
Abstract
Epidemiological studies have demonstrated that vitamin C decreases the risk of stroke, which has generally been ascribed to its function as antioxidant and free radical scavenger. However, whether there is a defined molecular target for vitamin C on stroke is unknown. Utilizing middle cerebral artery occlusion (MCAO) in rats as a model for ischemic stroke, we demonstrated that long-term, low-dose administration of vitamin C prior to MCAO could exert significant neuroprotective effect on the brain damage. The long-term, low-dose vitamin C pretreated rats had decreased brain infarct size and decreased neurological deficit score compared with the vehicle or single high dose pretreated MCAO rats. Furthermore, electrophysiological experiments using patch clamp technique showed that vitamin C increased the whole-cell current of the large-conductance Ca2+-activated K+ (BKCa) channel. Moreover, vitamin C increased the open probability of the channel without change its amplitude. Importantly, blockade of the BKCa channels abolished the neuroprotective effect of vitamin C on MCAO. Therefore, this study shows that long-term, low-dose pretreatment with vitamin C could reduce MCAO-induced brain damage through activation of the BKCa channels, suggesting that the BKCa channel is a molecular target of vitamin C on stroke.
Introduction
Vitamin C (also known as Ascorbic acid) is best known for its role as an essential nutrient in humans. A number of epidemiologic studies have shown that dietary intake of vitamin C or high plasma vitamin C concentrations are associated with a lower risk of stroke or stroke-related mortality (Ascherio et al., Citation1999; Del Rio et al., Citation2011; Gale et al., Citation1995). It has been shown more than two decades ago that vitamin C concentration is strongly related to the risk of death from stroke but not from coronary heart disease in elderly people (Gale et al., Citation1995). This association has been confirmed in the subsequent studies. From a prospective study of 20,649 participants aged 40–79 years with an average follow-up of 9.5 years, it has been shown that the risk of stroke over 10 years of follow-up was 42% lower in subjects with baseline plasma vitamin C levels ≥66 µmol.L−1 compared to those with vitamin C levels <41 µmol.L−1 (Myint et al., Citation2008). In addition, a meta-analysis of prospective studies has also demonstrated that both dietary vitamin C intake and circulating vitamin C are significantly inversely associated with the risk of stroke in a dose-response manner (Chen et al., Citation2013a). In consistence with the beneficial effect of vitamin C on stroke, multiple lines of evidence also support that a high consumption of fruits and vegetables is associated with a reduction in the risk of stroke (Dauchet et al., Citation2005; Joshipura et al., Citation1999; Johnsen et al., Citation2003; Lakkur & Judd, Citation2015; Padayatty & Levine, Citation2008).
On the other hand, several phase III randomized controlled trials have suggested that vitamin C supplementation has no effect on stroke prevention (Myung et al., Citation2013). Animal experiments also showed that vitamin C treatment was not associated with significant improvements in either infarct size or neurological function (Huang et al., Citation2001). These results are seemingly contradictory to the above epidemiologic studies. However, dietary intake represents long-term habitual exposure of vitamin C before the onset of stroke, whereas supplementation is generally characterized by shorter duration and higher dose. Thus, if vitamin C plays a preventive but not therapeutic role in stroke, the trials enrolling high-risk participants may not be able to detect any significant associations (Chen et al., Citation2013a). For example, it has been shown that administration of vitamin C (500 mg.day−1, iv) to ischemic stroke patients for 10 days beginning on day 1 after ischemic stroke did not substantially improve the clinical and functional status of patients after 3 months (Lagowska-Lenard et al., Citation2010).
In the present study, to study whether vitamin C has protective but not therapeutic effect on ischemic stroke, we administrate vitamin C intraperitoneally once a day for 3 weeks to rats before middle cerebral artery occlusion (MCAO). Furthermore, we provided several lines of evidence to confirm whether there is a clear molecular target for the effect of vitamin C on ischemic stroke.
Experimental procedure
Cell culture and transfection
Chinese hamster ovary (CHO-K1) cells were cultured in Ham’s F12 nutrient mixture supplemented with 10% FBS at 37 °C in a 5% CO2 incubator. Cells were plated onto poly-L-lysine-coated coverslips in 35 mm dishes at 60–80% confluence. Then they were transiently cotransfected with the full length BKCa channel gene and GFP (Clontech, Palo Alto, CA, USA). Transfection was performed with LipofectAMINE Plus reagent (Invitrogen) following the manufacturer's instructions. Cells were used for electrophysiological recordings 1–2 days after the transfection.
Whole-cell recording
Whole-cell currents were measured using a conventional tight seal whole-cell recording technique at room temperature. Pipettes had resistance at a range of 2–5 MΩ in recording solution containing (in mM) 145 KCl, 10 EGTA, 10 HEPES (pH 7.3 with KOH). The intracellular free Ca2+ concentration was 1 μM. The extracellular solution was Hanks’ balanced salt solution (HBSS, Sigma, in mmol.L−1): 1.3 CaCl2, 0.8 MgSO4, 5.4 KCl, 0.4 KH2PO4, 136.9 NaCl, 0.3 Na2PO4, 10 D-glucose and 4.2 NaHCO3. Compensation for cell capacitance and series resistance was made automatically using EPC-10 patch-clamp amplifiers (HEKA, Lambrecht, Germany); only recordings with stable series resistance less than 25 MΩ were included in the study. Currents were sampled at 2–5 kHz and filtered at 1.5–2.9 kHz using a 4-pole low-pass Bessel filter. Program packages Patchmaster 2.1 and TAC 4.1 (HEKA) were used for data acquisition and analysis. The rundown of the whole cell current of the BKCa channels is apparent at first 10 min after formation of the whole-cell configuration. To overcome this problem, each of the chemicals was added to judge its effect on the current only after the current was stable for 3 min period, which usually takes around 15 min.
Single-channel recording
The single-channel recordings were carried out at inside-out patch-clamp configuration using an EPC-10 amplifier (HEKA Elektronic, Lambrecht, Germany) at room temperature. Data were acquired at 2–5 kHz and low-pass filtered at 1 kHz. Patch electrodes were pulled from thin walled borosilicate capillary tubes using a Sutter programable puller (model P-97, Sutter Instrument, Novato, CA, USA) and fire-polished to achieve an electrical resistance in a range of 3–7 MΩ. An Ag/AgCl wire bath electrode was used as ground electrode. The pipette solution was HBSS. The bath solution consisted of (in mmol.L−1): 145 KCl, 10 EGTA, 2 Mg-ATP, 10 HEPES (pH 7.3 with KOH). The Po was simply calculated from the total time spent in the open state divided by the total time of recording for the patches containing a single channel. Continuous recordings of 2000–4000 ms were used to estimate Po. The Po in a patch with multiple channels was calculated by using TAC 4.1 (HEKA, Germany), based on the equation: Po = (1 − PC1/N), where Pc is the probability that all channels are in the closed state, N is the number of channels in the patch, which is estimated from the maximum number of channels observed.
Measurement of vitamin C concentration in serum and brain tissue
To obtain the serum sample, whole blood of each rat was sampled in a vacutainer tube. Serum was generated by centrifugation at 3000 g in 20 min at 4 °C and immediately frozen to −80 °C until further handled and analyzed. At the time of analysis, the serum sample (200 μl) was homogenized in a centrifuge tube with 500 μl vitamin C extracting solvent (0.35 mol.L−1 perchloric acid, 0.27 mmol.L−1 EDTA-Na and 0.1% dithiothreitol). After centrifugation at 1000 g in 5 min at 4 °C, the supernate was filtrated by a 0.22 μm membrane filter. To obtain samples of the brain tissue, 100 mg brain tissue from each rat was homogenized in a mixture of 1.0 ml dithiothreitol (20 mg.ml−1). After addition of 9.0 ml metaphosphoric acid (3%), the brain samples were eddied, sonicated for 10 min and centrifuged at 4000 g for 5 min after addition of 5 ml n-hexane. Then, the aqueous phase was filtrated by a 0.22 μm membrane filter. Vitamin C concentrations of the above samples were analyzed by an Ultra Performance Liquid Chromatography (UltiMate 3000, Dionex company, USA). Chromatographic separation was achieved by using an Agilent XDB-C18 column (5 μm, 4.6 × 150 mm, USA). A 10 μl sample (in 5% methanol) was injected into the column at a flow rate of 0.8 ml.min−1. The compounds were eluted using 95% mobile phase A (a solution of acetic acid at PH3) and 5% mobile phase B (100% methanol). The detection wavelength was set at 254 nm and the time of equilibrium was 30 min. The vitamin C concentrations of the samples were determined from the standard curves generated from vitamin C standards.
Transient Middle cerebral artery occlusion
A total of 297 male Sprague-Dawley rats (weighing 200–250 g, Xiamen University animal experiment center) were used throughout the study. Among them 17 rats died during experiment, which were excluded from the study. Details on the number of the rats in each experiment were shown in the corresponding figures. The transient middle cerebral artery occlusion (MCAO) was induced by using intraluminal filament insertion technique as previously described (Chi et al., Citation2010). Briefly, the filament thread (with a blunt tip of 0.36 ± 0.02 mm in diameter, type 2636-A3, Beijing Cinontech, China) was inserted through the left common carotid artery and advanced into the internal carotid artery to occlude the origin of the middle cerebral artery (approximately 18–20 mm) for 24 hours. Body temperatures were monitored and controlled at 37 ± 0.5 °C with a homoeothermic blanket. The sham-operated rats were treated identically, except that the middle cerebral artery was not occluded after the neck incision. All the rats were given unrestricted access to food and water. The animals were sacrificed 24 hr post-MCAO. Brains were rapidly removed, sectioned coronally at 2 mm intervals, and stained by 2,3,5-triphenyltetrazolium hydrochloride (TTC). The slices were photographed, and analyzed with Image-J software (NIH freeware) and presented with Photoshop software (Adobe Systems Inc., San Jose, California, USA). Infarct size in all slices was expressed as a percentage of the contralateral hemisphere after correcting for edema. All manipulations and analyses were performed by individuals blinded to all groups. Animal care and experiments were performed in accordance with procedures approved by the Animal Care and Use Committee of Xiamen University (accreditation number XMULAC20170250).
Drug administration
Vitamin C diluted in 0.9% saline was administrated intraperitoneally. Rats in the control group were given an equal volume of the vehicle (0.9% saline). Penitrem A (PA) and Paxilline (Pax) were dissolved in DMSO, diluted in 0.9% saline and then were applied intraperitoneally.
Neurological evaluation
Neurological deficit scores were evaluated after 24 hr of MCAO according to the Longa 5-point scale scoring system (Longa et al., Citation1989): grade 0, no deficit; grade 1, failure to fully extend the right forepaw; grade 2, spontaneous circling or walking to a contralateral side; grade 3, walking only when stimulated; grade 4, unresponsive to stimulation and a depressed level of consciousness; and grade 5, death. Grades of the neurological score were evaluated by an investigator blinded to the treatment protocol.
Statistics
The results were presented as mean ± SEM. The Origin 7.0 (Originlab, Northampton, MA, USA) was used for statistical analyses and plotting graphs. Statistical comparisons were made by using one-way ANOVA, followed by Bonferroni’s post hoc test. Differences at p < 0.05 were considered to be statistically significant.
Results
Preventive effect of long-term, low-dose vitamin C on MCAO rats
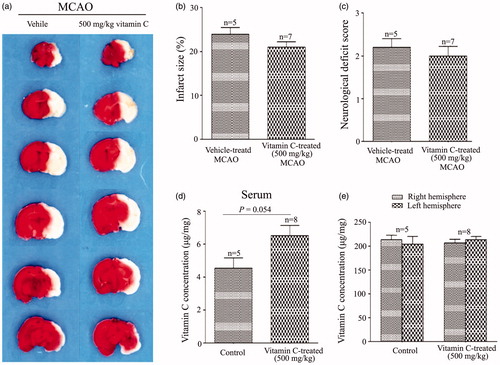
To investigate whether vitamin C has preventive effect on focal cerebral ischemia, we tested effect of long-term (once a day for 3 weeks) intraperitoneal administration of vitamin C on the rats prior to MCAO. At 24 hr post-MCAO, the brain slices were stained with TTC (), which stains healthy tissue red and leaves infarct tissue pale white. Statistical results showed that the percentage of infarct size was 22.77 ± 0.61% in the vehicle-treated MCAO rats, and the percentages of the infarct size were reduced to 20.83 ± 1.15%, 19.58 ± 0.89%, 20.46 ± 0.67%, 17.85 ± 1.93% for the rats treated with 0.5, 5, 20 and 50 mg.kg−1.day−1 vitamin C, respectively (). In addition, the neurological deficit score () was 2.03 ± 0.12 for the vehicle-treated MCAO rats, and 1.87 ± 0.09, 1.60 ± 0.10, 1.69 ± 0.13, 1.57 ± 0.20 for the rats treated with 0.5, 5, 20 and 50 mg.kg−1.day−1 vitamin C, respectively. As vitamin C at a dosage of 5 mg.kg−1.day−1 gave the significant protective effect on both infarct size and neurological deficit score, the dosage of 5 mg.kg−1 was used in most of the following studies. To test whether more frequently administration of vitamin C could have more beneficial effect on MCAO, we compared the effect of administration of 2.5 mg.kg−1 vitamin C twice a day with that of 5 mg.kg−1 once a day on MCAO. Representative images of TTC stained brain slices were present in . Statistical results showed that even though administration of 2.5 mg.kg−1 vitamin C twice a day had beneficial effect on infarct size () and neurological deficit score (), there was no significant difference between the groups administrated with 2.5 mg.kg−1 twice a day and 5 mg.kg−1 once a day, suggesting that more frequently administration of vitamin C does not have more beneficial effect on MCAO. Next, we measured vitamin C concentration in the serum and brain tissue after administration of vitamin C for three weeks. Before MCAO (24 hr after the last administration of vitamin C), the concentration of vitamin C in both serum () and brain tissue () was significantly increased from the groups administrated with 5 mg.kg−1 vitamin C once a day or 2.5 mg.kg−1 vitamin C twice a day. However, there was no significant difference between the groups administrated with 5 mg.kg−1 vitamin C once a day and the group administrated with 2.5 mg.kg−1 vitamin C twice a day. After 24 hr post-MCAO (i.e., 48 hr after the last administration of vitamin C), vitamin C concentration in the serum was significantly increased for both MCAO and vitamin C treated MCAO groups (vs. Sham operated group) (). Interestingly, the vitamin C concentration in the ipsilateral hemisphere was significant higher in the vitamin C treated MCAO group than that in the vehicle treated MCAO group (), even though there were no significant difference in that of the contralateral hemisphere (). The big differences of vitamin C levels in contralateral and ipsilateral brain might due to the influence of tissue damage. In addition, we found that administration of a single high dose (500 mg.kg−1) vitamin C has no effect on MCAO () and did not significantly change the vitamin C concentration in the serum () and brain tissue () before MCAO (24 hr after the last administration of vitamin C). These results suggested that pretreament with long-term, low-dose vitamin C could reduce the MCAO induced brain damage, while a single high dose had no effect.
Figure 1. Effect of long-term, low-dose vitamin C on MCAO induced infarct size and neurological deficit score. (a) Representative images of TTC stained brain slices obtained at 24 hr post-MCAO from MCAO rats pretreated with vitamin C at a dose of 0, 0.5, 5, 20 and 50 mg.kg−1.day−1 for 3 weeks. (b,c) Bars showing the relative infract size (b) or neurological deficit score (c) of MCAO rats pretreated with vitamin C at a dose of 0, 0.5, 5, 20 and 50 mg.kg−1.day−1 for 3 weeks. *p < 0.05; **p < 0.01; ***p < 0.005.
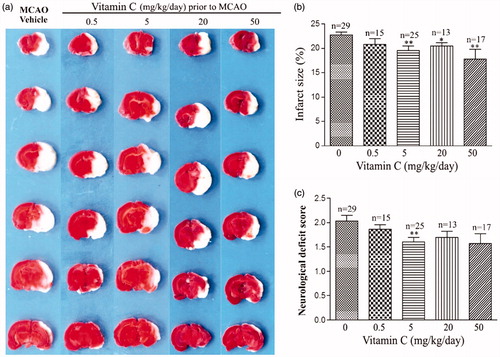
Figure 2. Application of vitamin C twice a day does not have more beneficial effect on ischemic stroke than that applied once a day. (a) Representative images of brain slices obtained at 24 hr post-MCAO from MCAO rats pretreated with vehicle as well as pretreated with vitamin C at a dose of 5 mg.kg−1 once a day or 2.5 mg.kg−1 twice a day for 3 weeks. (b,c) Bars showing the relative infract size (b) and neurological deficit score (c) of MCAO rats pretreated with vehicle and vitamin C at a dose of 5 mg.kg−1 once a day or 2.5 mg.kg−1 twice a day (2*2.5 mg.kg−1.day−1) for 3 weeks. *p < 0.05; **p < 0.01; ***p < 0.005.
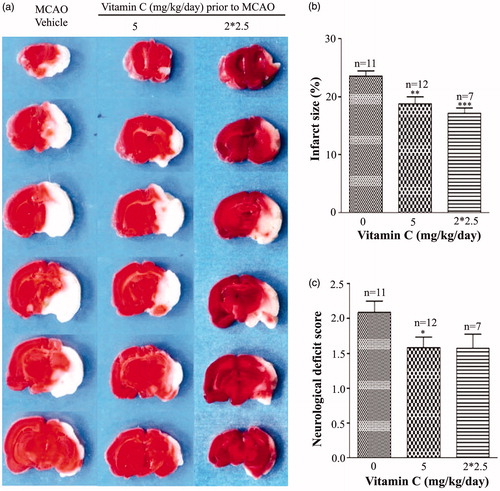
Figure 3. Effect of long-term and low dose pretreatment with vitamin C on the concentration of vitamin C in the serum and brain. Concentration of vitamin C in the serum (a) or right and left brain (b) for the rats treated with vehicle or treated with vitamin C at a dose of 5 mg.kg−1 once a day or 2.5 mg.kg−1 twice a day for three weeks. The concentration of vitamin C was measured 24 hr after the last administration of vitamin C. **p < 0.01, ***p < 0.005. (c–e) Concentration of vitamin C in the serum (c) and contralateral side (d) or ipsilateral side (e) of the brain for sham-operated, vehicle pretreated MCAO rats and vitamin C pretreated MCAO rats. Vitamin C was intraperitoneally administrated at a dose of 5 mg.kg−1 once a day or 2.5 mg.kg−1 twice a day for three weeks prior to MCAO. The concentration of vitamin C was measured 48 hr after the last administration of vitamin C.
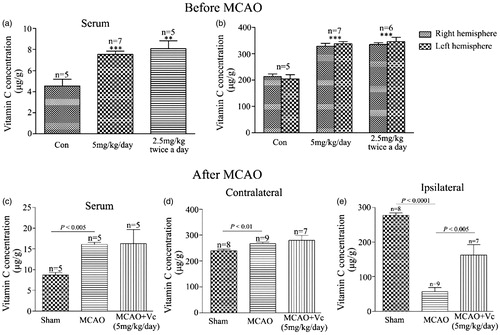
Figure 4. A single high dose vitamin C has no effect on MCAO. (a) Representative images of brain slices obtained at 24 hr post-MCAO from rats treated with vehicle or treated with a single high dose vitamin C (500 mg.kg−1) 24 hr before MCAO. (b,c) Bar graphs showing the relative infract size (b) and neurological deficit score (c) for a single high dose vitamin C-treated (500 mg.kg−1) vs vehicle-treated MCAO rats. (d,e) Vitamin C concentration in the serum (d) or brain tissue (e) of rats after 24 hr treament with vehicle or a single high dose vitamin C (500 mg.kg−1).
Vitamin C activates BKCa channels
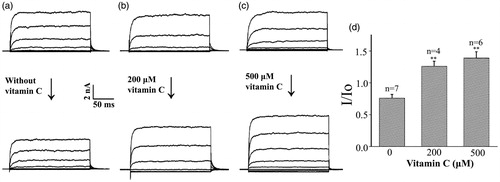
Many researches (Chi et al., Citation2010; Gribkoff et al., Citation2001; Hewawasam et al., Citation2003; Li et al., Citation2014; Su et al., Citation2017) have shown that BKCa channel is one of the molecular targets of the ischemic stroke. Therefore, we tested whether vitamin C could activate the BKCa channel. First, we investigated the effect of vitamin C on the whole-cell currents of the BKCa channels expressed in CHO cells. Under control condition, for which the procedure was the same except that no vitamin C was added, the whole cell BKCa current was decreased after 10 min, probably due to the rundown of the current (). Addition of 200 and 500 μM vitamin C into the bath solution clearly increased the BKCa channel current (). Statistical summary showed that the ratio of the whole cell BKCa channel current at a voltage of + 60 mV before and after 10 min of vitamin C treatment was 0.76 ± 0.06 in control condition. In contrast, the ratio values increased to 1.26 ± 0.08 and 1.39 ± 0.10 after 10 min addition of 200 and 500 μM vitamin C into the extracellular side of the cells ().
Figure 5. Vitamin C activates BKCa channels at the whole-cell level. (a–c) Representative whole-cell BKCa channel currents before (upper panel) and after 10 min (lower panel) application of 0 (a), 200 (b) and 500 (c) μM vitamin C in the bath solution at Ca2+ concentration of 1 μM. The currents were evoked by step depolarization from a hold potential of -80 mV to test potentials between −80 and +60 mV for 200 ms in 20 mV increments. (d) Ratio values (I/Io) of the whole-cell BKCa channel current at + 60 mV before (Io) and after 10 min (I) application of different concentration of vitamin C in the bath solution. **p < 0.01.
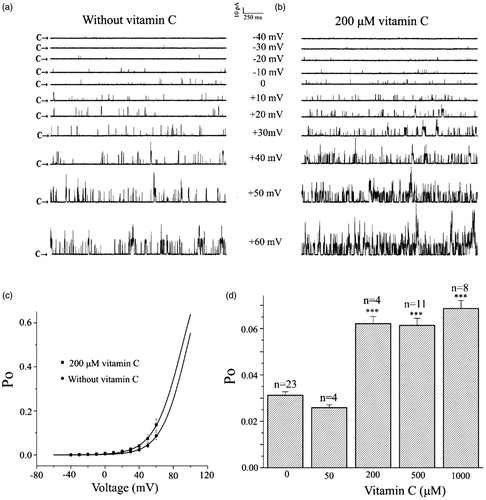
The above result indicated that vitamin C could increase the whole cell current of the BKCa channel. However, it is not known whether cytosolic factors are involved in the channel activation, whether the increased current is due to an increase in the single channel open probability (Po) or the amplitude of the single channel. To address these questions, we turned to the inside–out patch-clamp configuration. showed the single channel current traces at membrane potential ranging from −40 to + 60 mV without vitamin C, indicating that the Po of channels increased with the depolarization of the membrane potential. In contrast, application of 200 μM vitamin C further increased the Po of the channel at every corresponding membrane potential without changing the single channel amplitude (), suggesting that the increase in the whole cell current by vitamin C was due to increase in Po of the single channel but not its single channel amplitude. Next, we studied the effect of vitamin C on the voltage dependent activation of the BKCa channel. The Po of the BKCa channels as a function of the membrane potential with or without vitamin C was fitted by the Boltzmann equation: Po = 1/{1 + e[(V1/2 – V)/k]}, where V1/2 is the voltage required for half-maximal activity of the channel and k is the slope factor of the curve. The best fit to the data was obtained with values of 96.7 mV for V1/2 and 15.4 mV for k in the absence of vitamin C and with values of 90.7 mV for V1/2 and 16.4 mV for k in the presence of 200 μM vitamin C (). Therefore, vitamin C shifted the voltage activation curve toward less positive membrane potentials without affecting the slope factor of the curve, which suggests that vitamin C does not alter the voltage-sensitivity of the channel. showed the statistical data on the Po of the channel in the control condition and in the presence of vitamin C. In the control condition, Po was 0.031 ± 0.002, it was slightly decreased to 0.026 ± 0.004 in the presence of 50 μM vitamin C. In contrast, the Po was significantly increased to 0.062 ± 0.006, 0.061 ± 0.008 and 0.068 ± 007 when vitamin C concentration was increased to 200, 500 and 1000 μM at the intracellular side of the patch, respectively. Because inside-out patches are detached from the cell, the result indicated that cytosolic factors were not involved in the activation of the BKCa channel by vitamin C.
Figure 6. Vitamin C activates BKCa channels at the single-channel level. (a,b) Representative single channel current traces of the BKCa channels in the inside-out configuration at membrane potentials ranging from −80 mV to +60 mV with 20 mV increment before (a) and after application of 200 μM vitamin C (b) in the bath solution. Arrows indicate the level that all the channels are in their closed state. (c) The relationships between the single channel open probability (Po) of the BKCa channels and the membrane potentials in the absence and presence of 200 μM vitamin C are fitted with the Boltzmann equation (see text for detail). (d) Statistical data on Po of the channel at a voltage of +20 mV in the absence and presence of 50, 200, 500 and 1000 μM vitamin C. ***p < 0.001.
Blockade of BKCa channels abolishes the neuroprotective effect of vitamin C on MCAO
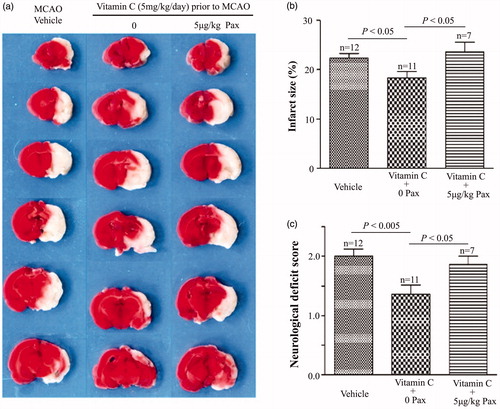
If the BKCa channel is the molecular target of vitamin C, then its specific blockers should suppress the neuroprotective effect of vitamin C on MCAO. Representative images in showed that penitrem A (PA), a lipid soluble specific BKCa channel blocker, could suppress the effect of vitamin C on the infarct size of the MCAO rats. showed that the percentage of the infarct size was 23.60 ± 0.92% in the vehicle-treated MCAO rats. Application of vitamin C (5 mg.kg−1.day−1 for three weeks) before MCAO decreased the percentage of the infarct size to 18.26 ± 1.01%. In contrast, application of vitamin C could not decrease the percentage of the infarct size when PA was given immediately at post-MCAO: the percentage of the infarct size was 21.5 ± 1.24% and 22.84 ± 2.18% when PA at a dose of 0.4 and 4 μg.kg−1 was applied immediately after MCAO (). Statistical data showed that application of PA also abolished the effect of vitamin C on the neurological deficit score (). The neurological deficit score was 2.09 ± 0.16 for vehicle-treated MCAO rats. Application of vitamin C decreased this value to 1.58 ± 0.15, while 0.4 and 4 μg.kg−1 PA-treatment increased this value to 1.93 ± 0.19 and 2.14 ± 0.14. To further confirm whether BKCa channel is the molecular target, we investigated the effect of another blocker of BKCa channel, paxilline (Pax), on the MCAO rats (). The percentage of the infarct size was 22.33 ± 0.98% in the vehicle-treated MCAO rats. Vitamin C treatment decreased the value to 18.39 ± 1.23%. In contrast, the value was 23.58 ± 1.99% when Pax was applied immediately after MCAO in the vitamin C pretreated group (). In addition, the neurological deficit score was 2.00 ± 0.12 in the vehicle-treated group, 1.36 ± 0.15 in the vitamin C-treated group, and 1.86 ± 0.14 in the vitamin C-treated group with i.p injection of Pax (). These results showed that vitamin C could protect MCAO-induced brain damage via activating the BKCa channels, suggesting that the BKCa channel is the molecular target of vitamin C.
Figure 7. BKCa channel blocker Penitrem A (PA) suppresses the effect of vitamin C on MCAO rats. (a) Representative images of brain slices for vehicle-treated MCAO rats and vitamin C pretreated (5 mg.kg−1.day−1 for three weeks prior to MCAO) MCAO rats at 24 hr post-MCAO in the absence and presence of PA (0.4 and 4 μg.kg−1). (b,c) Bar graphs showing relative infarct size (b) and neurological deficit score (c) in vehicle-treated and vitamin C pretreated MCAO rats at 24 hr post-MCAO in the absence and presence of PA.
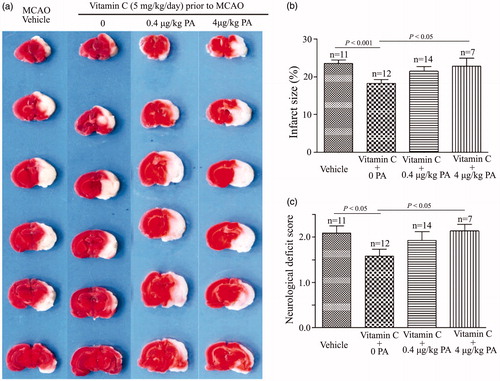
Figure 8. BKCa channel blocker paxiiline (Pax) suppresses the effect of vitamin C on MCAO rats. (a) Representative images of brain slices for vehicle-treated MCAO rats and vitamin C pretreated (5 mg.kg−1.day−1 for three weeks prior to MCAO) MCAO rats at 24 hr post-MCAO in the absence and presence of Pax (5 μg.kg−1). (b,c) Bar graphs showing relative infarct size (b) and neurological deficit score (c) in vehicle-treated and vitamin C pretreated MCAO rats at 24 hr post-MCAO in the absence and presence of Pax.
Discussion
A limitation of this study is that the results were limited to an animal model and cannot be generalized to humans. Another limitation of the current study is that the neurological deficit score and infarct size were assessed only at one timepoint, the day after stroke, and this is only based on a neurological deficit score, and not on any functional measures. Despite of these limitations, in the present study, we showed that pretreatment with vitamin C at a dose of 5 mg.kg−1 per day or 2.5 mg.kg−1 twice a day for three weeks significantly reduced the infarct size induced by MCAO, while single, high dose administration of vitamin C (500 mg.kg−1) had no effect, suggesting that long-term, low-dose intake of vitamin C has beneficial effect on the ischemic stroke. In consistence with this result, it has been reported that pretreatment with vitamin C (100 mg.kg−1, p.o.) on rat with diabetic state for two weeks significantly suppressed the exacerbation of cerebral ischemic injury (Iwata et al., Citation2014). However, administration of vitamin C (500 mg.day−1, iv) to ischemic stroke patients for 10 days beginning on day 1 after ischemic stroke did not substantially improve the clinical and functional status of patients (Lagowska-Lenard et al., Citation2010). Animal experiment also showed that there was no significant improvements in either infarct size or neurological function when vitamin C at a single dose of 500 mg.kg−1 was administered 15 min after the ischemic insult (Huang et al., Citation2001). These results suggest that vitamin C plays preventive but not therapeutic role in stroke. This is in good agreement with the conclusion that a high consumption of fruits and vegetables is associated with a reduction in the risk of stroke (Dauchet et al., Citation2005; Joshipura et al., Citation1999; Johnsen et al., Citation2003; Lakkur & Judd, Citation2015; Padayatty & Levine, Citation2008), because dietary intake represents long-term habitual exposure of vitamin C before the onset of stroke. Besides, it has been demonstrated that high level of plasma vitamin C could significantly reduce the risk of stroke (Chen et al., Citation2013a; Gale et al., Citation1995; Myint et al., Citation2008).
There is evidence that a rapid increase in the production of reactive oxygen species immediately after acute ischemic stroke rapidly overwhelm antioxidant Defense, causing further brain damage (Rodrigo et al., Citation2013). Vitamin C acts as part of the intracellular antioxidant network (Padayatty et al., Citation2003; Rice, Citation2000). Therefore, the effect of vitamin C on stroke is usually ascribed to its function as antioxidant and free radical scavenger. On the other hand, there is also growing evidence that inflammation plays a key role in the ischemic brain stroke (Anrather & Iadecola, Citation2016; McColl et al., Citation2007; Vidale et al., Citation2017). Thus, the effect of vitamin C on ischemic stroke may be due to the antiinflammatory function of vitamin C (Wannamethee et al., Citation2006). In addition, vitamin C intake has also been demonstrated to be inversely associated with blood pressure (Block et al., Citation2008; Juraschek et al., Citation2012). Hence, vitamin C may reduce stroke risk through its pressure-lowering effects. These putative mechanisms could somehow explain the protective effect of vitamin C on the ischemic brain stroke. However, it is not known whether there is a defined molecular target for the effect of vitamin C on the ischemic stroke. In this report, we show that the BKCa channel is the molecular target of vitamin C on the ischemic brain stroke, which is supported by several pieces of evidence. First, whole-cell recordings showed that vitamin C could enhance whole-cell BKCa current from extracellular side. Second, single channel recordings indicated that vitamin C could increase single channel open probability without change its amplitude of the BKCa channel. Most importantly, specific blockers of the BKCa channel abolished the effect of vitamin C on the MCAO rats.
Excess Ca2+ influx into the cells is one of the major pathophysiological mechanisms important for ischemic brain stroke. The BKCa channels are activated by either the membrane depolarization or the increase in intracellular Ca2+. They are expressed abundantly in the brain (Knaus et al., Citation1996; Sailer et al., Citation2006; Tseng-Crank et al., Citation1994). Their activation could prevent excessive Ca2+ buildup and abnormal glutamate release (Chi et al., Citation2010; Gribkoff et al., Citation2001) and are thus one of the candidates to protect against ischemic brain stroke (Tano & Gollasch, Citation2014). Indeed, it has been shown that activation of the BKCa channel could exerts neuroprotective effect on the ischemic brain damage, which could be antagonized by the BKCa channel inhibitors (Chi et al., Citation2010; Gribkoff et al., Citation2001; Hewawasam et al., Citation2003; Li et al., Citation2014; Su et al., Citation2017). In organotypic hippocampal slice cultures, BKCa channel opener could reduce cell death induced by oxygen and glucose deprivation or glutamate exposion, which could be reversed by the BKCa channel blockers (Piwońska et al., Citation2016; Runden-Pran et al., Citation2002). In addition, it has been found that MCAO produced larger infarct volume, more severe neurological deficits, and higher post-ischemic mortality in BK(-/-) mice than that of the WT littermates (Liao et al., Citation2010). These results suggest that neuronal BKCa channels are important for protection against ischemic brain damage. In the present study, we showed that vitamin C could activate the BKCa channel. It is worth to mention that the activation of the BKCa channel by vitamin C is not as potent as many known BKCa channel openers. This might be beneficial to the ischemic brain damage, because it has been shown that the over-activity of the BKCa channels could result in more severe hippocampal neuronal damage (Chen et al., Citation2013b).
Taken together, the present study showed that long-term, low-dose administration of vitamin C could reduce the MCAO induced brain damage. Therefore, long-term, low level activation of the BKCa channels might be responsible for the neuroprotective effect of vitamin C on the ischemic brain stroke, suggesting that long-term, low-dose intake of vitamin C may serve as a potential strategy to protect against ischemic brain stroke in humans.
Contributors
Z.Q. designed research; L.L., S.L, C.H., L.Z. and Y.Z. performed research; Z.Q., M.Y. and L.L. analyzed data; Z.Q. and L.L. wrote the manuscript. All authors approved the final manuscript.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Funding
References
- Anrather J, Iadecola C. 2016. Inflammation and stroke: an overview. Neurotherapeutics 13:661–670.
- Ascherio A, Rimm EB, Hernan MA, Giovannucci E, Kawachi I, Stampfer MJ, Willett WC. 1999. Relation of consumption of vitamin E, vitamin C, and carotenoids to risk for stroke among men in the United States. Ann Intern Med 130:963–970.
- Block G, Jensen CD, Norkus EP, Hudes M, Crawford PB. 2008. Vitamin C in plasma is inversely related to blood pressure and change in blood pressure during the previous year in young Black and White women. Nutr J 7:35.
- Chen GC, Lu DB, Pang Z, Liu QF. 2013a. Vitamin C intake, circulating vitamin C and risk of stroke: a meta-analysis of prospective studies. J Am Heart Assoc 2:e000329.
- Chen M, Sun HY, Hu P, Wang CF, Li BX, Li SJ. 2013b. Activation of BKca channels mediates hippocampal neuronal death after reoxygenation and reperfusion. Mol Neurobiol 48:794–807.
- Chi S, Cai W, Liu P, Zhang Z, Chen X, Gao L. 2010. Baifuzi reduces transient ischemic brain damage through an interaction with the STREX domain of BKCa channels. Cell Death Dis 1:e13.
- Dauchet L, Amouyel P, Dallongeville J. 2005. Fruit and vegetable consumption and risk of stroke: a meta-analysis of cohort studies. Neurology 65:1193–1197.
- Del Rio D, Agnoli C, Pellegrini N, Krogh V, Brighenti F, Mazzeo T, et al. 2011. Total antioxidant capacity of the diet is associated with lower risk of ischemic stroke in a large Italian cohort. J Nutr 141:118–123.
- Gale CR, Martyn CN, Winter PD, Cooper C. 1995. Vitamin C and risk of death from stroke and coronary heart disease in cohort of elderly people. BMJ 310:1563–1566.
- Gribkoff VK, Starrett JE Jr, Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, et al. 2001. Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med 7:471–477.
- Hewawasam P, Ding M, Chen N, King D, Knipe J, Pajor L, et al. 2003. Synthesis of water-soluble prodrugs of BMS-191011: a maxi-K channel opener targeted for post-stroke neuroprotection. Bioorg Med Chem Lett 13:1695–1698.
- Huang J, Agus DB, Winfree CJ, Kiss S, Mack WJ, McTaggart RA, et al. 2001. Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc Natl Acad Sci USA 98:11720–11724.
- Iwata N, Okazaki M, Xuan M, Kamiuchi S, Matsuzaki H, Hibino Y. 2014. Orally administrated ascorbic acid suppresses neuronal damage and modifies expression of SVCT2 and GLUT1 in the brain of diabetic rats with cerebral ischemia-reperfusion. Nutrients 6:1554–1577.
- Johnsen SP, Overvad K, Stripp C, Tjonneland A, Husted SE, Sorensen HT. 2003. Intake of fruit and vegetables and the risk of ischemic stroke in a cohort of Danish men and women. Am J Clin Nutr 78:57–64.
- Joshipura KJ, Ascherio A, Manson JE, Stampfer MJ, Rimm EB, Speizer FE, et al. 1999. Fruit and vegetable intake in relation to risk of ischemic stroke. JAMA 282:1233–1239.
- Juraschek SP, Guallar E, Appel LJ, Miller ER. 2012. Effects of vitamin C supplementation on blood pressure: a meta-analysis of randomized controlled trials. Am J Clin Nutr 95:1079–1088.
- Knaus HG, Schwarzer C, Koch RO, Eberhart A, Kaczorowski GJ, Glossmann H, et al. 1996. Distribution of high-conductance Ca(2+)-activated K + channels in rat brain: targeting to axons and nerve terminals. J Neurosci 16:955–963.
- Lagowska-Lenard M, Stelmasiak Z, Bartosik-Psujek H. 2010. Influence of vitamin C on markers of oxidative stress in the earliest period of ischemic stroke. Pharmacol Rep 62:751–756.
- Lakkur S, Judd SE. 2015. Diet and stroke: recent evidence supporting a Mediterranean-style diet and food in the primary prevention of stroke. Stroke 46:2007–2011.
- Li HJ, Zhang YJ, Zhou L, Han F, Wang MY, Xue MQ, Qi Z. 2014. Chlorpromazine confers neuroprotection against brain ischemia by activating BKCa channel. Eur J Pharmacol 735:38–43.
- Liao Y, Kristiansen AM, Oksvold CP, Tuvnes FA, Gu N, Runden-Pran E, et al. 2010. Neuronal Ca2+-activated K + channels limit brain infarction and promote survival. PLoS One 5:e15601.
- Longa EZ, Weinstein PR, Carlson S, Cummins R. 1989. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91.
- McColl BW, Allan SM, Rothwell NJ. 2007. Systemic inflammation and stroke: aetiology, pathology and targets for therapy. Biochem Soc Trans 35:1163–1165.
- Myint PK, Luben RN, Welch AA, Bingham SA, Wareham NJ, Khaw KT. 2008. Plasma vitamin C concentrations predict risk of incident stroke over 10 y in 20 649 participants of the European Prospective Investigation into Cancer Norfolk prospective population study. Am J Clin Nutr 87:64–69.
- Myung SK, Ju W, Cho B, Oh SW, Park SM, Koo BK, Park BJ. 2013. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ 346:f10.
- Padayatty SJ, Katz A, Wang Y, Eck P, Kwon O, Lee JH, et al. 2003. Vitamin C as an antioxidant: evaluation of its role in disease prevention. J Am Coll Nutr 22:18–35.
- Padayatty SJ, Levine M. 2008. Fruit and vegetables: think variety, go ahead, eat! Am J Clin Nutr 87:5–7.
- Piwońska M, Szewczyk A, Schröder UH, Reymann KG, Bednarczyk I. 2016. Effectors of large-conductance calcium-activated potassium channel modulate glutamate excitotoxicity in organotypic hippocampal slice cultures. Acta Neurobiol Exp (Wars) 76:20–31.
- Rice ME. 2000. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci 23:209–216.
- Rodrigo R, Fernandez-Gajardo R, Gutierrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W. 2013. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets 12:698–714.
- Runden-Pran E, Haug FM, Storm JF, Ottersen OP. 2002. BK channel activity determines the extent of cell degeneration after oxygen and glucose deprivation: a study in organotypical hippocampal slice cultures. Neuroscience 112:277–288.
- Sailer CA, Kaufmann WA, Kogler M, Chen L, Sausbier U, Ottersen OP, et al. 2006. Immunolocalization of BK channels in hippocampal pyramidal neurons. Eur J Neurosci 24:442–454.
- Su F, Guo AC, Li WW, Zhao YL, Qu ZY, Wang YJ, et al. 2017. Low-dose ethanol preconditioning protects against oxygen-glucose deprivation/reoxygenation-induced neuronal injury by activating large conductance, Ca2+-activated K + channels in vitro. Neurosci Bull 33:28–40.
- Tano JY, Gollasch M. 2014. Hypoxia and ischemia-reperfusion: a BiK contribution? Am J Physiol Heart Circ Physiol 307:H811–H817.
- Tseng-Crank J, Foster CD, Krause JD, Mertz R, Godinot N, DiChiara TJ, et al. 1994. Cloning, expression, and distribution of functionally distinct Ca(2+)-activated K + channel isoforms from human brain. Neuron 13:1315–1330.
- Vidale S, Consoli A, Arnaboldi M, Consoli D. 2017. Postischemic inflammation in acute stroke. J Clin Neurol 13:1–9.
- Wannamethee SG, Lowe GD, Rumley A, Bruckdorfer KR, Whincup PH. 2006. Associations of vitamin C status, fruit and vegetable intakes, and markers of inflammation and hemostasis. Am J Clin Nutr 83:567–574; quiz 726–567.