at a fast rate, and new advances take time to penetrate into algal phylogenetic studies. This is most likely attributable to the statistical and computational nature of the primary literature, and the fact that it usually takes time for new advances to be implemented in user-friendly software. The goal of this paper is to review those advances in phylogenetic and evolutionary analysis that we deem relevant to algal systematic studies and should, in our opinion, be more widely used among algal systematists.
Phylogenetic analysis techniques come in many flavours, each method having its own set of assumptions, merits and drawbacks. We do not aim to review all inference techniques; several excellent synopses and textbooks serve this purpose (e.g. Holder & Lewis, Citation2003; Felsenstein, Citation2004). Inference techniques are commonly subdivided into parametric techniques, which infer trees based on a model of sequence evolution (e.g. ML, BI), and non-parametric techniques, which do not assume such a model. Although MP is often thought of as a non-parametric technique, it comes with some implicit assumptions. For example, equal weights are assigned to all types of nucleotide substitutions in Fitch parsimony (Fitch, Citation1971). The merits and drawbacks of the most common phylogenetic methods have been extensively debated. It has become clear that although MP can yield more accurate results than model-based methods (ML, BI) on certain simulated datasets (Siddall, Citation1998; Pol & Siddall, Citation2001; Kolaczkowski & Thornton, Citation2004), model-based methods outperform MP over a wide range of conditions (e.g. Gaut & Lewis, Citation1995; Huelsenbeck, Citation1995; Swofford et al., Citation2001; Philippe et al., Citation2005a). For that reason, this review will mostly centre on techniques that make explicit use of statistical models of sequence evolution.
ML inference sets out to find the phylogenetic configuration of species and set of model parameters with the highest likelihood of having produced the observed DNA data matrix under the assumed model of sequence evolution. Likelihood analyses evaluate different trees one at a time and identify the set of parameter values that optimize the likelihood for each tree. The tree with the highest overall likelihood score is retained. Several reviews about ML inference, details about likelihood calculation and tree searching shortcuts are available (e.g. Swofford et al., Citation1996; Whelan et al., Citation2001; Holder & Lewis, Citation2003; Felsenstein, Citation2004; Yang, Citation2006).
Bayesian inference techniques are related to ML but work differently. They look for hypotheses (trees and sets of model parameters) with high posterior probabilities. The posterior probability of a hypothesis is proportional to the product of its prior probability and the probability of observing the dataset given the hypothesis (i.e. the likelihood of the hypothesis). The prior probability of different hypotheses is derived from previous knowledge, but because one does not usually want to introduce a bias towards one or another tree or set of model parameters, prior probabilities are usually chosen to be vague, i.e. giving the same prior probability to all hypotheses. This way the likelihood of the hypotheses will determine their posterior probabilities.
Because of the complexity of phylogenetic likelihood functions, posterior probability distributions cannot be calculated analytically. Instead, they are approximated using Markov chain Monte Carlo (MCMC) simulation. At each step in the chain (generation), a change of a parameter is proposed. These parameters include the topology, the branch lengths and the model parameters. If the proposed change increases the posterior, it is accepted and forms the starting point for the next step in the chain. If the change decreases the posterior, it may be accepted or rejected, with the probability of acceptance depending on the amount of change. Whereas small decreases are often accepted, large decreases are usually rejected. During the initial stages of the MCMC, parameters are usually not near their optimal values and proposed changes are accepted very often until parameter values approach their optimal values. These initial stages of MCMC are called the burn-in. Running an MCMC for millions of generations after the burn-in generates a large set of trees that have a high likelihood.
A ‘Bayesian tree’ is calculated by summarizing the MCMC trees. A popular way of doing this is by generating a majority rule consensus of the trees visited during the MCMC after the burn-in. Alternatively, the topology at the highest peak of the posterior probability distribution, commonly called MAP tree, can be calculated. It should be noted that in contrast to the MAP tree, a tree obtained with the majority rule consensus method is not necessarily optimal. Instead, it reflects the most common combination of branches encountered in the MCMC run. MrBayes reports the MAP tree when summarizing the posterior distribution (first tree in the. trprobs file). We refer to the literature for more elaborate introductions to Bayesian phylogenetic inference (e.g. Huelsenbeck et al., Citation2001; Huelsenbeck et al., Citation2002a; Yang, Citation2006).
Many software applications carry out model-based phylogenetic inference, a selection of which is listed in . A comprehensive list of phylogeny programs is maintained by Joe Felsenstein and can be found at: http://evolution.genetics.washington.edu/phylip/software.html
Table 1. A selection of software for Bayesian and maximum likelihood phylogenetic inference. The second column lists the most recent released version at the time of writing and the specified features apply to this version. The listed properties are the method of tree inference (BI or ML), whether the program supports data partitioning, the implemented models of sequence evolution, and the options to deal with rate variation across sites. The last two columns specify whether the program can be run in parallel (i.e. can make use of multiple processors to speed up analyses and/or do more thorough tree searches) and the availability of the program (web means that analyses can be run remotely on a web server). Note that in addition to the DNA based models listed here, most programs implement additional models for analysis of amino-acid, binary and n-state discrete data types.
Model selection
The trees obtained from phylogenetic analyses form the foundation of all further interpretations. It is therefore essential that the obtained trees reflect the evolutionary history of the used marker as closely as possible. The use of statistical models to infer phylogenies follows from the knowledge that the DNA sequences of extant species reflect the evolutionary processes that have acted on them. The parameters of the model of sequence evolution specify in a statistical way how past changes have led to the present diversity of DNA sequences. Models of sequence evolution are manifold and diverse, and choosing one that is suitable for the data at hand is crucial in obtaining reliable phylogenies. In this section, we highlight the most important aspects of common models of sequence evolution and techniques for selecting a suitable model.
The following section (Systematic error) deals with the problems that can occur when the model of sequence evolution deviates too much from the evolutionary processes that have generated the dataset. Obviously, model selection and systematic error are tightly interwoven and we chose to treat them separately because this better reflects natural progress in a phylogenetic analysis. Whereas the aspects of model selection that are treated in the present section are normally considered before the phylogenetic analysis, the next section deals with less obvious aspects of molecular evolution that manifest themselves as errors after a phylogenetic analysis has been carried out.
Basic model elements
It is well-known that certain types of substitutions occur more commonly than others (e.g. transitions vs. transversions, synonymous vs. non-synonymous). The common models of sequence evolution include parameters describing the relative rates of change between different bases (the rate matrix). Some models have only one parameter to distinguish between transition and transversion rates, but in many cases the general time reversible (GTR) model is used. This model describes the relative substitution rates between all combinations of bases (AC, AG, AT, CG, CT, and GT) with five parameters. The base frequencies are a second important component of the model. Sometimes they are simply calculated from the dataset (‘empirical’ base frequencies) but they can also be regarded as parameters of the model. Because sites in an alignment evolve at different rates (e.g. different codon positions), rate variation across sites is usually accounted for in the model, most commonly by assuming that the site rates follow a gamma distribution and/or by incorporating a proportion of invariable sites. Using a discrete gamma distribution (+Γ) and a proportion of invariable sites (+I) each add an extra parameter to the model of sequence evolution, making it more complex.
Partitioning strategies
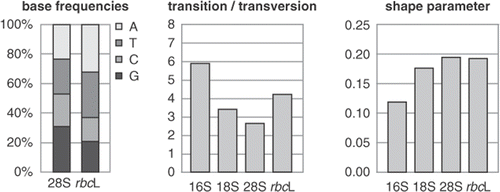
Some datasets are composed of parts that have evolved under different evolutionary processes. For example, when an alignment is composed of multiple markers, parameter estimates of the model of sequence evolution typically differ among them (). If the differences between the evolutionary processes are sufficiently large, partitioning the data into its component markers and allowing each marker to have its own set of model parameters can be expected to result in better fit of the model to the data. Simulation studies and analysis of empirical data have shown that choosing an appropriate partitioning strategy is important for obtaining accurate phylogenetic results from a composite dataset (Brandley et al., Citation2005; Brown & Lemmon, Citation2007). Both over- and under-partitioning yield suboptimal results, under-partitioning leading to the strongest deviations from the expectations.
Fig. 1. Model parameters often differ among markers in a multi-marker dataset. This graph illustrates differences between model parameters for a red algal multi-marker dataset. Base frequencies show lower AT content in 28S than rbcL, while the ratio between transitions and transversions, and the shape parameter of the Γ distribution (α) also differ between markers.
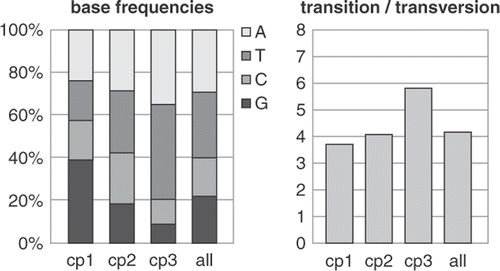
It must be noted that even within a single marker, groups of characters can evolve under different processes (e.g. the codon positions in protein-coding genes, different regions of rRNA molecules, coding vs. non-coding parts) and globally estimated model parameters are often not representative for the different codon positions (). This can be overcome by partitioning the gene into codon positions and uncoupling model parameters among partitions. The heterogeneous model resulting from this practice is called a codon-position model and often provides a much closer fit to the data than a global, homogeneous model (Shapiro et al., Citation2006). Codon position models have been shown to outperform global models in algal datasets, too (Alverson et al., Citation2007; Le Gall & Saunders, Citation2007; Verbruggen et al., Citation2007).
Fig. 2. Model parameters usually differ quite strongly among codon positions of protein-coding genes. These graphs show differences between model parameters for a green algal dataset comprising atpB and rbcL sequences. The base frequencies graph shows marked differences in base composition among codon positions (cp1, cp2, cp3), with a strong AT bias at third codon positions. The fourth column represents the global frequencies, which are not representative for first and third codon positions.
Models for interdependent sites
A number of more exotic models of sequence evolution can be useful to analyse certain datasets or partitions. Such models are usually based on biochemical characteristics of the type of data under consideration. Ribosomal RNA, for example, has a secondary structure composed of stems and loops. Nucleotides in the stems form base pairs and, because there is a selective pressure for maintenance of the rRNA secondary structure, their evolution is interdependent (compensatory base changes). Because phylogenetic inference techniques typically assume independence of characters in an alignment, it would be more correct to include this non-independence in the model of sequence evolution (Schöniger & Von Haeseler, Citation1995; Lewis, Citation2001a). This can be done by partitioning the rRNA into stems and loops and applying a doublet-model to the stems (Schöniger & Von Haeseler, Citation1994; Telford et al., Citation2005; Erpenbeck et al., Citation2007). This model merges paired nucleotides into doublets, and uses those doublets instead of the individual nucleotides as characters for tree inference. The model describes patterns of changes between paired nucleotides (e.g. compensatory vs. non-compensatory changes) and has been applied successfully to algal datasets, yielding a higher fit to the data than standard four-state models (Murray et al., Citation2005; Alverson et al., Citation2007; Leliaert et al., Citation2007).
A similar approach can be used to overcome heterogeneous processes among codon positions. In codon substitution models (Goldman & Yang, Citation1994; Muse & Gaut, Citation1994; Yang et al., Citation2000), triplets of nucleotides are considered as a single character and the substitution models describe changes between such triplets, taking into account that some changes are more likely to occur than others (e.g. synonymous vs. non-synonymous changes). Models that take biochemical characteristics of the data into account fit more closely to the data and yield more accurate results (Schöniger & Von Haeseler, Citation1995; Telford et al., Citation2005; Erpenbeck et al., Citation2007). The downside of such models is that more parameters have to be estimated, resulting in substantially higher computational demands.
Rationale of model selection
It is important to realize that none of the available models of sequence evolution reflect all aspects of the evolutionary history that has resulted in the set of sequences under study. All models are therefore wrong, but models that are sufficiently close to the ‘true model’ will yield accurate results (Posada & Buckley, Citation2004). Methods of model selection aim to identify a model yielding a good trade-off between the fit of the data to the model and the number of model parameters that need to be estimated from the data. Parameter-rich models always yield a better fit to the data, but this comes at a price: more parameters have to be estimated from the same amount of data, resulting in higher computational requirements and less accurate parameter estimates. In this respect, ML and BI behave slightly differently, though. Bayesian analyses are much more sensitive to model under-specification than ML, leading to the recommendation that for BI, “the model should be as complex as possible while still allowing parameters to be identified” (Huelsenbeck & Rannala, Citation2004). This recommendation does not render model selection superfluous but encourages more extensive model selection strategies (see section above: Partitioning strategies; Models for interdependant sites). The necessity of model selection and the methods to achieve it have been reviewed thoroughly (Posada & Buckley, Citation2004; Sullivan & Joyce, Citation2005).
Performing model selection
The most commonly used model selection procedures start by generating a guide tree using a fast method (usually a distance-based algorithm). This tree is taken to be an approximate estimate of the relationships among the taxa in the dataset. Subsequently, the log-likelihood of the guide tree is calculated by estimating the parameter values of the model using ML optimization. Finally, the log-likelihood values are used to calculate the fit of the different models to the data. For this final step, several options have been proposed. The most common method uses hierarchical likelihood ratio tests (LRT) to decide between nested pairs of models. LRTs have been shown to be inferior to other methods (Posada & Buckley, Citation2004) and will not be given further consideration. Instead, information criteria can be used to rank the different models based on their fit to the data.
Information criteria are statistics that incorporate a term proportional to the likelihood of the data under the model (i.e. the fit of the model) and a term that penalizes model complexity. Commonly used criteria are the Akaike information criterion (AIC), the second-order Akaike information criterion (AICc) and the Bayesian information criterion (BIC). The criteria differ mainly in the degree of penalty given to model complexity, AIC having the lowest, AICc an intermediate, and BIC the highest penalty. Whereas AIC only uses the number of model parameters in its penalty, AICc and BIC also include the alignment length so as to penalize situations in which many parameters have to be estimated from a small number of characters. Thus, for a given dataset, AIC will tend to prefer more complex models than AICc and BIC.
The performance-based model selection procedure proposed by Minin et al. (Citation2003) is even more stringent than these information criteria by penalizing models that yield branch length estimates deviating from those of other models in the comparison. The plethora of model selection procedures to choose from can be confusing, especially when they yield different results. Consensus about which method to use in which situation has not yet emerged. Given the different behaviour of ML and BI in relation to model complexity (see section above: Rationale of model selection), it may be advisable to use a less stringent selection procedure to select a model for Bayesian analyses and a more stringent one for ML inferences.
An alternative and increasingly popular method for model selection and partitioning strategies compares the performance of different models using the Bayes factor (Nylander et al., Citation2004; Brandley et al., Citation2005; Brown & Lemmon, Citation2007). The Bayes factor is a measure that can be used for comparing the relative fit of two models to a dataset and is not conditional on a guide tree (Kass & Raftery, Citation1995; Brown & Lemmon, Citation2007). To calculate it, Bayesian analyses have to be run using the two competing models, implying considerably higher computation times. The factor is the ratio of marginal likelihoods from two competing models, which can be calculated using different methods (Suchard et al., Citation2001; Nylander et al., Citation2004; Brandley et al., Citation2005; Lartillot & Philippe, Citation2006). It is compared against a table of cut-off values (Kass & Raftery, Citation1995; Nylander et al., Citation2004). Although this may seem rather arbitrary, the statistical validity of the cut-off values has been implied in simulation studies (Brown & Lemmon, Citation2007). It is important to note that different methods of calculating the Bayes factor can lead to different results. When marginal likelihoods are computed by harmonic mean estimation (e.g. MrBayes, Tracer), overly complex models are selected (Lartillot & Philippe, Citation2006). The computational requirements of the recently proposed alternative, thermodynamic integration, are very high (Lartillot & Philippe, Citation2006). Additional research will be needed to make Bayesian model selection a viable alternative to the ML-based methods.
Several software applications carry out model selection in a more or less automated way (). Unfortunately, most of them are set up only to compare the general time-reversible model with its simpler derivatives, rendering them of limited utility for dealing with exotic models and composite datasets. Automated model selection becomes difficult when composite datasets yield hundreds of combinations of partitioning strategies and models (but see Tanabe, Citation2007). In such cases model testing requires some manual work. Model selection using the Bayes factor can be carried out by running Bayesian analyses using several combinations of models and partitions. MCMC output serves as the starting point to calculate Bayes factors, either manually or with PhyloBayes or Tracer (Lartillot et al., Citation2007; Rambaut & Drummond, Citation2007). In case one prefers to work with one of the selection criteria, the log likelihood and corresponding AIC, AICc or BIC scores of a guide tree need to be calculated under many combinations of models and partitions (). A suitable guide tree can be obtained with MP or NJ in PAUP* (Swofford, Citation2003) or fast ML implementations in PhyML, GARLI, TreeFinder or RAxML (Guindon & Gascuel, Citation2003; Jobb et al., Citation2004; Stamatakis, Citation2006; Zwickl, Citation2006). We find TreeFinder, HyPhy and PAML particularly powerful applications for evaluating the fit of complex models using the inferred guide tree (Jobb et al., Citation2004; Kosakovsky Pond et al., Citation2005; Yang, Citation2007).
Fig. 3. Manual selection of models using the Akaike Information Criterion. The scheme illustrates the four steps that need to be taken to calculate the AIC score of a set of user-specified combinations of partitions and models. The combination receiving the lowest AIC score can be used in further analyses. The table shows the fit of various partitioning strategies and models to a dataset of two plastid genes (rbcL and atpB) for representatives of the Viridiplantae. The AIC scores are represented with colour codes, red indicating high scores (poor fit to the data) and green indicating low scores (good fit), Whereas partitioning into genes does not improve model fit, partitioning into codon positions yields a significant increase. Adding among-site rate variation to the models (+Γ4) also yields considerable increase in model fit. The lowest score, however, is that obtained with a simplified version of the GY94 codon substitution model, illustrating that models with extra biochemical realism better fit the data than standard and partitioned nucleotide models.
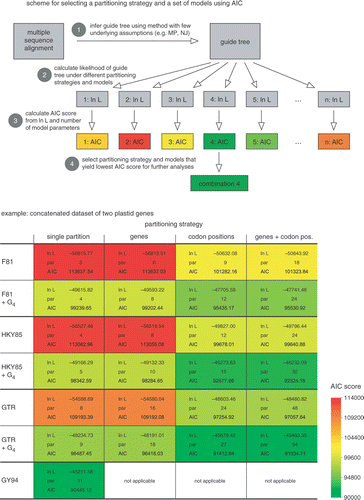
Table 2. Selection of software that performs model selection in a more or less automated way.
Systematic error
Because every dataset contains noise in addition to the phylogenetic signal, inference methods can yield topologies that do not reflect the true phylogenetic relationships due to stochastic error when they are operating on a small amount of data. As more data are added, the methods will normally converge onto the correct result. However, in some cases they do not – a problem known as systematic error, phylogenetic bias or method inconsistency. All inference methods are consistent when their assumptions are met and they all become inconsistent when their assumptions are violated. In a parametric setting, bias is caused by misspecification (usually oversimplification) of the model of DNA sequence evolution.
Common causes of systematic error
The main causes for phylogenetic bias can be subdivided into three classes. First, non-independence among sites can cause systematic error (Schöniger & Von Haeseler, Citation1995). Paired bases in RNA stems and different bases within codons are well-known examples. Interdependence among sites can be countered by using models of sequence evolution that incorporate this autocorrelation (e.g. codon and doublet models; Goldman & Yang, Citation1994; Schöniger & Von Haeseler, Citation1994).
Second, substitution rates, base frequencies and other model parameters can differ across sites in an alignment (site-heterogeneity) and failing to model such variation may cause systematic error (Yang, Citation1994a; Yang, Citation1996; Lartillot & Philippe, Citation2004; Pagel & Meade, Citation2004; Stefankovic & Vigoda, Citation2007). Inference problems caused by process heterogeneity among sites are relatively easy to detect and correct for. Incorporating rate heterogeneity across sites has become common practice using the discrete gamma distribution and/or a proportion of invariant sites (Yang, Citation1994a; Gu et al., Citation1995; Yang, Citation1996). Among-site heterogeneity in other aspects of the processes of sequence evolution can be countered by partitioning the data and applying appropriate models to each of the partitions (see section above: Partitioning strategies) or using mixture models, which accommodate among-site heterogeneity but do not require prior partitioning of the data (Lartillot & Philippe, Citation2004; Pagel & Meade, Citation2004).
Third, substitution rates, base frequencies and other model parameters can change along the tree (tree-heterogeneity). Long-branch attraction, a form of phylogenetic bias in which long branches cluster together even though they are not related because evolutionary rates differ strongly among lineages, is the best-known example of systematic error due to tree-heterogeneity (reviewed by Bergsten, Citation2005). Biases can also occur when base frequencies or the substitution rate matrix do not remain constant along the tree (Lockhart et al., Citation1998; Conant & Lewis, Citation2001; Lopez et al., Citation2002; Rosenberg & Kumar, Citation2003; Kolaczkowski & Thornton, Citation2004; Philippe et al., Citation2005a; Baele et al., Citation2006; Lockhart et al., Citation2006; Ruano-Rubio & Fares, Citation2007). Obviously, site- and tree-heterogeneity can both be present in a dataset. A special case where site-specific rates vary across the tree is known as heterotachy (Lopez et al., Citation2002). Systematic error resulting from tree-heterogeneity is much more difficult to identify and overcome than bias due to site-heterogeneity. Examination of its occurrence usually ensues from observing unexpected relationships or indications for long-branch attraction and is rarely carried out by default (but see, e.g. Shalchian-Tabrizi et al., Citation2006).
Branch-specific rate shifts can usually be identified in a preliminary tree, either visually or more formally with the relative-rates or Tajima test (Tajima, Citation1993). Compositional heterogeneity can be visualized with SeqVis (Ho et al., Citation2006). Other methods for assessing compositional heterogeneity are reviewed by Jermiin et al. (Citation2004). A few procedures to detect heterotachy have been proposed (Lockhart et al., Citation1998; Baele et al., Citation2006; Ruano-Rubio & Fares, Citation2007). Because both the misleading signal and the correct phylogenetic signal are present in the dataset, they may also be detectable using spectral analysis and network methods (Kennedy et al., Citation2005 and references therein). A number of explorative experiments have been proposed to examine whether observed heterogeneities influence the topology. First, excluding some of the deviant branches from the analysis may change the position of the remaining deviant branches (e.g. Rodríguez-Ezpeleta et al., Citation2007a). Second, exploring the exclusion of fast sites (see section below: Data saturation) may indicate problems relating to tree-heterogeneity (Bergsten, Citation2005; Rodríguez-Ezpeleta et al., Citation2007a). Third, parametric simulation may be used to assess whether the tree-heterogeneity is strong enough to mislead the tree inference method (e.g. Huelsenbeck, Citation1997; Foster, Citation2004).
The various solutions that have been proposed to overcome phylogenetic bias due to tree-heterogeneity can be subdivided in three main classes. First, tree-heterogeneous models of sequence evolution can be used. The covarion model (usually called covariotide when applied to nucleotides) is a relatively simple and quite commonly used tree-heterogeneous model. It models a special form of heterotachy where characters can switch between an on-state, during which they evolve according to a regular model (e.g. GTR) and an off-state, during which they do not change (e.g. Penny et al., Citation2001). Incorporating tree-heterogeneity of base composition or substitution rate matrices is also possible, but such models are very parameter-rich, processor-intensive, and rarely used. They are implemented in HyPhy (Kosakovsky Pond et al., Citation2005) and p4 (Foster, Citation2004). Second, more taxa can be included in the analysis (e.g. Graybeal, Citation1998). Expanded taxon sampling does not decrease the tree-heterogeneity but the extra information may provide the tree-inference methods with the necessary clues to recover a better phylogeny (see section below: Taxon sampling). Third, it may be possible to mitigate systematic error due to tree heterogeneity by reducing the amount of substitutional saturation in the dataset. Reducing the level of saturation can be done by coding the characters differently (e.g. RY-coding) or by removing a fraction of fast-evolving characters, a technique known as site stripping. Data saturation is dealt with in more detail below.
Combinations of model misspecifications can affect phylogenetic analyses in complex ways (e.g. Ho & Jermiin, Citation2004). It is important to note that analyses of distantly related organisms are more susceptible to phylogenetic bias because data saturation enhances phylogenetic bias. It has been shown that systematic error becomes especially problematic when internal branches are short compared with terminal branches (Ho & Jermiin, Citation2004; Jermiin et al., Citation2004). Consequently, when one wishes to resolve rapid, ancient radiations (e.g. in genome-scale studies), the interaction between saturation and systematic error becomes a major issue that has to be dealt with in detail (Philippe et al., Citation2005b; Rodríguez-Ezpeleta et al., Citation2007a). Because the algae consist of some ancient groups, extreme care should be taken to avoid phylogenetic biases (Müller et al., Citation2001; Rodríguez-Ezpeleta et al., Citation2007b). Systematic error has also been suggested to be at play at lower taxonomic levels in algae (Leliaert et al., Citation2007; Verbruggen et al., Citation2007), so it is advisable to be alert to this problem in all phylogenetic endeavours.
Data saturation
Because nucleotide characters possess only four states, fast-evolving sites that undergo multiple changes along the branches of a tree become saturated with convergent substitutions and state reversals. In the absence of model violations, increasing amounts of saturation mask the remaining phylogenetic signal, resulting in loss of resolution in the obtained phylogenies and decreasing accuracy (Ho & Jermiin, Citation2004). In the more realistic case that the chosen model of sequence evolution deviates from the true evolutionary processes that have acted on the sequences, saturation enhances systematic error, which can lead to inference of a strongly supported but wrong tree (Ho & Jermiin, Citation2004; Jeffroy et al., Citation2006; Rodríguez-Ezpeleta et al., Citation2007a).
Whether saturation is present in a dataset depends on the marker's rate of evolution and the age of the group of organisms under study. Saturation is mainly a concern for phylogenetic inferences between more distantly related organisms; it is rarely an issue for studies of closely related organisms. Fast markers are more likely than slow markers to show saturation. It should be noted that even within a single marker, characters usually evolve at different rates (e.g. codon positions within genes), and saturation of fast characters may mask the historical signal present in slower characters.
Several approaches have been suggested to detect saturation in empirical datasets. The most commonly used one is making a scatterplot of uncorrected versus corrected genetic distances of all taxon pairs (). The corrected distance (on the x-axis) between two taxa is the patristic distance based on a model of sequence evolution that takes into account that multiple substitutions can happen along a branch. The uncorrected distance (on the y-axis) is the fraction of sites that differ between the two taxa. When no saturation is present one would expect both distances to be equal (dashed line), whereas in the presence of saturation the plot would be expected to level off with increasing distance. The degree to which the curve levels off indicates the amount of patristic distance that is not represented in the uncorrected distances, in other words, it is a measure for the amount of saturation (). In empirical studies, the slope of a linear regression through the plot is sometimes used as a measure of saturation (; Jeffroy et al., Citation2006; Rodríguez-Ezpeleta et al., Citation2007a).
Fig. 4. Visual methods for detecting saturation in molecular phylogenetic datasets. The three graphs on the left show how plotting uncorrected versus corrected pairwise genetic distances allow assessment of the degree of substitutional saturation in a dataset. The dashed line indicates the expected correlation in the absence of saturation (i.e. uncorrected distances equal corrected distances). The datasets in the three plots were generated by simulating markers evolving at different rates along the same tree, facilitating comparison between the three panels. The top panel represents the slowest marker and does not deviate far from the dashed line. The centre plot shows the results for a marker evolving at an intermediate rate. The bottom panel shows the strongest deviation from the dashed line, indicating strong saturation in this fast marker. Note the different scales along the x-axis. The top right panel illustrates how the slope of the linear regression through the saturation curve can be used as a measure of the amount of saturation in a dataset. The data in this plot are for the slow and intermediate markers from the previous graphs. The triangles in the lower right of the figure illustrate likelihood mapping. The left panel shows the parts of the graph indicating tree-like signal (corners, indicated with +), conflicting signal (along the sides) or the lack of signal (in the centre). The centre panel shows the application of this technique to a red algal rbcL dataset of 20-taxa (Hommersand et al., Citation2006). A great majority of points are located in the corners, indicating that the quartets in this dataset are tree-like. When only third codon positions are considered (right panel), a substantially larger amount of the quartets were unresolved, indicating moderate saturation at third codon positions.
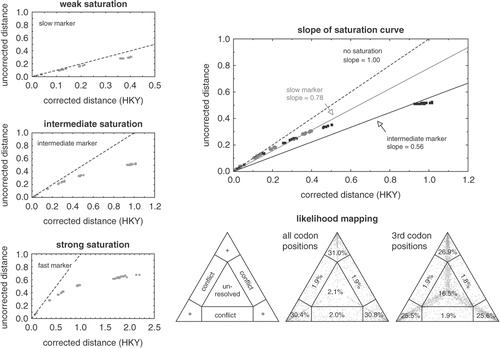
Likelihood mapping is another method to visualize the amount of signal vs. noise in a dataset (Strimmer & von Haeseler, Citation1997) and is implemented in Tree-Puzzle (Schmidt et al., Citation2002). This approach visualizes the tree-likeness of quartets of taxa in a triangular graph. Quartets that are unresolved are plotted as a dot near the centre of the triangle, those that yield strong support for one topology are plotted in one of the corners, and when there is support for two conflicting topologies, the quartet is plotted near a side of the triangle (). High densities of dots near the centre and/or sides indicate high levels of noise. High densities in the corners indicate strong phylogenetic signal in the quartets. Although this is usually indicative of strong phylogenetic signal in the complete dataset, this is not guaranteed because quartets may be in conflict with one another. Treeness-triangles are another triangle-based method to visualize the tree-likeness of a phylogenetic dataset but have not yet been extensively used (White et al., Citation2007).
One flaw of the visual exploration methods is that they do not allow an objective assessment of whether or not the amount of saturation present in the data is problematic for tree inference. To counter this problem, an index to measure substitution saturation and a test to determine the usefulness of a dataset for phylogenetic analysis have been proposed (Xia et al., Citation2003) and implemented in DAMBE (Xia & Xie, Citation2001). The index is a measure of entropy and is rooted in information theory rather than phylogenetic theory, but the measure is clearly related to the amount of noise in a dataset. Despite the fact that this method has not been thoroughly tested yet, it has gained some popularity in recent years.
The amount of saturation in a dataset can be reduced by removing saturated characters, a technique known as site stripping. In many cases, a coarse and largely subjective approach toward data removal is used, e.g. exclusion of third codon positions. A more objective technique is to measure the evolutionary rate of all characters and re-run analyses with increasing amounts of fast characters removed (Ruiz-Trillo et al., Citation1999; Burleigh & Mathews, Citation2004; Rodríguez-Ezpeleta et al., Citation2007a). Site stripping has been used to resolve some ancient nodes crucial to the understanding of algal evolution (Lemieux et al., Citation2007; Rodríguez-Ezpeleta et al., Citation2007a; Rodríguez-Ezpeleta et al., Citation2007b). It must be noted that despite the fact that site stripping is gaining popularity, we do not know of any studies validating the approach through simulation. Similarly, removing blocks of ambiguously aligned data also discards fast-evolving parts of the sequences in which homology is hard to assess (Talavera & Castresana, Citation2007). The packages Gblocks and SOAP can be used to detect and remove ambiguously aligned alignment regions (Castresana, Citation2000; Loytynoja & Milinkovitch, Citation2001).
As an alternative or supplement to direct exclusion of data, saturation can also be mitigated by using different character coding strategies. Because transitions are more common than transversions, they are more likely to cause saturation. This can be overcome by recoding the characters into puRines (A & G → R) and pYrimidines (C & T → Y), a process known as RY-coding (Phillips & Penny, Citation2003). This recoding has two important consequences: (i) only transversions are considered in phylogenetic analyses and (ii) potential GC-biases are removed, reducing the potential for systematic error. Similarly, protein-coding sequences can be analysed as amino-acid sequences instead of nucleotide sequences, mitigating the saturation that can occur by excessive synonymous substitutions. Like site-stripping, RY and amino-acid coding reduce the total amount of information in the dataset, but it is done by modifying the character space instead of removing characters. Finally, the application of codon models changes the parameter space to mitigate saturation at degenerate sites while retaining all information in the dataset.
A comment on substitutional saturation at third codon positions seems appropriate to conclude this section. On average, third positions evolve much faster than first and second positions () due to the fact that substitutions at third positions are largely silent. There has been considerable debate about whether third positions should be included or excluded from phylogenetic analyses, with pleas for their exclusion (e.g. Swofford et al., Citation1996; Blouin et al., Citation1998) and defences of their inclusion (Björklund, Citation1999; Källersjö et al., Citation1999; Müller et al., Citation2006). The topic has received substantial attention in the algal literature (Daugbjerg & Andersen, Citation1997; Siemer et al., Citation1998; McIvor et al., Citation2002; Goertzen & Theriot, Citation2003; De Clerck et al., Citation2006; Le Gall & Saunders, Citation2007). Despite indications for saturation, third codon positions commonly outperform first and second codon positions in phylogenetic analyses (Simmons et al., Citation2006).
Fig. 5. Graph showing distribution of substitution rates at first (cp1), second (cp2) and third (cp3) codon positions in a green algal dataset composed of the plastid rbcL and atpB genes. Whereas first and second positions evolve slowly, third codon positions show a broad rate distribution.
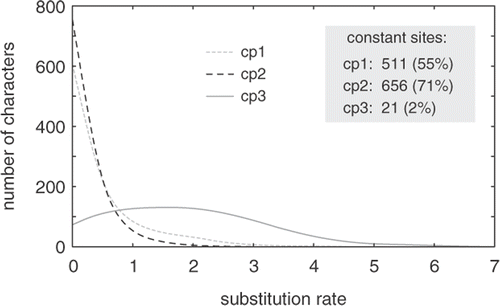
As far as character rate distributions are concerned, the performance of third codon positions agrees with theoretical expectations: compared to first and second codon positions, the majority of which are very slow and can be expected to yield sparse information about relatively deep nodes, evolutionary rates of third codon positions vary widely and can be expected to yield information about nodes across a considerable time span (Townsend, Citation2007). Obviously, the whole equation largely depends on the age of the group of organisms under study, and therefore requires a case by case evaluation. Furthermore, one is not compelled to make the drastic choice between inclusion and exclusion of third codon positions. Intermediate solutions (i.e. moderate site stripping) and alternative approaches such as the use of codon models or character recoding may turn out to be worthy substitutes.
Tree rooting
Determining the correct location of the root (i.e. the oldest point) of a tree is fundamental to the interpretation of the branching order of the taxa under study and is a prerequisite for inferring and interpreting the historical patterns of biologically relevant characters. It has long been appreciated that tree rooting is one of the most delicate aspects of phylogenetic analysis (Smith, Citation1994; Swofford et al., Citation1996), and algae are no exception to this rule (e.g. Saunders et al., Citation2002; Withall & Saunders, Citation2006; Leliaert et al., Citation2007; Verbruggen et al., Citation2007).
The most common way of inferring the root of a tree is by including one or a few outgroup taxa in addition to the organisms of interest. Although this method may work in a majority of cases, it is important to realize that outgroup rooting introduces one or several significantly more distantly related sequences, potentially exposing the phylogenetic analysis to systematic error. It has been well-documented that outgroups can attach to a wrong ingroup branch and even disrupt the relationships among ingroup taxa (Holland et al., Citation2003; Shavit et al., Citation2007).
Two methods that do not require the use of outgroups have been proposed for rooting phylogenetic trees. The first method is based on the use of non-reversible models of sequence evolution. Standard models of sequence evolution assume reversibility of the substitution process, meaning that the probability of any type of substitution is equal to the probability of the inverse substitution (e.g. Pr (A→T)=Pr (T→A)). When this assumption is not made, the model is called non-reversible, and trees inferred under such models are automatically rooted (Yang, Citation1994b). The second type of method relies to some degree on the assumption of clock-like evolution. The rationale behind this approach is that, if evolution is clock-like, the root of the tree is situated at exactly the same distance from each terminal taxon. Phylogenetic inference under a uniform molecular clock model automatically roots the tree at its oldest point. Tree inference with a relaxed molecular clock method has the same effect but does not require strict clock-like evolution (Drummond et al., Citation2006). Mid-point rooting starts from an unrooted phylogenetic tree of ingroup taxa inferred using a standard model. It finds the two most divergent taxa in the phylogeny and places the root at the midpoint of the path connecting these two taxa (Farris, Citation1972). This method also assumes a certain degree of clock-like evolution, but in this case only the most divergent lineages are assumed to have evolved at the same rate.
Simulation studies have shown that outgroup rooting is very accurate if the outgroup is closely related to the ingroup and that its accuracy decreases with increasing genetic distance between the ingroup and the outgroup (Huelsenbeck et al., Citation2002b; Holland et al., Citation2003). In other words, the ideal outgroup sequence would be that of the latest common ancestor of the ingroup, and the further the chosen outgroup is from this latest common ancestor, the lower the chances that outgroup rooting will yield the correct root position. One should thus strive for selecting the closest possible outgroup in terms of genetic distance. In most cases this will be the immediate sister clade. However, if the immediate sister clade has experienced increased rates of evolution, an earlier branching, slower evolving clade may serve better (Lyons-Weiler et al., Citation1998). Using multiple outgroup taxa yields more accurate results than using a single outgroup taxon (Shavit et al., Citation2007), but these taxa should preferably come from within a single, closely related lineage. Sampling outgroups by taking one species from each of a set of increasingly distant lineages has been shown to be a poor strategy (Smith, Citation1994).
The molecular clock rooting method was shown to be a valuable alternative to outgroup rooting, being robust to the problems affecting outgroup rooting (Huelsenbeck et al., Citation2002b). The drawback of molecular clock rooting is that its accuracy decreases with the degree of violation of the molecular clock assumption (Huelsenbeck et al., Citation2002b). Although it has not been tested in detail, the same can be expected from the mid-point rooting method. Relaxed clock models should provide accurate estimates of the root position under a wider range of rate variation than the strict molecular clock method (Drummond et al., Citation2006), but this assertion awaits verification. Using standard non-reversible models of sequence evolution for inferring the root position was shown to be inaccurate (Huelsenbeck et al., Citation2002b) but a more recent study shows that small alterations of the model architecture may yield much better inferences about the root position (Yap & Speed, Citation2005). This method clearly has potential, but it has not yet been extensively tested and non-reversible models are rarely implemented in tree inference programs. Molecular clock models are much more commonly implemented. The uniform (strict) molecular clock is implemented in several ML and BI programs. The BEAST package allows inferences under strict and relaxed molecular clock models (Drummond & Rambaut, Citation2007).
A general recommendation to the rooting problem is to use the method whose assumptions are least likely to be violated by your data. If a nearby outgroup is available, outgroup rooting is a good option. If the data do not violate the molecular clock too strongly, inferences under a strict or relaxed molecular clock model will yield a good root position. When in doubt about the suitability of a method or an outgroup, the golden rule is to explore the position of the root with different methods and different outgroups. When using the outgroup method, it is also advisable to establish the effect of outgroup sequences on the ingroup topology by re-running the analysis without the outgroup sequences. If the ingroup topology differs between analyses, the result without the outgroup is more trustworthy (Holland et al., Citation2003; Shavit et al., Citation2007).
Experimental design
As in other branches of science, experimental design is of great importance for phylogenetic studies. Despite the attention and debate about ‘adding taxa or characters’ (Hillis et al., Citation2003 and references therein), such aspects of experimental design are often overlooked in empirical studies. Yet it is important to realize that the number of taxa one includes, the phylogenetic spectrum that they cover, the choice of markers and the selection of characters are decisions that can influence the outcome of a phylogenetic analysis.
Taxon sampling
Many studies have shown that choice of taxa may strongly affect the inferred phylogeny (e.g. Pollock et al., Citation2002; Zwickl & Hillis, Citation2002; Goertzen & Theriot, Citation2003). In the worst case, sparse and uneven sampling of taxa can result in long-branch attraction or other biases. Improved taxon sampling can ameliorate many of the problems affecting phylogenetic inference. While this makes intuitive sense, the literature presents conflicting claims (e.g. Rosenberg & Kumar, Citation2001 vs. Pollock et al., Citation2002). Many of these conflicts can be resolved when one considers the two components to taxon sampling: the number of taxa sampled and the distribution of taxa along long and short branches. Generally speaking, cases in which increasing the number of taxa decreased the accuracy of phylogenetic inference have generally either increased the taxon sampling outside the scope of interest (i.e. included taxa distantly related to the ingroup) or added taxa that are very closely related to those already in the analysis (Graybeal, Citation1998; Rosenberg and Kumar, Citation2001, Citation2003; Pollock et al., Citation2002; Zwickl and Hillis, Citation2002; Hillis et al., Citation2003; Hedtke et al., Citation2006). In summary, the consensus is that increasing the number of taxa sampled in the ingroup, particularly where those taxa intersect long branches, improves phylogenetic accuracy. These results are relevant for researchers with limited processing power at their disposal. Because model-based analyses are demanding in terms of computing power, such researchers may be tempted to lower the number of taxa in their parametric analyses, which may be counter-productive.
Another practical consideration is whether the systematist includes more taxa or more characters. The answer is likely to be study-specific (Poe & Swofford, Citation1999; Hedtke et al. Citation2006). A recent genome-scale analysis has led to the claim that adding enough genes can significantly improve accuracy (Rokas et al., Citation2003). This specific claim was flawed because bootstrap values were used to measure accuracy, while little attention was paid to accommodating the complexity of the data in the analyses. When data complexity is insufficiently accounted for, phylogenetic inference methods are prone to statistical inconsistency, and increased amounts of data can yield high support values for incorrect groups. Genome-scale studies are likely to converge on some answer whether it reflects phylogenetic history or biases in the data, underscoring advice we have given in previous sections, to understand and explore one's data through various means.
At present, we see no alternative to a somewhat ad hoc and recursive approach to taxon sampling: primary studies should focus on increasing the number of taxa, evaluate for long-branch and bias problems, and then add taxa and/or characters as might be suggested by analogous situations (in the above cited and similar studies). While this is not entirely satisfactory, Geuten et al. (Citation2007) have made an initial foray into a more objective approach into this problem and the advice given there is very similar to that of Graybeal (Citation1998) and Poe (Citation2003): trees are constructed most accurately if new taxa are added towards the base of long branches and least accurately if new taxa are added close to the tips of long branches. In conclusion, the focus should not lie exclusively on the number of taxa included in the study. Rather, one should attempt to obtain a set of taxa that maximizes the phylogenetic diversity within the bounds of financial and practical possibilities (Pardi & Goldman, Citation2007).
Marker choice
Given the reality of limited funding, only one or a few markers can be sequenced for the majority of systematic studies. As a consequence, the question poses which DNA marker(s) to choose. A number of factors usually determine this choice. The marker's use in previous studies may be important, because using previously published information can reduce the cost. The ease of amplification, sequencing and alignment, and the copy number of the marker are other factors to consider. The main focus of marker choice, however, should be on the utility of the marker to resolve the question at hand. Markers often have different properties and, as a consequence, some are more useful for a certain purpose than others. Important aspects of the phylogenetic utility of a marker are its rate of evolution and the distribution of rates across characters (Graybeal, Citation1994; Townsend, Citation2007). It is intuitively simple that fast characters will be more useful for resolving recent divergences and slow characters will prove more effective for older divergences. Actually, the ideal rate of a character to resolve a polytomy is inversely related to the age of the polytomy (Townsend, Citation2007). The rate one wishes a marker to have depends on the age of the group under study, and the distribution of rates across characters ideally spans the range of relationships that need to be resolved.
Despite its consequential role in phylogenetic experimental design, the choice of markers for investigating phylogenetic questions at a given taxonomic level is more often based on common belief than determined on the basis of objective criteria. A number of studies highlight the different approaches that can be taken to investigate theoretical and practical aspects of marker informativeness (Graybeal, Citation1994; Goldman, Citation1998; Yang, Citation1998; Shpak & Churchill, Citation2000; Müller et al., Citation2006; Townsend, Citation2007). However, such techniques are rarely used and have, to our knowledge, not yet been applied in large-scale surveys of phylogenetic information content of commonly used markers for algal systematics.
Marker combination
Analysing datasets composed of multiple markers is an issue closely related to the previous one. Additional markers can increase the phylogenetic signal; yet using different markers also introduces data heterogeneity that may hamper the analysis. Initially, two opposed approaches were advocated: simultaneous vs. separate analysis. The total evidence approach recommends simultaneous analysis of all evidence. In molecular phylogenetics, this would come down to analysis of a concatenated dataset containing all available markers (supermatrix approach) (Kluge, Citation1989). The opposite approach consists of analysing individual loci separately and combining the resulting trees using consensus or supertree approaches. Using consensus trees was largely abandoned because it did not retain information about support for the individual trees (Eernisse & Kluge, Citation1993) but supertree approaches have become relatively popular despite the reservations that exist about these methods (Bininda-Emonds, Citation2004). The simultaneous vs. separate analysis issue has been strongly debated (reviewed by, e.g. Nixon & Carpenter, Citation1996), with arguments often varying depending on the particular circumstance of concern. The debate has largely settled and alternative, more sensible approaches have gained popularity.
The practice of conditional combination was an early intermediate between both extremes, which comes down to performing a statistical test to evaluate whether the data are sufficiently homogeneous to be analysed simultaneously (Bull et al., Citation1993; Huelsenbeck et al., Citation1996). If homogeneous, the data are analysed simultaneously; otherwise, they are analysed separately. Detecting homogeneity in practice is difficult. The incongruence-length difference test (Farris et al., Citation1994), also known as partition homogeneity test, has been used for many years but is a poor indicator of data combinability (Barker & Lutzoni, Citation2002; Darlu & Lecointre, Citation2002). A parametric method has been proposed (Huelsenbeck & Bull, Citation1996; Huelsenbeck et al., Citation1996) but has not been used much, probably because there are no user-friendly implementations of this test. In addition to these early methods, a few other approaches have been used but so far, no standard has emerged (Hipp et al., Citation2004; Planet, Citation2006; Struck et al., Citation2006; Chen et al., Citation2007; Sung et al., Citation2007).
As mentioned above (Model selection, Systematic error), data heterogeneity can also be incorporated in phylogenetic inference by using partitioned or mixture models. This comes down to applying the total evidence approach but allowing the evolutionary process to vary among loci. Nonetheless, it is important to stress that tree inference under such models still assumes that all loci evolved along a common species tree. Violation of this assumption, for example by lateral gene transfer, may lead to unstable phylogenies and, more fundamentally, inferring trees from data that are known to violate this assumption is quite nonsensical. In the case of lateral gene transfer, it may be more sensible to use methods that can simultaneously infer the species tree and lateral gene transfer events, given that a sufficiently large number of genes are available (Galtier, Citation2008). Methods have also been designed to infer the species tree in the presence of incomplete lineage sorting (Carstens & Knowles, Citation2007).
Uncertainty about results
The tree resulting from a phylogenetic analysis is often seen as a point estimate, a single and best result. This view may be misleading for a number of reasons. First, it is possible that shortcuts in the algorithms lead to a suboptimal solution (see section below: Exploration of tree space). Second, one should be aware that there are usually several, similar topologies whose likelihood values hardly differ. Considering this, it may be useful to obtain and study a set of trees that explain the data almost equally well. Several techniques have been developed to quantify topological uncertainty. At the level of individual nodes, these include non-parametric bootstrapping and interior branch tests (reviewed by e.g. Felsenstein, Citation2004; Yang, Citation2006). At the level of whole trees, one can use likelihood tests to compare different topologies (see section below: Likelihood tests of topologies) or posterior probabilities from Bayesian inferences (see section below: Posterior distribution of trees).
Exploration of tree space
The set of possible topologies for a given set of taxa is called the tree space. The tree space grows incredibly fast with increasing taxon numbers. In order to obtain the ML tree of a set of taxa, the likelihood score has to be calculated for every possible topology (exhaustive search). This becomes impossible even for moderate-sized datasets because of computational limitations. Instead, shortcuts (heuristic algorithms) are used to walk through tree space in search for the ML tree (Felsenstein, Citation2004; Yang, Citation2006; Whelan, Citation2007). Most heuristics are hill-climbing algorithms, meaning that they start from a given tree, create a set of neighbouring trees by making modifications to the start tree, evaluate the likelihood of the neighbouring trees, and pick the one with the highest likelihood as the starting point for a next modification step. This procedure is repeated until a likelihood optimum is reached. If the modifications to the start tree are small, only a limited part of tree space surrounding the start tree is explored. If the true ML tree is separated from the start tree by a set of intermediate trees that have a relatively low likelihood, the true ML tree may never be visited during the search. In this case, the analysis is said to get stuck on a local optimum. In order to avoid this problem one can perform multiple heuristic searches from different start trees. If the start trees are sufficiently spaced out, this yields a strong increase of the probability of finding the true ML tree. It is important to note that especially the programs that allow likelihood inference using complex models are liable to becoming stuck on local optima. Using complex models and performing intensive tree searches are both computationally expensive, and in order to yield acceptable running times, there is often a trade-off between them. RAxML and TreeFinder both choose model complexity at the expense of tree space coverage, and, as mentioned in these programs' manuals, it is critical to start searches from multiple start trees.
In contrast to the hill-climbing methods used in ML inference, the MCMC used in Bayesian analysis does allow moderate decreases in likelihood along the chain (see section above: Introduction), which reduces the chances of getting stuck on local PP peaks. Furthermore, the popular BI program MrBayes (Ronquist & Huelsenbeck, Citation2003) implements Metropolis-coupled MCMC (MCMCMC or MC3), in which several chains are run in parallel. The first chain is called the cold chain and the other chains are incrementally heated. Heating chains flattens out the posterior distribution, making it easier to hover through tree space and find distant regions with high PP. After each generation, chains can be swapped, resulting in a situation where heated chains can become colder when they arrive in a high-PP region of tree space. Only the output from the cold chain is used to summarize the posterior distribution and, thanks to chain swapping, this chain will contain a more complete image of the high-PP regions of tree space. MC3 comes at a considerable computational cost because several chains have to be run in parallel (Altekar et al., Citation2004; Beiko et al., Citation2006).
Even though BI was initially portrayed as a fast alternative to ML (e.g. Larget & Simon, Citation1999), it seems that this view needs to be re-evaluated. The fact that several chains have to be run in parallel for MC3, that one should preferably perform several independent runs, and that chains need to be run sufficiently long to achieve convergence of the parameter estimates and obtain a sizable posterior sample can imply long running times. Because computations are much slower for large datasets and complex models, many users may be tempted to run shorter chains to have a result within an acceptable amount of time. This is bad practice because longer chains are often needed to achieve convergence in larger datasets and with complex models. So, chain lengths should be increased rather than decreased when analysing large or complex datasets.
A number of visual tools and statistics can be used to investigate convergence of runs. These tools cannot be used to prove convergence but they can be used to diagnose the lack thereof (Nylander et al., Citation2007). The program Tracer (Rambaut & Drummond, Citation2007) draws traces of all parameter values optimized during an MCMC. When multiple runs are loaded, the traces can be viewed in a single graph for rapid visual assessment of convergence between runs. Tracer also calculates the effective sample size (ESS) for each parameter. This statistic is calculated for each run separately and indicates whether the parameter in question has been sampled sufficiently during the run. The application Are we there yet? (AWTY) focuses on comparing tree files between parallel MCMC runs (Nylander et al., Citation2007). It includes various visual methods to investigate topological convergence between the runs.
Likelihood tests of topologies
Competing hypotheses are fairly common in algal systematics. One example concerns the relationships between the three green algal classes, Ulvophyceae, Trebouxiophyceae and Chlorophyceae comprising what has become known as the UTC clade. Based on ultrastructural observations, one would expect the Ulvophyceae to branch first, leaving Trebouxiophyceae and Chlorophyceae as sisters (Mattox & Stewart, Citation1984). Evidence from the chloroplast genome, however, suggests that Chlorophyceae branch first, leaving Ulvophyceae and Trebouxiophyceae as sisters (Pombert et al., Citation2005). The two competing topologies can be compared in a likelihood framework using the Kishino-Hasegawa (KH) test (Kishino & Hasegawa, Citation1989). The test assesses whether or not the topological hypotheses have a significantly different likelihood, given the data and model of sequence evolution. The KH test assumes that the topologies being compared were both pre-specified. For our green algal example this would mean that it would be inappropriate to use the KH test to test both hypotheses against a chloroplast genome dataset because the chloroplast genome hypothesis is derived from such a dataset. This illustrates that molecular systematists are more often interested in evaluating whether certain pre-specified topologies are significantly different from (i.e. worse than) the tree obtained by ML analysis. The Shimodaira-Hasegawa (SH) and approximately unbiased (AU) tests can be used for this purpose (Shimodaira & Hasegawa, Citation1999; Shimodaira, Citation2002). For our green algal example, the hypothesis based on the ultrastructural evidence turned out to be significantly worse than the ML tree (Pombert et al., Citation2005).
The KH and SH tests are implemented in PAUP* (Swofford, Citation2003). For applications that require more complex models, the TreeFinder or PAML implementations of the SH test may be used (Jobb et al., Citation2004; Yang, Citation2007). The consel package implements a variety of tests, including the KH, SH and AU tests (Shimodaira & Hasegawa, Citation2001); it takes input from several phylogeny programs, so the models used can be as complex as those programs allow.
Parametric simulation has also been proposed to compare topological hypotheses (Huelsenbeck & Bull, Citation1996; Swofford et al., Citation1996). Due to the computational burden, high error levels, and model sensitivity associated with such tests (Goldman et al., Citation2000; Buckley, Citation2002), they are not used often.
Posterior distribution of trees
Bayesian inference is naturally suited to handle topological uncertainty. The posterior probability (PP) of each tree and of individual branches in the tree can be calculated from the post-burn-in sample of trees. A tree's PP is estimated by the number of generations the MCMC has spent on that tree. The interpretation of a tree's PP is straightforward: it is the probability that the tree is correct given the data, model and prior. In taxon-rich empirical datasets, posterior probabilities of whole trees can be very low because the data usually contain noise, yielding support for a set of similar trees.
The posterior distribution makes comparing topological hypotheses straightforward: one can sum the PP of all posterior trees that support each hypothesis and compare the resulting probabilities (Buckley, Citation2002). Although useful, this Bayesian approach for tree comparison has been shown to be highly sensitive to model mis-specification, particularly under-specification (Buckley, Citation2002). Posterior probabilities of topological hypotheses can be calculated by defining them as a constraint in PAUP* and filtering the post-burn-in MCMC tree output using PAUP*'s filter command. The proportion of trees retained corresponds to the posterior probability of the hypothesis of interest. If one wishes to attach significance values to a comparison of competing topological hypotheses, Bayes factors may be used (Aris-Brosou, Citation2003).
Posterior probabilities and bootstrap values
Clade support can also be calculated from the posterior distribution. Posterior clade probability is calculated as the proportion of MCMC trees in which the clade is present. Again, its interpretation is simple: it is the probability that the clade is correct given the data, model and prior. Bayesian PPs are often extremely high compared with ML bootstrap values and there has been considerable debate about how to compare them (reviewed by Alfaro & Holder, Citation2006). Both measures come with their drawbacks. Bootstrap values lack a straightforward statistical interpretation (Berry & Gascuel, Citation1996; Soltis & Soltis, Citation2003). The systematist's interest is usually in knowing how accurate an inferred branch is.
Bootstrap values have been shown to correlate with accuracy to some extent in simulation studies (e.g. Hillis & Bull, Citation1993; Efron et al., Citation1996). However, this conclusion should be interpreted with care, as they depend on the assumption that the correct model of sequence evolution is being used. It is not clear how robust this inference is to violations of model assumptions. As mentioned above, Bayesian PPs are easier to interpret. However, they are very sensitive to model mis-specification (particularly under-specification) and choice of priors (Buckley, Citation2002; Huelsenbeck & Rannala, Citation2004; Lemmon & Moriarty, Citation2004; Yang & Rannala, Citation2005).
Consequently, interpretation of bootstrap values and PPs in empirical studies remains problematic. Both seem to be good estimators of accuracy when the chosen model of sequence evolution is identical to the model that generated the data, but this deviates from the reality confronting empirical workers. Many systematists use bootstrap values of 70 or more as strong support and of 80 or more as very strong support. These thresholds, originally suggested by Hillis & Bull (Citation1993), are often used without consideration of the conditions under which they were inferred (symmetric trees, rate homogeneity throughout the trees, and small divergences). Similarly, PPs higher than 0.95 are often used to indicate strong support and PPs of 1.00 are considered evidence for very strong support, but without consideration of the sensitivity of PP to model parameterization. Because of these issues, it is considered good practice to compare support using different models and inference methods.
Visualizing uncertainty
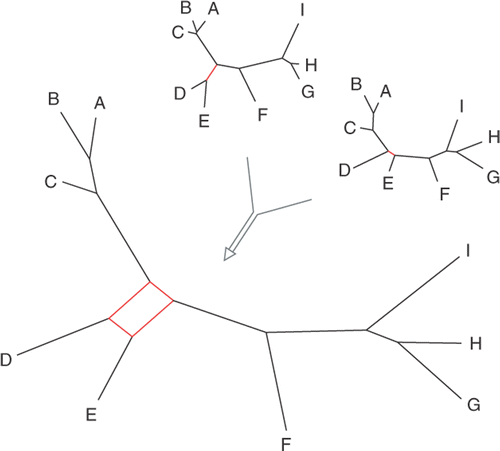
It can be quite useful to incorporate topological uncertainty in the presentation of phylogenetic results, as this allows quick evaluation of confidence. One commonly used method is to present bootstrap values or PPs at the nodes of a tree. When node support is reflected in the thickness of the branches preceding them, it allows immediate visual inspection of the credibility of different parts of the tree. The previous approach shows only one topology. Another way to visualize noise in a dataset or conflict between datasets is to show multiple topological configurations simultaneously in a single graph, i.e. a split network (; Holland et al., Citation2004; Holland et al., Citation2006). SplitsTree is a useful program for this purpose (Huson & Bryant, Citation2006).
Fig. 6. Consensus networks are useful to visualize conflict between two or more trees in a single graph.
Dating trees
Phylogenetic trees that are calibrated in time yield much richer evolutionary interpretations than trees in which this is not the case. Among other things, they allow juxtaposition of the branching events with earth's history, augmenting biogeographic interpretations. When taxon sampling is close to complete, rates of cladogenesis can be calculated in different time frames or in different lineages of the tree, leading to new hypotheses about the evolutionary diversification of the organisms under study.
Calibrating trees in absolute time requires a firm knowledge and sound interpretation of the fossil record. When fossils can be placed in a phylogenetic framework inferred from extant species, the methods described below will estimate the ages of all nodes in the tree. Placing fossils in a phylogenetic tree inferred from DNA sequences of extant species requires an excellent knowledge of the morphology of the extant lineages, identification of unique characters and unambiguous identification of these characters (or combinations of characters) in the fossil record. Once this has been established and the ages of the fossils are known, these can be used as minimum ages for the corresponding lineages. They are minimum ages because newly evolved character states do not usually fossilize until they become relatively common.
The situation outlined above, with multiple fossil calibration points, is an ideal scenario that is seldom encountered. Most algal groups do not fossilize well because they lack hard parts. Furthermore, their relatively simple, highly plastic morphologies do not facilitate identifying clear-cut synapomorphies. However, even in the absence of fossil calibration points, using the techniques below can be profitable because not all of the evolutionary interpretations cited above require an absolute time-frame.
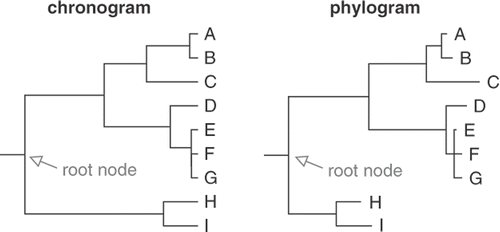
Dated trees are chronograms; i.e. their branch-lengths are proportional to time (). Such trees are ultrametric, i.e. all root-to-tip path lengths are equal. Because substitution rates of nucleotide markers are never entirely constant in time, phylogenetic analysis under standard models yields non-ultrametric phylograms ().
Fig. 7. Comparison of a chronogram and a phylogram. In a chronogram, branch lengths are proportional to time and root-to-tip path lengths are equal. In a phylogram inferred from sequence data, branch lengths are proportional to the number of substitutions along the branches and root-to-tip path lengths are usually unequal.
We will give a brief, hence incomplete overview of the types of methods and their implementation in software applications. More detailed reviews of the methods for obtaining dated trees are available (Bromham & Penny, Citation2003; Magallón, Citation2004; Sanderson et al., Citation2004; Welch & Bromham, Citation2005; Rutschmann, Citation2006; Yang, Citation2006).
In the simplest case of all, no changes in the rate of molecular evolution are present along the tree. Under the assumption of such a uniform (strict) molecular clock, all branches of a phylogenetic tree have the same substitution rate and the phylogram corresponds to a chronogram, allowing easy temporal interpretation. Unfortunately, in empirical datasets, substitution rates almost always vary among branches and, for that reason, the strict molecular clock is seldom used. The clock-likeness of a dataset can be checked with a hierarchical likelihood ratio test in several programs (e.g. PAUP*, HyPhy) or using the Bayes factor (e.g. MrBayes).
Many solutions have been proposed to deal with the fact that rates of molecular evolution vary along the tree. The first class of solutions consists of local molecular clock methods, which assume a small number of rate changes along the tree and rate homogeneity within large chunks of the tree (Yoder & Yang, Citation2000). This method is implemented in PAML (Yang, Citation2007).
A second, much more popular class of methods, known as relaxed molecular clock methods, allow for many changes in the rate of molecular evolution along the tree (as opposed to only a few in the local molecular clock methods). Methods from this class assume that small rate changes are more likely to occur than large rate changes and optimize rates along the branches of the tree. This assumption is known as rate autocorrelation because under this assumption, rates on parent branches and daughter branches are positively correlated. Implementations of this basic idea are manifold. There are non-parametric (NPRS=non-parametric rate smoothing: Sanderson, Citation1997), semi-parametric (PL=penalized likelihood: Sanderson, Citation2002), or completely parametric approaches (Bayesian methods: Thorne et al., Citation1998; Kishino et al., Citation2001; Thorne & Kishino, Citation2002; Lepage et al., Citation2007). These methods differ in various ways, yielding similar or divergent results (Aris-Brosou & Yang, Citation2002; Yang & Yoder, Citation2003; Pérez-Losada et al., Citation2004), and the accuracy of these methods over a range of realistic conditions has not been studied sufficiently. One recent study brought a significant advance in this area by testing the fit of a multitude of relaxed clock models to a series of representative empirical datasets, concluding that models that assume rate autocorrelation outperform those that do not (Lepage et al., Citation2007).
The NPRS and PL approaches are implemented in the r8s software (Sanderson, Citation2003). Bayesian methods are implemented in multidivtime (Thorne & Kishino, Citation2003) and PhyloBayes (Lartillot et al., Citation2007). The latter program includes a large variety of relaxed clock models and allows evaluating these models using Bayes factors (Lepage et al., Citation2007).
In contrast to the methods described above, which all start from a fixed tree and optimize branch lengths to be proportional to time, relaxed molecular clock models can also be applied at the tree inference stage. This is, to our knowledge, currently only possible using the Bayesian inference software BEAST (Drummond & Rambaut, Citation2007), which implements models in which the rate on each branch of the tree is drawn independently from an underlying rate distribution (Drummond et al., Citation2006). So, unlike the previously cited relaxed molecular clock methods, this approach does not assume autocorrelation of rates. In addition to their merits for molecular dating, analyses with relaxed molecular clock models have also been shown to yield more accurate topologies under certain circumstances (Drummond et al., Citation2006).
Ancestral character-state estimation
Phylogenetic hypotheses are often used to study the origin of morphological and/or molecular characters, physiological adaptations, etc. Starting with information on contemporary species only, one aims to make inferences about the character states of ancestral taxa. This is achieved by ancestral character state estimation methods, which map the character of interest onto a phylogenetic tree, constraining inferences about the conditions at internal nodes by the shape of the tree and the character states observed in contemporary species. The general methods to estimate character states are comparable to those used to build trees (Schluter et al., Citation1997; Mooers & Schluter, Citation1999; Pagel, Citation1999b; Pagel et al., Citation2004).
A wide variety of traits, both discrete and continuous, are of interest to evolutionary biologists. The states of discrete characters are fixed values, with no intermediate values possible. DNA sequence data, for example, are by their nature discrete. They occur in states that are unique and non-overlapping: A, C, G or T (U in the RNA molecule). Phenotypic data such as physiological or morphological traits, however, naturally occur as features that can be quantified in one of two ways, as a measurement along some continuum or as counts of abundance. For such data, the concept of ‘discrete’ is intuitively understood to mean characters whose states are separated by discontinuities, even though there may be variation within each state. Model-based ancestral state estimation of discrete traits usually assumes a continuous-time Markov model to describe the evolution of the trait (Pagel, Citation1994; Lewis, Citation2001b). Much like the situation for DNA (see section above: Basic model elements), characters can change states at any given moment and a matrix describing the rates of change between different states is estimated from the data.
Although algal systematists tend to work mainly with discrete morphological data, several studies stress the relevance of morphometric data for taxonomic purposes (Edgar & Theriot, Citation2004; Verbruggen et al., Citation2005; Neustupa & Stastny, Citation2006). Because most morphometric variables are continuous (measurements of a structure or variables derived from e.g. landmark analysis), they are less commonly studied in a phylogenetic framework. Furthermore, other types of continuous variables, such as ecological or physiological features, are rarely studied in a phylogenetic framework despite their evolutionary relevance. Several methods have been proposed to study the evolution of continuous characters along a phylogeny. These include squared-change parsimony (Maddison, Citation1991), an ML method in which the character is modelled to evolve according to Brownian motion (Schluter et al., Citation1997) and the general least squares (GLS) method, which allows more flexibility in model assumptions (Hansen & Martins, Citation1996; Martins & Hansen, Citation1997; Pagel, Citation1997).
As was the case for phylogenetic inference, ancestral state estimation of fast-evolving characters can pose problems in the sense that higher degrees of uncertainty are associated with ancestral states compared to slow-evolving characters (Schluter et al., Citation1997; Martins, Citation1999; Oakley & Cunningham, Citation2000). Similarly, inferred ancestral states can be biased if the estimation method's assumptions are violated (e.g. Wiens et al., Citation2007). Because likelihood-based inferences take branch lengths and rates of evolution into account whereas MP does not, the former should be more resistant to inference error (Pagel, Citation1999a). Because of such errors, estimated ancestral states should be interpreted with caution. It is often tempting to infer correlated evolution between characters by observing their reconstructions along a phylogeny but this is bad practice. In such cases, model testing can be used to see how well models that do and do not assume interdependent evolution of the traits fit the data (Pagel, Citation1994). Model fitting can also be used to gain insight into the evolution of individual characters. For example, one could wonder whether change in a character is associated with evolutionary time (gradual change) or with speciation events (punctuational change). Such questions and many others can be answered using model fitting approaches.
Parsimony reconstruction of discrete data can be carried out with most of the available tree construction software. Mesquite (Maddison & Maddison, Citation2007) and its predecessor MacClade (Maddison & Maddison, Citation2000) provide a graphical user interface and visualization tools for such analyses. Model-based inferences can be made with Mesquite (ML: Maddison & Maddison, Citation2007), ape (ML: Paradis et al., Citation2004) and BayesTraits (ML & BI: Pagel & Meade, Citation2006). The squared-change parsimony method for continuous data is implemented in Mesquite (Maddison & Maddison, Citation2007). ML methods for continuous data are available in ancml and ape (Schluter, Citation1997; Paradis et al., Citation2004). The GLS method is implemented in compare and ape (Martins, Citation2004; Paradis et al., Citation2004). BayesTraits includes a Bayesian implementation of ancestral state estimation for discrete characters, allowing for uncertainty about the phylogeny, model parameters and ancestral states. Model comparison can be carried out with most model-based ancestral state estimation software. Additionally, comet and geiger allow a variety of models to be fitted (Lee et al., Citation2006; Harmon et al., Citation2008).
Closing remarks
Phylogenetic analysis is and will continue to be a prominent aspect of algal systematic research. Thanks to their power and flexibility, the role of model-based techniques in phylogenetic reconstruction will continue to increase. Like all statistical methods, using phylogenetic inference methods necessitates understanding their assumptions and examining whether the data meet these assumptions. A key requirement for obtaining accurate phylogenetic results from molecular sequences is that the model of sequence evolution is sufficiently close to the processes that have generated the sequence data. Identifying a suitable model requires knowledge about the marker and application of model selection techniques. Once a phylogenetic result is obtained, further scrutiny is needed because saturation and process heterogeneity may have misled the phylogenetic inference. In other words, if one aims to infer accurate phylogenetic trees, extensive exploration of the dataset is needed to understand various aspects of the molecular evolution of the marker under study, and trees obtained at the click of a mouse need to be interpreted with extreme caution. Furthermore, it is important to realize that, although ML analyses normally yield a single tree, considerable phylogenetic uncertainty surrounds this tree in most empirical studies.
Model-based phylogenetic analyses can be computationally demanding, which may be a limiting factor for some researchers. Yet among the trends in molecular phylogenetic analysis is one towards remote execution. Several phylogenetic programs have a web submission form that can be used to run analyses on remote computing clusters. In addition to several small-scale initiatives offering execution of a single program, two supercomputing centres offer remote execution for a variety of inference programs. At the time of writing, the Computational Biology Service Unit (CBSU) at Cornell University offered remote execution of BEAST and MrBayes jobs (http://cbsu.tc.cornell.edu/). The Cyberinfrastructure for Phylogenetic Research (CIPRES) project currently offers GARLI, RAxML and PAUP*, with several other applications on the way (http://www.phylo.org/).
Finally, it is important to realize that the capacity of phylogenies extends far beyond reporting the relationships between species. A good phylogeny is the starting point for a wide array of inferences about character evolution and diversification of the organisms in spatial, temporal, ecological and physiological dimensions. Why are some taxonomic groups more species-rich than others? Did certain features promote diversification? Did traits evolve gradually or did bursts of change alternate with periods of stasis? How can evolution and ecology explain current distribution patterns? The questions one can ask are innumerable and so are the methods to answer them (e.g. Pagel, Citation1997; Yesson & Culham, Citation2006; Moore & Donoghue, Citation2007; Paradis, Citation2007). The algae show an astonishing diversity of life histories, ecologies and physiologies, making them unequalled case studies for learning about certain processes of evolution. Yet many questions about the evolution of algae remain unanswered, and this is where, in our opinion, there is a bright future for algal systematists.
Acknowledgements
We thank Wytze Stam, Paul Lewis, Olivier De Clerck and Frederik Leliaert for commenting on previous versions of the manuscript. Funding was provided by FWO-Flanders (post-doctoral fellowship to HV). ET was supported by NSF EF 0629410, the Tony Institute, and the Jane and Roland Blumberg Centennial Professorship in Molecular Evolution.
References
- Alfaro , ME and Holder , MT . 2006 . The posterior and the prior in Bayesian phylogenetics . Ann. Rev. Ecol. Evol. Systemat. , 37 : 19 – 42 .
- Altekar , G , Dwarkadas , S , Huelsenbeck , JP and Ronquist , F . 2004 . Parallel metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference . Bioinformatics , 20 : 407 – 415 .
- Alverson , AJ , Jansen , RK and Theriot , EC . 2007 . Bridging the Rubicon: phylogenetic analysis reveals repeated colonizations of marine and fresh waters by thalassiosiroid diatoms . Mol. Phylogenet. Evol. , 45 : 193 – 121 .
- Aris-Brosou , S . 2003 . How Bayes tests of molecular phylogenies compare with frequentist approaches . Bioinformatics , 19 : 618 – 624 .
- Aris-Brosou , S and Yang , ZH . 2002 . Effects of models of rate evolution on estimation of divergence dates with special reference to the metazoan 18S ribosomal RNA phylogeny . Syst. Biol. , 51 : 703 – 714 .
- Baele , G , Raes , J , Van de Peer , Y and Vansteelandt , S . 2006 . An improved statistical method for detecting heterotachy in nucleotide sequences . Mol. Biol. Evol. , 23 : 1397 – 1405 .
- Barker , FK and Lutzoni , FM . 2002 . The utility of the incongruence length difference test . Syst. Biol. , 51 : 625 – 637 .
- Beiko , RG , Keith , JM , Harlow , TJ and Ragan , MA . 2006 . Searching for convergence in phylogenetic Markov chain Monte Carlo . Syst. Biol. , 55 : 553 – 565 .
- Bergsten , J . 2005 . A review of long-branch attraction . Cladistics , 21 : 163 – 193 .
- Berry , V and Gascuel , O . 1996 . On the interpretation of bootstrap trees: appropriate threshold of clade selection and induced gain . Mol. Biol. Evol. , 13 : 999 – 1011 .
- Bininda-Emonds , ORP . 2004 . Phylogenetic Supertrees: combining Information to Reveal the Tree of Life , Dordrecht, , Germany : Kluwer .
- Björklund , M . 1999 . Are third positions really that bad? A test using vertebrate cytochrome b . Cladistics , 15 : 191 – 197 .
- Blouin , MS , Yowell , CA , Courtney , CH and Dame , JB . 1998 . Substitution bias, rapid saturation, and the use of mtDNA for nematode systematics . Mol. Biol. Evol. , 15 : 1719 – 1727 .
- Brandley , MC , Schmitz , A and Reeder , T . 2005 . Partitioned Bayesian analyses, partition choice, and the phylogenetic relationships of scincid lizards . Syst. Biol. , 54 : 373 – 390 .
- Brodie , J and Lewis , J . 2007 . Unravelling the Algae: the Past, Present and Future of Algal Systematics , Boca Raton, , USA : CRC Press .
- Bromham , L and Penny , D . 2003 . The modern molecular clock . Nat. Rev. Genet. , 4 : 216 – 224 .
- Brown , JM and Lemmon , AR . 2007 . The importance of data partitioning and the utility of Bayes factors in Bayesian phylogenetics . Syst. Biol. , 56 : 643 – 655 .
- Buckley , TR . 2002 . Model misspecification and probabilistic tests of topology: evidence from empirical data sets . Syst. Biol. , 51 : 509 – 523 .
- Bull , JJ , Huelsenbeck , JP , Cunningham , CW , Swofford , DL and Waddell , PJ . 1993 . Partitioning and combining data in phylogenetic analysis . Syst. Biol. , 42 : 384 – 397 .
- Burleigh , JG and Mathews , S . 2004 . Phylogenetic signal in nucleotide data from seed plants: implications for resolving the seed plant tree of life . Am. J. Bot. , 91 : 1599 – 1613 .
- Carstens , BC and Knowles , LL . 2007 . Estimating species phylogeny from gene-tree probabilities despite incomplete lineage sorting: an example from Melanoplus grasshoppers . Syst. Biol. , 56 : 400 – 411 .
- Castresana , J . 2000 . Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis . Mol. Biol. Evol. , 17 : 540 – 552 .
- Chen , D , Burleigh , GJ and Fernández-Baca , D . 2007 . Spectral partitioning of phylogenetic data sets based on compatibility . Syst. Biol. , 56 : 623 – 632 .
- Conant , GC and Lewis , PO . 2001 . Effects of nucleotide composition bias on the success of the parsimony criterion in phylogenetic inference . Mol. Biol. Evol. , 18 : 1024 – 1033 .
- Darlu , P and Lecointre , G . 2002 . When does the incongruence length difference test fail? . Mol. Biol. Evol. , 19 : 432 – 437 .
- Daugbjerg , N and Andersen , RA . 1997 . A molecular phylogeny of the heterokont algae based on analyses of chloroplast-encoded rbcL sequence data . J. Phycol. , 33 : 1031 – 1041 .
- De Clerck , O , Leliaert , F , Verbruggen , H , Lane , CE , De Paula , JC , Payo , DA and Coppejans , E . 2006 . A revised classification of the Dictyoteae (Dictyotales, Phaeophyceae) based on rbcL and 26S ribosomal DNA sequence analyses . J. Phycol. , 42 : 1271 – 1288 .
- Drummond , AJ and Rambaut , A . 2007 . BEAST: bayesian evolutionary analysis by sampling trees . BMC Evol. Biol. , 7 : 214
- Drummond , AJ , Ho , SYW , Phillips , MJ and Rambaut , A . 2006 . Relaxed phylogenetics and dating with confidence . PLoS Biol. , 4 : e88
- Edgar , SM and Theriot , EC . 2004 . Phylogeny of Aulacoseira (Bacillariophyta) based on molecules and morphology . J. Phycol. , 40 : 772 – 788 .
- Eernisse , DJ and Kluge , AG . 1993 . Taxonomic congruence versus total evidence, and amniote phylogeny inferred from fossils, molecules, and morphology . Mol. Biol. Evol. , 10 ( 6 ) : 1170 – 1195 .
- Efron , B , Halloran , E and Holmes , S . 1996 . Bootstrap confidence levels for phylogenetic trees . Proc. Natl. Acad. Sci. USA , 93 : 13429 – 13434 .
- Erpenbeck , D , Nichols , S , Voigt , O , Dohrmann , M , Degnan , B , Hooper , J and Wörheide , G . 2007 . Phylogenetic analyses under secondary structure-specific substitution models outperform traditional approaches: case studies with diploblast LSU . J. Mol. Evol. , 64 : 543 – 557 .
- Farris , JS . 1972 . Estimating phylogenetic trees from distance matrices . Am. Nat. , 106 : 645 – 668 .
- Farris , JS , Kallersjo , M , Kluge , AG and Bult , C . 1994 . Testing significance of incongruence . Cladistics , 10 : 315 – 319 .
- Felsenstein , J . 2004 . Inferring Phylogenies , Sunderland, , USA : Sinauer Associates .
- Fitch , WM . 1971 . Toward defining the course of evolution: minimum change for a specific tree topology . Syst. Zool. , 20 : 406 – 416 .
- Foster , PG . 2004 . Modeling compositional heterogeneity . Syst. Biol. , 53 : 485 – 495 .
- Galtier , N . 2008 . “ Paper presented at: statistical and computational challenges in molecular phylogenetics and evolution ” . In Phylogenomic Analyses with Horizontal Gene Transfer , London, UK : Royal Society .
- Gaut , BS and Lewis , PO . 1995 . Success of maximum likelihood phylogeny inference in the four-taxon case . Mol. Biol. Evol. , 12 : 152 – 162 .
- Geuten , K , Massingham , T , Darius , P , Smets , E and Goldman , N . 2007 . Experimental design criteria in phylogenetics: where to add taxa . Syst. Biol. , 56 : 609 – 622 .
- Goertzen , LR and Theriot , EC . 2003 . Effect of taxon sampling, character weighting, and combined data on the interpretation of relationships among the heterokont algae . J. Phycol. , 39 : 423 – 439 .
- Goldman , N . 1998 . Phylogenetic information and experimental design in molecular systematics . Proc. Roy. Soc. B – Biol. Sci. , 265 : 1779 – 1786 .
- Goldman , N and Yang , ZH . 1994 . Codon-based model of nucleotide substitution for protein-coding DNA sequences . Mol. Biol. Evol. , 11 : 725 – 736 .
- Goldman , N , Anderson , JP and Rodrigo , AG . 2000 . Likelihood-based tests of topologies in phylogenetics . Syst. Biol. , 49 : 652 – 670 .
- Graybeal , A . 1994 . Evaluating the phylogenetic utility of genes: a search for genes informative about deep divergences among vertebrates . Syst. Biol. , 43 : 174 – 193 .
- Graybeal , A . 1998 . Is it better to add taxa or characters to a difficult phylogenetic problem? . Syst. Biol. , 47 : 9 – 17 .
- Gu , X , Fu , YX and Li , WH . 1995 . Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites . Mol. Biol. Evol. , 12 : 546 – 557 .
- Guindon , S and Gascuel , O . 2003 . A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood . Syst. Biol. , 52 : 696 – 704 .
- Hansen , TF and Martins , EP . 1996 . Translating between microevolutionary process and macroevolutionary patterns: the correlation structure of interspecific data . Evolution , 50 : 1404 – 1417 .
- Harmon , LJ , Weir , JT , Brock , CD , Glor , RE and Challenger , W . 2008 . GEIGER: investigating evolutionary radiations . Bioinformatics , 24 : 129 – 131 .
- Hedtke , S , Townsend , T and Hillis , D . 2006 . Resolution of phylogenetic conflict in large data sets by increased taxon sampling . Syst. Biol. , 55 ( 3 ) : 522 – 529 .
- Hillis , DM and Bull , JJ . 1993 . An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis . Syst. Biol. , 42 : 182 – 192 .
- Hillis , DM , Pollock , DD , McGuire , JA and Zwickl , DJ . 2003 . Is sparse taxon sampling a problem for phylogenetic inference? . Syst. Biol. , 52 : 124 – 126 .
- Hipp , AL , Hall , JC and Sytsma , KJ . 2004 . Congruence versus phylogenetic accuracy: revisiting the incongruence length difference test . Syst. Biol. , 53 : 81 – 89 .
- Ho , SYW and Jermiin , LS . 2004 . Tracing the decay of the historical signal in biological sequence data . Syst. Biol. , 53 : 623 – 637 .
- Ho , JWK , Adams , CE , Lew , JB , Matthews , TJ , Ng , CC , Shahabi-Sirjani , A , Tan , LH , Zhao , Y , Easteal , S , Wilson , SR and Jermiin , LS . 2006 . SeqVis: visualization of compositional heterogeneity in large alignments of nucleotides . Bioinformatics , 22 : 2162 – 2163 .
- Holder , M and Lewis , PO . 2003 . Phylogeny estimation: traditional and Bayesian approaches . Nat. Rev. Genet. , 4 : 275 – 284 .
- Holland , BR , Penny , D and Hendy , MD . 2003 . Outgroup misplacement and phylogenetic inaccuracy under a molecular clock – A simulation study . Syst. Biol. , 52 : 229 – 238 .
- Holland , BR , Huber , KT , Moulton , V and Lockhart , PJ . 2004 . Using consensus networks to visualize contradictory evidence for species phylogeny . Mol. Biol. Evol. , 21 : 1459 – 1461 .
- Holland , BR , Jermiin , LS and Moulton , V . 2006 . Improved consensus network techniques for genome-scale phylogeny . Mol. Biol. Evol. , 23 : 848 – 855 .
- Hommersand , MH , Freshwater , DW , Lopez-Bautista , JM and Fredericq , S . 2006 . Proposal of the Euptiloteae Hommersand et Fredericq, trib. nov and transfer of some Southern Hemisphere Ptiloteae to the Callithamnieae (Ceramiaceae, Rhodophyta) . J. Phycol. , 42 : 203 – 225 .
- Huelsenbeck , JP . 1995 . Performance of phylogenetic methods in simulation . Syst. Biol. , 44 : 17 – 48 .
- Huelsenbeck , JP . 1997 . Is the Felsenstein zone a fly trap? . Syst. Biol. , 46 : 69 – 74 .
- Huelsenbeck , JP and Bull , JJ . 1996 . A likelihood ratio test to detect conflicting phylogenetic signal . Syst. Biol. , 45 : 92 – 98 .
- Huelsenbeck , JP and Rannala , B . 2004 . Frequentist properties of Bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models . Syst. Biol. , 53 : 904 – 913 .
- Huelsenbeck , JP , Bull , JJ and Cunningham , CW . 1996 . Combining data in phylogenetic analysis . Trends Ecol. Evol. , 11 : 152 – 158 .
- Huelsenbeck , JP , Ronquist , F , Nielsen , R and Bollback , JP . 2001 . Bayesian inference of phylogeny and its impact on evolutionary biology . Science , 294 : 2310 – 2314 .
- Huelsenbeck , JP , Larget , B , Miller , RE and Ronquist , F . 2002a . Potential applications and pitfalls of Bayesian inference of phylogeny . Syst. Biol. , 51 : 673 – 688 .
- Huelsenbeck , JP , Bollback , JP and Levine , AM . 2002b . Inferring the root of a phylogenetic tree . Syst. Biol. , 51 : 32 – 43 .
- Huson , DH and Bryant , D . 2006 . Application of phylogenetic networks in evolutionary studies . Mol. Biol. Evol. , 23 : 254 – 267 .
- Jeffroy , O , Brinkmann , H , Delsuc , F and Philippe , H . 2006 . Phylogenomics: the beginning of incongruence? . Trends Genet. , 22 : 225 – 231 .
- Jermiin , LS , Ho , SYW , Ababneh , F , Robinson , J and Larkum , AW . 2004 . The biasing effect of compositional heterogeneity on phylogenetic estimates may be underestimated . Syst. Biol. , 53 : 638 – 643 .
- Jobb , G , von Haeseler , A and Strimmer , K . 2004 . TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics . BMC Evol. Biol. , 4 : 18
- Källersjö , M , Albert , VA and Farris , JS . 1999 . Homoplasy increases phylogenetic structure . Cladistics , 15 : 91 – 93 .
- Kass , RE and Raftery , AE . 1995 . Bayes factors . J. Am. Stat. Assoc. , 90 : 773 – 795 .
- Kennedy , M , Holland , BR , Gray , RD and Spencer , HG . 2005 . Untangling long branches: identifying conflicting phylogenetic signals using spectral analysis, neighbor-net, and consensus networks . Syst. Biol. , 54 : 620 – 633 .
- Kishino , H and Hasegawa , M . 1989 . Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea . J. Mol. Evol. , 29 : 170 – 179 .
- Kishino , H , Thorne , JL and Bruno , WJ . 2001 . Performance of a divergence time estimation method under a probabilistic model of rate evolution . Mol. Biol. Evol. , 18 : 352 – 361 .
- Kluge , AG . 1989 . A concern for evidence and a phylogenetic hypothesis of relationships among Epicrates (Boidae, Serpentes) . Syst. Zool. , 38 : 7 – 25 .
- Kolaczkowski , B and Thornton , JW . 2004 . Performance of maximum parsimony and likelihood phylogenetics when evolution is heterogeneous . Nature , 431 : 980 – 984 .
- Kosakovsky Pond , SL , Frost , SDW and Muse , SV . 2005 . HyPhy: hypothesis testing using phylogenies . Bioinformatics , 21 : 676 – 679 .
- Larget , B and Simon , DL . 1999 . Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees . Mol. Biol. Evol. , 16 : 750 – 759 .
- Lartillot , N and Philippe , H . 2004 . A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process . Mol. Biol. Evol. , 21 : 1095 – 1109 .
- Lartillot , N and Philippe , H . 2006 . Computing Bayes factors using thermodynamic integration . Syst. Biol. , 55 : 195 – 207 .
- Lartillot , N , Blanquart , S and Lepage , T . 2007 . PhyloBayes. v2.3. http://www.lirmm.fr/mab/article.php3?id_article=329
- Le Gall , L and Saunders , GW . 2007 . A nuclear phylogeny of the Florideophyceae (Rhodophyta) inferred from combined EF2, small subunit and large subunit ribosomal DNA: establishing the new red algal subclass Corallinophycidae . Mol. Phylogenet. Evol. , 43 : 1118 – 1130 .
- Lee , C , Blay , S , Mooers , AO , Singh , A and Oakley , TH . 2006 . CoMET: a Mesquite package for comparing models of continuous character evolution on phylogenies . Evolutionary Bioinformatics Online , 2 : 193 – 196 .
- Leliaert , F , De Clerck , O , Verbruggen , H , Boedeker , C and Coppejans , E . 2007 . Molecular phylogeny of the Siphonocladales (Chlorophyta: Cladophorophyceae) . Mol. Phylogenet. Evol. , 44 : 1237 – 1256 .
- Lemieux , C , Otis , C and Turmel , M . 2007 . A clade uniting the green algae Mesostigma viride and Chlorokybus atmophyticus represents the deepest branch of the Streptophyta in chloroplast genome-based phylogenies . BMC Biol. , 5 : 2
- Lemmon , AR and Moriarty , EC . 2004 . The importance of proper model assumption in Bayesian phylogenetics . Syst. Biol. , 53 : 265 – 277 .
- Lepage , T , Bryant , D , Philippe , H and Lartillot , N . 2007 . A general comparison of relaxed molecular clock models . Mol. Biol. Evol. , 24 : 2669 – 2680 .
- Lewis , PO . 2001a . Phylogenetic systematics turns over a new leaf . Trends Ecol. Evol. , 16 : 30 – 37 .
- Lewis , PO . 2001b . A likelihood approach to estimating phylogeny from discrete morphological character data . Syst. Biol. , 50 : 913 – 925 .
- Lockhart , PJ , Steel , MA , Barbrook , AC , Huson , DH , Charleston , MA and Howe , C . 1998 . A covariotide model explains apparent phylogenetic structure of oxygenic photosynthetic lineages . Mol. Biol. Evol. , 15 : 1183 – 1188 .
- Lockhart , P , Novis , P , Milligan , BG , Riden , J , Rambaut , A and Larkum , T . 2006 . Heterotachy and tree building: a case study with plastids and eubacteria . Mol. Biol. Evol. , 23 : 40 – 45 .
- Lopez , P , Casane , D and Philippe , H . 2002 . Heterotachy, an important process of protein evolution . Mol. Biol. Evol. , 19 : 1 – 7 .
- Loytynoja , A and Milinkovitch , MC . 2001 . SOAP, cleaning multiple alignments from unstable blocks . Bioinformatics , 17 : 573 – 574 .
- Lyons-Weiler , J , Hoelzer , GA and Tausch , RJ . 1998 . Optimal outgroup analysis . Biol. J. Linn. Soc. , 64 : 493 – 511 .
- Maddison , WP . 1991 . Squared-change parsimony reconstructions of ancestral states for continuous-valued characters on a phylogenetic tree . Syst. Zool. , 40 : 304 – 314 .
- Maddison , DR and Maddison , WP . 2000 . MacClade 4 , Sunderland, , Massachusets : Sinauer .
- Maddison , WP and Maddison , DR . 2007 . Mesquite: a modular system for evolutionary analysis. v2.0. http://mesquiteproject.org
- Magallón , SA . 2004 . Dating lineages: molecular and paleontological approaches to the temporal framework of clades . Int. J. Plant Sci. , 165 : S7 – S21 .
- Maggs , CA , Verbruggen , H and De Clerck , O . 2007 . “ Molecular systematics of red algae: building future structures on firm foundations ” . In Unravelling the Algae: the Past, Present, and Future of Algal Systematics , Edited by: Brodie , J and Lewis , J . 103 – 121 . Boca Raton, , USA : CRC Press .
- Mann , DG and Evans , KM . 2007 . “ Molecular genetics and the neglected art of diatomics ” . In Unravelling the Algae: the Past, Present, and Future of Algal Systematics , Edited by: Brodie , J and Lewis , J . 231 – 266 . Boca Raton, , USA : CRC Press .
- Martins , EP . 1999 . Estimation of ancestral states of continuous characters: a computer simulation study . Syst. Biol. , 48 : 642 – 650 .
- Martins , EP . 2004 . COMPARE, version 4.6b. Computer programs for the statistical analysis of comparative data. http://compare.bio.indiana.edu/
- Martins , EP and Hansen , TF . 1997 . Phylogenies and the comparative method: a general approach to incorporating phylogenetic information into the analysis of interspecific data . Am. Nat. , 149 : 646 – 667 .
- Mattox , KR and Stewart , KD . 1984 . “ Classification of the green algae: a concept based on comparative cytology ” . In Systematics of the Green Algae , Edited by: Irvine , DEG and John , DM . 29 – 72 . London, UK : Academic Press .
- McIvor , L , Maggs , CA and Stanhope , MJ . 2002 . rbcL sequences indicate a single evolutionary origin of multinucleate cells in the red algal tribe Callithamnieae . Mol. Phylogenet. Evol. , 23 : 433 – 446 .
- Minin , V , Abdo , Z , Joyce , P and Sullivan , J . 2003 . Performance-based selection of likelihood models for phylogeny estimation . Syst. Biol. , 52 : 674 – 683 .
- Mooers , AO and Schluter , D . 1999 . Reconstructing ancestor states with maximum likelihood: support for one- and two-rate models . Syst. Biol. , 48 : 623 – 633 .
- Moore , BR and Donoghue , MJ . 2007 . Correlates of diversification in the plant clade Dipsacales: geographic movement and evolutionary innovations . Am. Nat. , 170 : S28 – S55 .
- Müller , KM , Oliveira , MC , Sheat , RG and Bhattacharya , D . 2001 . Ribosomal DNA phylogeny of the Bangiophycidae (Rhodophyta) and the origin of secondary plastids . Am. J. Bot. , 88 : 1390 – 1400 .
- Müller , KF , Borsch , T and Hilu , KW . 2006 . Phylogenetic utility of rapidly evolving DNA at high taxonomical levels: contrasting matK, trnT-F, and rbcL in basal angiosperms . Mol. Phylogenet. Evol. , 41 : 99 – 117 .
- Murray , S , Jorgensen , MF , Ho , SYW , Patterson , DJ and Jermiin , LS . 2005 . Improving the analysis of dinoflagellate phylogeny based on rDNA . Protist , 156 : 269 – 286 .
- Muse , SV and Gaut , BS . 1994 . A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome . Mol. Biol. Evol. , 11 : 715 – 724 .
- Neustupa , J and Stastny , J . 2006 . The geometric morphometric study of Central European species of the genus Micrasterias (Zygnematophyceae, Viridiplantae) . Preslia , 78 : 253 – 263 .
- Nixon , KC and Carpenter , JM . 1996 . On simultaneous analysis . Cladistics , 12 : 221 – 241 .
- Nylander , JAA , Ronquist , F , Huelsenbeck , JP and Nieves-Aldrey , JL . 2004 . Bayesian phylogenetic analysis of combined data . Syst. Biol. , 53 : 47 – 67 .
- Nylander , JAA , Wilgenbusch , JC , Warren , DL and Swofford , DL . 2007 . AWTY (Are We There Yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics . Bioinformatics , btm388
- Oakley , TH and Cunningham , CW . 2000 . Independent contrasts succeed where ancestor reconstruction fails in a known bacteriophage phylogeny . Evolution , 54 : 397 – 405 .
- Pagel , M . 1994 . Detecting correlated evolution on phylogenies: a general method for the comparative analysis of discrete characters . Proc. Roy. Soc. B, Biol. Sci. , 255 : 37 – 45 .
- Pagel , M . 1997 . Inferring evolutionary processes from phylogenies . Zoologica Scripta , 26 : 331 – 348 .
- Pagel , M . 1999a . Inferring the historical patterns of biological evolution . Nature , 401 : 877 – 884 .
- Pagel , M . 1999b . The maximum likelihood approach to reconstructing ancestral character states of discrete characters on phylogenies . Syst. Biol. , 48 : 612 – 622 .
- Pagel , M and Meade , A . 2004 . A phylogenetic mixture model for detecting pattern-heterogeneity in gene sequence or character-state data . Syst. Biol. , 53 : 571 – 581 .
- Pagel , M and Meade , A . 2006 . BayesTraits http://www.evolution.rdg.ac.uk/BayesTraits.html
- Pagel , M , Meade , A and Barker , D . 2004 . Bayesian estimation of ancestral character states on phylogenies . Syst. Biol. , 53 : 673 – 684 .
- Paradis , E . 2007 . Analysis of Phylogenetics and Evolution with R , New York, , USA : Springer .
- Paradis , E , Claude , J and Strimmer , K . 2004 . APE: analyses of phylogenetics and evolution in R language . Bioinformatics , 20 : 289 – 290 .
- Pardi , F and Goldman , N . 2007 . Resource-aware taxon selection for maximizing phylogenetic diversity . Syst. Biol. , 56 : 431 – 444 .
- Penny , D , McComish , BJ , Charleston , MA and Hendy , MD . 2001 . Mathematical elegance with biochemical realism: the covarion model of molecular evolution . J. Mol. Evol. , 53 : 711 – 723 .
- Pérez-Losada , M , Høeg , JT and Crandall , KA . 2004 . Unraveling the evolutionary radiation of the thoracican barnacles using molecular and morphological evidence: a comparison of several divergence time estimation approaches . Syst. Biol. , 53 : 244 – 264 .
- Philippe , H , Zhou , Y , Brinkmann , H , Rodrigue , N and Delsuc , F . 2005a . Heterotachy and long-branch attraction in phylogenetics . BMC Evol. Biol. , 5 : 50
- Philippe , H , Delsuc , F , Brinkmann , H and Lartillot , N . 2005b . Phylogenomics . Ann. Rev. Ecol. Evol. Syst. , 36 : 541 – 562 .
- Phillips , MJ and Penny , D . 2003 . The root of the mammalian tree inferred from whole mitochondrial genomes . Mol. Phylogenet. Evol. , 28 : 171 – 185 .
- Planet , PJ . 2006 . Tree disagreement: measuring and testing incongruence in phylogenies . J. Biomed. Informat. , 39 : 86 – 102 .
- Poe , S . 2003 . Evaluation of the strategy of long-branch subdivision to improve the accuracy of phylogenetic methods . Syst. Biol. , 52 ( 3 ) : 423 – 428 .
- Poe , S and Swofford , DL . 1999 . Taxon sampling revisited . Nature , 398 : 300 – 301 .
- Pol , D and Siddall , ME . 2001 . Biases in maximum likelihood and parsimony: a simulation approach to a 10-taxon case . Cladistics , 17 : 266 – 281 .
- Pollock , DD , Zwickl , DJ , McGuire , JA and Hillis , DM . 2002 . Increased taxon sampling is advantageous for phylogenetic inference . Syst. Biol. , 51 : 664 – 671 .
- Pombert , JF , Otis , C , Lemieux , C and Turmel , M . 2005 . The chloroplast genome sequence of the green alga Pseudendoclonium akinetum (Ulvophyceae) reveals unusual structural features and new insights into the branching order of chlorophyte lineages . Mol. Biol. Evol. , 22 : 1903 – 1918 .
- Posada , D and Buckley , TR . 2004 . Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests . Syst. Biol. , 53 : 793 – 808 .
- Rambaut , A and Drummond , AJ . 2007 . Tracer. v1.4. http://beast.bio.ed.ac.uk/tracer
- Rodríguez-Ezpeleta , N , Brinkmann , H , Roure , B , Lartillot , N , Lang , BF and Philippe , H . 2007a . Detecting and overcoming systematic errors in genome-scale phylogenies . Syst. Biol. , 56 : 389 – 399 .
- Rodríguez-Ezpeleta , N , Philippe , H , Brinkmann , H , Becker , B and Melkonian , M . 2007b . Phylogenetic analyses of nuclear, mitochondrial, and plastid multigene data sets support the placement of Mesostigma in the Streptophyta . Mol. Biol. Evol. , 24 : 723 – 731 .
- Rokas , A , Williams , BL , King , N and Carroll , SB . 2003 . Genome-scale approaches to resolving incongruence in molecular phylogenies . Nature , 425 : 798 – 804 .
- Ronquist , F and Huelsenbeck , JP . 2003 . MrBayes 3: bayesian phylogenetic inference under mixed models . Bioinformatics , 19 : 1572 – 1574 .
- Rosenberg , MS and Kumar , S . 2001 . Incomplete taxon sampling is not a problem for phylogenetic inference . Proc. Natl. Acad. Sci. USA , 98 : 10751 – 10756 .
- Rosenberg , MS and Kumar , S . 2003 . Heterogeneity of nucleotide frequencies among evolutionary lineages and phylogenetic inference . Mol. Biol. Evol. , 20 : 610 – 621 .
- Ruano-Rubio , V and Fares , MA . 2007 . Artifactual phylogenies caused by correlated distribution of substitution rates among sites and lineages: the good, the bad, and the ugly . Syst. Biol. , 56 : 68 – 82 .
- Ruiz-Trillo , I , Riutort , M , Littlewood , DTJ , Herniou , EA and Baguna , J . 1999 . Acoel flatworms: earliest extant bilaterian metazoans, not members of Platyhelminthes . Science , 283 : 1919 – 1923 .
- Rutschmann , F . 2006 . Molecular dating of phylogenetic trees: a brief review of current methods that estimate divergence times . Divers. Distrib. , 12 : 35 – 48 .
- Sanderson , MJ . 1997 . A nonparametric approach to estimating divergence times in the absence of rate constancy . Mol. Biol. Evol. , 14 : 1218 – 1231 .
- Sanderson , MJ . 2002 . Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach . Mol. Biol. Evol. , 19 : 101 – 109 .
- Sanderson , MJ . 2003 . r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock . Bioinformatics , 19 : 301 – 302 .
- Sanderson , MJ , Thorne , JL , Wikström , N and Bremer , K . 2004 . Molecular evidence on plant divergence times . Am. J. Bot. , 91 : 1656 – 1665 .
- Saunders , GW , Chiovitti , A and Kraft , GT . 2002 . Small-subunit rDNA sequences from representatives of selected families of the Gigartinales and Rhodymeniales (Rhodophyta). 3. Delineating the Gigartinales sensu stricto . Can. J. Bot. , 82 : 43 – 74 .
- Schluter , D . 1997 . ANCML: ancestor states for continuous traits using maximum likelihood http://www.zoology.ubc.ca/∼schluter/ancml.html
- Schluter , D , Price , T , Mooers , AO and Ludwig , D . 1997 . Likelihood of ancestor states in adaptive radiation . Evolution , 51 : 1699 – 1711 .
- Schmidt , HA , Strimmer , K , Vingron , M and von Haeseler , A . 2002 . TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing . Bioinformatics , 18 : 502 – 504 .
- Schöniger , M and von Haeseler , A . 1994 . A stochastic model for the evolution of autocorrelated DNA sequences . Mol. Phylogenet. Evol. , 3 : 240 – 247 .
- Schöniger , M and von Haeseler , A . 1995 . Performance of the maximum likelihood, neighbor joining, and maximum parsimony methods when sequence sites are not independent . Syst. Biol. , 44 : 533 – 547 .
- Shalchian-Tabrizi , K , Skanseng , M , Ronquist , F , Klaveness , D , Bachvaroff , TR , Delwiche , CF , Botnen , A , Tengs , T and Jakobsen , KS . 2006 . Heterotachy processes in rhodophyte-derived secondhand plastid genes: implications for addressing the origin and evolution of dinoflagellate plastids . Mol. Biol. Evol. , 23 : 1504 – 1515 .
- Shapiro , B , Rambaut , A and Drummond , AJ . 2006 . Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences . Mol. Biol. Evol. , 23 : 7 – 9 .
- Shavit , L , Penny , D , Hendy , MD and Holland , BR . 2007 . The problem of rooting rapid radiations . Mol. Biol. Evol , 24 : 2400 – 2411 .
- Shimodaira , H . 2002 . An approximately unbiased test of phylogenetic tree selection . Syst. Biol. , 51 : 492 – 508 .
- Shimodaira , H and Hasegawa , M . 1999 . Multiple comparisons of log-likelihoods with applications to phylogenetic inference . Mol. Biol. Evol. , 16 : 1114 – 1116 .
- Shimodaira , H and Hasegawa , M . 2001 . CONSEL: for assessing the confidence of phylogenetic tree selection . Bioinformatics , 17 : 1246 – 1247 .
- Shpak , M and Churchill , GA . 2000 . The information content of a character under a Markov model of evolution . Mol. Phylogenet. Evol. , 17 : 231 – 243 .
- Siddall , ME . 1998 . Success of parsimony in the four-taxon case: long-branch repulsion by likelihood in the Farris Zone . Cladistics , 14 : 209 – 220 .
- Siemer , BL , Stam , WT , Olsen , JL and Pedersen , PM . 1998 . Phylogenetic relationships of the brown algal orders Ectocarpales, Chordariales, Dictyosiphonales, and Tilopteridales (Phaeophyceae) based on RUBISCO large subunit and spacer sequences . J. Phycol. , 34 : 1038 – 1048 .
- Simmons , MP , Zhang , LB , Webb , CT and Reeves , A . 2006 . How can third codon positions outperform first and second codon positions in phylogenetic inference? An empirical example from the seed plants . Syst. Biol. , 55 : 245 – 258 .
- Smith , AB . 1994 . Rooting molecular trees: problems and strategies . Biol. J. Linn. Soc. , 51 : 279 – 292 .
- Soltis , PS and Soltis , DE . 2003 . Applying the bootstrap in phylogeny reconstruction . Stat. Sci. , 18 : 256 – 267 .
- Stamatakis , A . 2006 . RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models . Bioinformatics , 22 : 2688 – 2690 .
- Stefankovic , D and Vigoda , E . 2007 . Pitfalls of heterogeneous processes for phylogenetic reconstruction . Syst. Biol. , 56 : 113 – 124 .
- Strimmer , K and von Haeseler , A . 1996 . Quartet puzzling: a quartet maximum-likelihood method for reconstructing tree topologies . Mol. Biol. Evol. , 13 : 964 – 969 .
- Strimmer , K and von Haeseler , A . 1997 . Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment . Proc. Natl. Acad. Sci. USA , 94 : 6815 – 6819 .
- Struck , TH , Purschke , G and Halanych , KM . 2006 . Phylogeny of Eunicida (Annelida) and exploring data congruence using a partition addition bootstrap alteration (PABA) approach . Syst. Biol. , 55 : 1 – 20 .
- Suchard , MA , Weiss , RE and Sinsheimer , JS . 2001 . Bayesian selection of continuous-time Markov chain evolutionary models . Mol. Biol. Evol. , 18 : 1001 – 1013 .
- Sullivan , J and Joyce , P . 2005 . Model selection in phylogenetics . Ann. Rev. Ecol. Evol. Syst. , 36 : 445 – 466 .
- Sung , GH , Sung , JM , Hywel-Jones , NL and Spatafora , JW . 2007 . A multi-gene phylogeny of Clavicipitaceae (Ascomycota, Fungi): identification of localized incongruence using a combinational bootstrap approach . Mol. Phylogenet. Evol. , 44 : 1204 – 1223 .
- Swofford , DL . 2003 . PAUP*: phylogenetic Analysis Using Parsimony (* and other methods). v4.0b10 ,
- Swofford , DL , Olsen , GJ , Waddell , PJ and Hillis , DM . 1996 . “ Phylogenetic inference ” . In Molecular Systematics , Edited by: Hillis , DM , Moritz , C and Mable , BK . 407 – 514 . Sunderland, , USA : Sinauer .
- Swofford , DL , Waddell , PJ , Huelsenbeck , JP , Foster , PG , Lewis , PO and Rogers , JS . 2001 . Bias in phylogenetic estimation and its relevance to the choice between parsimony and likelihood methods . Syst. Biol. , 50 : 525 – 539 .
- Tajima , F . 1993 . Simple methods for testing the molecular evolutionary clock hypothesis . Genetics , 135 : 599 – 607 .
- Talavera , G and Castresana , J . 2007 . Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments . Syst. Biol. , 56 : 564 – 577 .
- Tanabe , AS . 2007 . Kakusan: a computer program to automate the selection of a nucleotide substitution model and the configuration of a mixed model on multilocus data . Mol. Ecol. Notes , 7 : 962 – 964 .
- Telford , MJ , Wise , MJ and Gowri-Shankar , V . 2005 . Consideration of RNA secondary structure significantly improves likelihood-based estimates of phylogeny: examples from the bilateria . Mol. Biol. Evol. , 22 : 1129 – 1136 .
- Thorne , JL and Kishino , H . 2002 . Divergence time and evolutionary rate estimation with multilocus data . Syst. Biol. , 51 : 689 – 702 .
- Thorne , JL and Kishino , H . 2003 . Mutidivtime , http://statgen.ncsu.edu/thorne/multidivtime.html
- Thorne , JL , Kishino , H and Painter , IS . 1998 . Estimating the rate of evolution of the rate of molecular evolution . Mol. Biol. Evol. , 15 : 1647 – 1657 .
- Townsend , JP . 2007 . Profiling phylogenetic informativeness . Syst. Biol. , 56 : 222 – 231 .
- Verbruggen , H , De Clerck , O , Kooistra , W and Coppejans , E . 2005 . Molecular and morphometric data pinpoint species boundaries in Halimeda section Rhipsalis (Bryopsidales, Chlorophyta) . J. Phycol. , 41 : 606 – 621 .
- Verbruggen , H , Leliaert , F , Maggs , CA , Shimada , S , Schils , T , Provan , J , Booth , D , Murphy , S , De Clerck , O , Littler , DS , Littler , MM and Coppejans , E . 2007 . Species boundaries and phylogenetic relationships within the green algal genus Codium (Bryopsidales) based on plastid DNA sequences . Mol. Phylogenet. Evol. , 44 : 240 – 254 .
- Welch , JJ and Bromham , L . 2005 . Molecular dating when rates vary . Trends Ecol. Evol. , 20 : 320 – 327 .
- Whelan , S . 2007 . New approaches to phylogenetic tree search and their application to large numbers of protein alignments . Syst. Biol. , 56 : 727 – 740 .
- Whelan , S , Lio , P and Goldman , N . 2001 . Molecular phylogenetics: state-of-the-art methods for looking into the past . Trends Genet. , 17 : 262 – 272 .
- White , WT , Hills , SF , Gaddam , R , Holland , BR and Penny , D . 2007 . Treeness triangles: visualizing the loss of phylogenetic signal . Mol. Biol. Evol. , 24 : 2029 – 2039 .
- Wiens , JJ , Kuczynski , CA , Duellman , WE and Reeder , TW . 2007 . Loss and re-evolution of complex life cycles in marsupial frogs: does ancestral trait reconstruction mislead? . Evolution , 61 : 1886 – 1899 .
- Withall , RD and Saunders , GW . 2006 . Combining small and large subunit ribosomal DNA genes to resolve relationships among orders of the Rhodymeniophycidae (Rhodophyta): recognition of the Acrosymphytales ord. nov. and Sebdeniales ord. nov . Eur. J. Phycol. , 41 : 379 – 394 .
- Xia , X and Xie , Z . 2001 . DAMBE: software package for data analysis in molecular biology and evolution . J. Hered. , 92 : 371 – 373 .
- Xia , XH , Xie , Z , Salemi , M , Chen , L and Wang , Y . 2003 . An index of substitution saturation and its application . Mol. Phylogenet. Evol. , 26 : 1 – 7 .
- Yang , Z . 1994a . Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods . J. Mol. Evol. , 39 : 306 – 314 .
- Yang , Z . 1994b . Estimating the pattern of nucleotide substitution . J. Mol. Evol. , 39 : 105 – 111 .
- Yang , Z . 1996 . Among-site rate variation and its impact on phylogenetic analyses . Trends Ecol. Evol. , 11 : 367 – 372 .
- Yang , Z . 1998 . On the best evolutionary rate for phylogenetic analysis . Syst. Biol. , 47 : 125 – 133 .
- Yang , Z . 2006 . Computational Molecular Evolution , Oxford, UK : Oxford University Press .
- Yang , Z . 2007 . PAML 4: phylogenetic Analysis by Maximum Likelihood . Mol. Biol. Evol. , 24 : 1586 – 1591 .
- Yang , Z and Rannala , B . 2005 . Branch-length prior influences Bayesian posterior probability of phylogeny . Syst. Biol. , 54 : 455 – 470 .
- Yang , ZH , Nielsen , R , Goldman , N and Pedersen , AMK . 2000 . Codon-substitution models for heterogeneous selection pressure at amino acid sites . Genetics , 155 : 431 – 449 .
- Yang , ZH and Yoder , AD . 2003 . Comparison of likelihood and Bayesian methods for estimating divergence times using multiple gene loci and calibration points, with application to a radiation of cute-looking mouse lemur species . Syst. Biol. , 52 : 705 – 716 .
- Yap , VB and Speed , T . 2005 . Rooting a phylogenetic tree with nonreversible substitution models . BMC Evol. Biol. , 5 : 2
- Yesson , C and Culham , A . 2006 . Phyloclimatic modeling: combining phylogenetics and bioclimatic modeling . Syst. Biol. , 55 : 785 – 802 .
- Yoder , AD and Yang , ZH . 2000 . Estimation of primate speciation dates using local molecular clocks . Mol. Biol. Evol. , 17 : 1081 – 1090 .
- Zwickl , DJ . 2006 . Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion , http://www.bio.utexas.edu/faculty/antisense/garli/Garli.html
- Zwickl , DJ and Hillis , DM . 2002 . Increased taxon sampling greatly reduces phylogenetic error . Syst. Biol. , 51 : 588 – 598 .