Abstract
Prions are the causative agent of the transmissible spongiform encephalopathies, such as Creutzfeldt-Jakob disease in humans. In these prion diseases the normal cellular form of the prion protein (PrPC) undergoes a post-translational conformational conversion to the infectious form (PrPSc). PrPC associates with cholesterol- and glycosphingolipid-rich lipid rafts through association of its glycosyl-phosphatidylinositol (GPI) anchor with saturated raft lipids and through interaction of its N-terminal region with an as yet unidentified raft associated molecule. PrPC resides in detergent-resistant domains that have different lipid and protein compositions to the domains occupied by another GPI-anchored protein, Thy-1. In some cells PrPC may endocytose through caveolae, but in neuronal cells, upon copper binding to the N-terminal octapeptide repeats, the protein translocates out of rafts into detergent-soluble regions of the plasma membrane prior to endocytosis through clathrin-coated pits. The current data suggest that the polybasic region at its N-terminus is required to engage PrPC with a transmembrane adaptor protein which in turn links with the clathrin endocytic machinery. PrPC associates in rafts with a variety of signalling molecules, including caveolin-1 and Fyn and Src tyrosine kinases. The clustering of PrPC triggers a range of signal transduction processes, including the recruitment of the neural cell adhesion molecule to rafts which in turn promotes neurite outgrowth. Lipid rafts appear to be involved in the conformational conversion of PrPC to PrPSc, possibly by providing a favourable environment for this process to occur and enabling disease progression.
Introduction
Prions are the causative agent of the transmissible spongiform encephalopathies (TSEs). This group of diseases includes scrapie in sheep, bovine spongiform encephalopathy in cattle and Creutzfeldt-Jakob disease (CJD) in humans. In these diseases the cellular isoform of the prion protein (PrPC) is post-translationally misfolded into the infectious scrapie isoform (PrPSc). PrPC, encoded by the Prpn gene, is a cell surface glycosyl-phosphatidylinositol (GPI)-anchored protein expressed by a variety of cell types, but being particularly abundant in neurons. The protein consists of a flexible N-terminus, whose structure remains undetermined, and a C-terminal globular domain containing predominantly α-helical secondary structure Citation[1]. According to the protein only hypothesis Citation[2], an interaction between the pathogenic PrPSc and endogenous PrPC is sufficient to cause the template-driven formation of more PrPSc.
Although PrPC is essential for the development of prion disease Citation[3], the normal physiological function(s) of PrPC remains largely unknown. However, a growing number of studies implicate PrPC in the cellular resistance to oxidative stress Citation[4], in cell signalling Citation[5], in copper and zinc metabolism Citation[6], Citation[7] and in synaptic transmission Citation[8]. PrP is associated for much of its life cycle with cholesterol- and glycosphingolipid-rich lipid rafts and it is the purpose of this review to explore the role of lipid rafts in prion protein biology, both for the normal and disease-associated forms of the protein. In model membranes containing sphingomyelin, cholesterol and an unsaturated glycerophospholipid, the saturated lipids pack together in liquid ordered domains which are characterized by being insoluble in detergents, such as Triton X-100, at 4oC. Although extraction of membranes with cold non-ionic detergents has been extensively used to isolate ‘rafts’, a growing body of evidence suggests caution in identifying detergent-resistant membranes (DRMs) with rafts in cell membranes Citation[9]. However, as extraction of membranes with detergents has been widely used to study the raft association of PrP and other proteins, for the purpose of this review we have equated DRMs with rafts in the membrane.
Targeting of PrPC to lipid rafts
GPI anchor-dependent targeting
PrPC is attached to the lipid bilayer via a GPI anchor which is added to the serine residue at position 231 (murine PrP numbering) Citation[10] (). The GPI anchor is added rapidly to the protein on translocation into the endoplasmic reticulum (ER), following the cleavage of the C-terminal GPI anchor addition signal sequence. Both cleavage of the polypeptide and addition of the preformed GPI anchor are carried out by a transamidase complex whose active site is on the lumenal side of the ER membrane. The GPI anchor consists of a core tetrasaccharide (three mannose residues and a glucosamine) linked through ethanolamine phosphate to Ser-231. The glucosamine residue is linked to the headgroup of phosphatidylinositol whose acyl chains contain the fully saturated stearic acid (C18:0) Citation[10]. On transit to the cell surface a number of modifications and additions are made to the core GPI anchor structure, including the addition of sialic acid to one of the mannose residues Citation[11]. This latter modification has only been documented in two other GPI-anchored proteins, porcine membrane dipeptidase Citation[12] and human CD59 Citation[13].
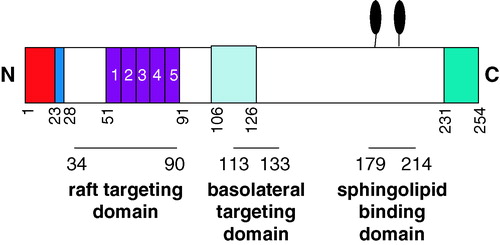
Figure 1. Schematic of PrPC. Nascent murine PrPC is a protein of 254 amino acids. On translocation into the ER the N-terminal signal peptide (chequered box; red online) and the C-terminal GPI anchor addition signal (wavy line box; green online) are removed and the latter replaced with a GPI anchor. The polybasic region (black; blue online; residues 23–28) at the N-terminus of mature PrPC, the copper-binding octapeptide repeats (stippled; purple online; residues 51-91), the central hydrophobic, neurotoxic domain (diagonal lines; light blue online; residues 106-126) and the two N-linked glycans (black lollipops; Asn-180 and Asn-196) are indicated. The positions of the raft targeting domain, the basolateral targeting domain and the sphingolipid binding domain (see text for details) are indicated below the protein. This figure is reproduced in colour in Molecular Membrane Biology online.
GPI anchors have the propensity to target proteins to lipid raft domains due to their saturated acyl chains preferentially associating with the saturated sphingolipids rather than the unsaturated glycerophospholipids Citation[14], Citation[15]. In their seminal article, Brown and Rose Citation[16] showed that GPI-anchored proteins become detergent-insoluble as they traverse the Golgi due to their association with detergent-insoluble, cholesterol-rich lipid rafts. The association of PrPC with rafts was assumed to occur in the Golgi, as several studies only detected the mature protein in DRMs Citation[17–19]. Recently, however, it was reported that the immature diglycosylated precursor of PrPC associates with cholesterol-rich rafts in the ER Citation[20]. This association was required to facilitate correct folding of the protein, as cholesterol depletion led to its misfolding. These authors suggested that association with cholesterol in the ER is required to target the immature PrPC to rafts or, perhaps by functioning as a lipochaperone within the ER, cholesterol directly affects the folding of PrPCCitation[20].
The association of the GPI anchor with raft domains in the secretory pathway has been proposed to be the mechanism by which GPI anchored proteins are sorted and targeted to the apical surface of polarized epithelial cells Citation[21], although for some GPI-anchored proteins it is the N-linked glycans, not the GPI anchor, that targets the protein to the apical surface Citation[22]. Interestingly, when transfected into polarized Fischer rat thyroid (FRT) or Madin Darby canine kidney (MDCK) cells, PrPC was targeted to the basolateral membrane Citation[23], Citation[24]. The basolateral targeting of PrPC was maintained after the cells were subjected to cholesterol depletion, indicating that the raft association of PrPC is not required for its exocytic transport Citation[23]. Deletion mutagenesis studies have revealed that the internal hydrophobic domain of PrPC (residues 113–133; ) confers basolateral sorting in a dominant manner Citation[24]. These data imply that the association of PrPC with rafts in the secretory pathway plays additional roles than simply membrane targeting.
Non GPI anchor-dependent targeting
Several studies have provided evidence that PrP can associate with rafts by means other than through its GPI anchor. Our group has shown that the raft association of PrPC is not solely dependent upon its GPI anchor Citation[25]. Using alternatively anchored forms of PrP expressed in human neuronal SH-SY5Y cells it was demonstrated that amino acid residues 23–90 of the flexible N-terminal domain () are necessary for this non-GPI-dependent raft association. Indeed, fusion of these residues to a non-raft resident protein was sufficient to redirect the protein into DRMs. These data are supported by the observation that the binding of GPI-deficient PrP to sphingolipid-cholesterol-rich raft-like liposomes (SCRLs) was significantly decreased after deletion of residues 34–94 Citation[26]. As all of the protein interacting partners of PrPC identified to date, such as the neural cell adhesion molecule (NCAM), stress inducible protein-1 and the 37 kDa/67 kDa laminin receptor, interact with PrPC C-terminal to residue 90 (for a review see Citation[27]), the identity of the lipid raft interacting partner for PrPC, be it protein or lipid, awaits determination.
Another lipid raft targeting motif identified within the prion protein is a sphingolipid binding domain Citation[28] (). This was identified through structural homology to a similar domain in the human immunodeficiency (HIV)-1 surface envelope protein gp120. It is also found in the Alzheimer amyloid-β peptide. Synthetic peptides derived from the predicted sphingolipid binding domains of these three proteins interacted with monomolecular films of galactosylceramide and sphingomyelin. Interestingly, the E200K mutation in PrP associated with some types of familial CJD occurs within this binding domain, which is within the disulphide-linked loop (Cys179–Cys214), and apparently interfered with sphingomyelin binding Citation[28]. However, as full-length PrP containing the E200K mutation is still found in DRMs Citation[29], the significance of this sphingolipid binding domain in the interaction of PrPC with rafts in cells is not clear.
Distribution of PrPC in rafts
In studies comparing the membrane microdomains occupied by PrPC and another GPI-anchored protein, Thy-1, it was found that the two proteins displayed differential detergent solubility Citation[30]. The sphingolipid-rafts containing PrPC were more detergent-soluble than those containing Thy-1, indicating that PrPC is likely to be present in less tightly ordered domains than Thy-1. Indeed, immunoaffinity purification studies suggested that domains containing PrPC border the edges of the more tightly ordered Thy-1 containing domains Citation[30]. Alternative explanations are that PrPC is located at the boundary between two domains or that PrPC and Thy-1 reside in distinct domains with different compositions and properties. In a further study, the same group showed that the lipid composition of the PrPC and Thy-1 containing DRMs differed markedly Citation[31]. The PrPC DRMs contained a larger proportion of unsaturated longer chain lipids than the Thy-1 domains. A higher proportion of unsaturated lipids would be expected to decrease the liquid order of the PrPC domains.
Doppel, the paralog of PrPC, which is encoded by the Prnd gene some 16 kb downstream of the Prnp gene, is a protein of 179 amino acids that is expressed mainly in the testis and not in the brain of adult mice Citation[32]. Doppel has sequence and structural similarity to the C-terminal half of PrP, but lacks the N-terminal Cu2 + -binding octapeptide repeats. It too is N-glycosylated and GPI anchored Citation[32]. Doppel is also found in DRMs, although one study reported that at detergent conditions in which membrane rafts were intact, it did not co-immunoprecipitate with PrPC, indicating that the two proteins are not present in the same raft domains Citation[33], while another study showed that the two proteins share common membrane microdomains and internalization pathways Citation[34].
A growing body of evidence is accumulating to indicate that rafts are heterogeneous in both protein and lipid composition, in their cellular localization, and, hence, in their biological function Citation[35]. The limited data available for PrPC would be consistent with this, and thus any consideration of the association of PrPC with rafts and raft components almost certainly has to consider the heterogeneity of such structures.
Endocytosis of PrPC
Originally, it was proposed by Harris and co-workers, in experiments using chicken PrP (which has only approx. 30% sequence homology to mammalian PrPs) transfected into mouse neuroblastoma cells, that PrPC recycles between the cell surface and an endocytic compartment with a transit time of approximately 60 minutes Citation[36]. The same group localized chicken PrPC to clathrin-coated pits and vesicles by electron microscopy Citation[37]. However, it was subsequently shown that endogenous PrPC in murine neuronal N2a and Chinese Hamster Ovary (CHO) cells was localized in caveolae or morphologically similar caveolae-like domains Citation[18], Citation[38], Citation[39]. Caveolae, flask-shaped invaginations of the plasma membrane that contain the coat protein caveolin-1, are involved in non-clathrin-dependent endocytosis Citation[40]. As caveolae appear to be a subset of cholesterol-rich lipid rafts, depletion of cellular cholesterol disrupts their endocytosis Citation[41]. In CHO cells that have endogenous caveolin-1, cryoimmunogold electron microscopy was used to show that at steady state PrPC was enriched in caveolae both at the plasma membrane and at the trans-Golgi network and in interconnecting chains of endocytic caveolae Citation[42]. Furthermore, the cholesterol-binding agent filipin prevented the Cu2 + -induced endocytosis of PrPC in both both neuronal and non-neuronal cells, leading to the conclusion that endocytosis is occurring through a caveolae-dependent mechanism Citation[43]. However, this conclusion is confused by the fact that filipin is known also to induce the shedding of PrPCCitation[43], Citation[44]. In addition, as most neuronal cells do not express caveolin-1 Citation[45], Citation[46] and lack morphologically distinguishable caveolae Citation[37], the role of caveolae in the endocytosis of PrPC in the brain has to be questioned.
Clathrin-coated pits are the most well studied endocytic mechanism and allow the endocytosis of transmembrane proteins that are recruited to the pits by interaction of targeting motifs in their cytoplasmic tails with various accessory proteins, including the adaptor protein AP-2, on the cytoplasmic face of the membrane Citation[47]. As the GPI-anchored PrPC does not contain transmembrane or cytoplasmic domains, it would have to associate with a transmembrane adaptor in order to engage the clathrin endocytic machinery Citation[48]. There is a precedent for this; the GPI-anchored urokinase-type plasminogen activator (uPA)-receptor when bound to uPA is internalized via interaction with the transmembrane low-density lipoprotein receptor-related protein-1 Citation[49]. However, the highly ordered nature of the saturated lipids in rafts will not permit the tight curvature of the membrane required for the formation of clathrin-coated pits Citation[50], Citation[51]. Therefore, for a GPI-anchored protein to be internalized through clathrin-coated pits it probably has to first translocate out of the rafts into non-raft regions of the membrane.
Sunyach et al. Citation[50] were the first to report this to occur during the endocytosis of endogenous PrPC in both primary sensory neurons and N2a cells. This was based on 50% of the PrPC in N2a cells being detergent soluble (where 99% of another GPI-anchored protein remained detergent insoluble) and the statistically significant co-localisation of PrPC with the transferrin receptor and the low density lipoprotein receptor (as shown by light and electron microscopy), two prototypical receptors endocytosed through clathrin-coated pits. A green fluoresecent protein (GFP)-tagged form of PrP was shown to internalize via a dynamin-dependent endocytic pathway, with the protein being targeted to the recycling endosomal compartment via Rab5-positive early endosomes Citation[52]. The steady state distribution of GFP-GPI between the plasma membrane and early endosomes was not affected by a Rab5 mutant, implying that the endocytosis of GFP-PrP is different from other GPI-anchored proteins and is not determined predominantly by the GPI anchor Citation[52].
PrPC contains four complete octapeptide repeats (PHGG(G/S)WGQ) in its N-terminal half that are capable of binding Cu2 + ions Citation[53], Citation[54] (). Cu2 + ions, at levels similar to those occurring naturally in the extracellular spaces of the brain, can stimulate the endocytosis of PrPC in neuronal cells Citation[6], Citation[55]. Recently, we determined the mechanism involved in the Cu2 + -stimulated endocytosis of PrPC in neuronal cells Citation[56] (). Under basal conditions PrPC was associated with DRMs, however, upon exposure of cells to Cu2 + a proportion of PrPC moved into detergent-soluble regions of the plasma membrane. The subsequent internalization of PrPC could be specifically blocked by selectively inhibiting clathrin-mediated endocytosis. This was achieved using a tyrosine analogue, tyrphostin A23, that specifically disrupts interactions between transmembrane protein cytoplasmic tail coated-pit targeting motifs and AP-2 and with a dominant negative mutant of the clathrin assembly protein AP180 Citation[56]. The deletion of large parts of the N-terminal region of PrP has revealed the critical importance of this region to its endocytosis Citation[57–59]. More specifically, point mutations within the polybasic region (residues 23–28; ) disrupted the constitutive endocytosis of PrPCCitation[50]. Using mutants of PrPC that either lacked the N-terminal polybasic region or the octapeptide repeats, we were able to show that copper binding, perhaps by driving a conformational change in the protein Citation[60], is required to dissociate PrPC from lipid rafts, while the polybasic region is required to mediate the endocytosis of PrP, possibly through interaction with a transmembrane adaptor protein Citation[56] (). Although we and others have shown that PrPC in neuronal cells can translocate out of rafts and be internalized via the clathrin-endocytic machinery, the involvement of other raft-based endocytic mechanisms in the internalization of PrPC in other cells cannot be ruled out.
Figure 2. PrPC is localized in rafts but translocates out of them before being endocytosed through clathrin-coated pits. PrPC is attached to the exoplasmic leaflet of the plasma membrane via its GPI anchor and localises within detergent-insoluble rafts through interactions between its N-terminal region (residues 23-90) and a raft-resident protein (grey; orange online) or lipid. Upon Cu2 + binding to the octapeptide repeats the protein undergoes a conformational change that dissociates it from the raft-resident partner and PrPC then moves laterally out of the rafts into detergent-soluble regions of the plasma membrane. The polybasic N-terminal region (blue online) then interacts with the ectodomain of a transmembrane protein (grey; turquoise online) that engages, via its cytoplasmic domain, with the adaptor protein AP-2 and the endocytic machinery of clathrin-coated pits. Modified from Citation[56]. This figure is reproduced in colour in Molecular Membrane Biology online.
![Figure 2. PrPC is localized in rafts but translocates out of them before being endocytosed through clathrin-coated pits. PrPC is attached to the exoplasmic leaflet of the plasma membrane via its GPI anchor and localises within detergent-insoluble rafts through interactions between its N-terminal region (residues 23-90) and a raft-resident protein (grey; orange online) or lipid. Upon Cu2 + binding to the octapeptide repeats the protein undergoes a conformational change that dissociates it from the raft-resident partner and PrPC then moves laterally out of the rafts into detergent-soluble regions of the plasma membrane. The polybasic N-terminal region (blue online) then interacts with the ectodomain of a transmembrane protein (grey; turquoise online) that engages, via its cytoplasmic domain, with the adaptor protein AP-2 and the endocytic machinery of clathrin-coated pits. Modified from Citation[56]. This figure is reproduced in colour in Molecular Membrane Biology online.](/cms/asset/41059f3f-43a3-424c-9aa9-9f68630d5585/imbc_a_144982_f0002_b.jpg)
PrPC, lipid rafts and signal transduction
In many cells, including neurons, lipid rafts provide a platform for signal transduction processes Citation[61]. Many-cell surface receptors and cytoplasmic signalling molecules, such as Src family kinases and trimeric and small GTPases, are concentrated in DRMs, and caveolin-1, acting as a scaffold protein, is essential in the regulation and formation of signalling complexes in rafts. For some years it has been known that cross-linking with antibodies of many diverse GPI-anchored proteins results in signal transduction that is characterized by: (i) transient elevation of cytoplasmic Ca2 + concentrations; (ii) tyrosine phosphorylation of cellular substrates; and (iii) triggering of T cell proliferation and differentiation Citation[62]. Therefore, in some ways, it was not too surprising when it was reported that antibody-mediated cross-linking of PrPC on the surface of differentiated murine 1C11 neuronal cells triggered a signal transduction cascade involving the caveolin-1-dependent coupling of PrPC to the intracellular tyrosine kinase Fyn Citation[5]. More recently in the same differentiated 1C11 cells, it has been reported that PrPC via caveolin-1 modulates serotonin receptor coupling to G-proteins, thereby acting as a protagonist contributing to the homeostasis of serotoninergic neurons Citation[63]. Further evidence for a role for PrPC in signal transduction came from the observation that PrPC interacts with the neuronal phosphoprotein synapsin Ib, the adaptor protein Grb2 and the prion interactor Pint1 Citation[64].
PrPC has been shown also to be a component of the multimolecular signalling complex involved in T cell activation. PrPC associates with gangliosides in DRMs from neural and lymphocytic cells Citation[65], and in T cells co-immunoprecipitates with Fyn and, after T cell activation, with the phosphorylation protein ZAP-70 Citation[66]. Antibody cross-linking of PrPC on T cells resulted in its coclustering with the caveolin-like raft proteins flotillins-1 and -2 (reggie-2 and -1) in polarized caps Citation[67]. Several signalling molecules, including Thy-1, CD3/TCR and LAT, were also directed to the cap and the cross-linking of PrPC provoked mitogen-activated protein (MAP) kinase activation and a brief elevation of the intracellular Ca2 + concentration Citation[67].
The activation of intracellular signals implies the existence of an extracellular ligand(s) capable of triggering activation of PrPC. One candidate as such a ligand is the stress-inducible protein 1 that has been shown to bind to cell surface PrPC and induce neuroprotective signals via a cAMP/protein kinase A-dependent pathway that rescues cells from apoptosis Citation[68], Citation[69]. Another candidate is the neural cell adhesion molecule (NCAM) which has been reported recently to interact directly both in cis (on the same cell membrane) and in trans (on neighbouring membranes) with PrPC at the neuronal cell surface Citation[70]. PrPC was shown to promote the recruitment of transmembrane isoforms of NCAM to rafts where the latter activates Fyn kinase and enhances neurite outgrowth Citation[70].
However, questions remain as to how the GPI-anchored PrPC, present on the extracellular face of the plasma membrane can directly interact with signalling proteins on the cytosolic face. One suggestion is that transmembrane forms of PrP could link directly to such cytosolic proteins Citation[64]. Another possibility is that PrPC links to such cytoplasmic signalling proteins via one or more transmembrane adaptors. Interestingly, antibodies against clathrin reduced, but did not ablate, the level of Fyn activation upon PrPC cross-linking, implicating clathrin in mediating, in part, the signal transduction from PrPCCitation[5]. Whether this is due to a proportion of PrPC engaging with a transmembrane adaptor protein within clathrin-coated pits (see above) remains to be determined. Clustering of PrPC with antibodies on the surface of GT1-7 neurohypothalamic cells resulted in a rapid and transient phosphorylation of the MAP kinases extracellular receptor kinases 1 and 2 (ERK1/2) and the microtubule-destabilizing protein stathmin Citation[71], the latter protein being phosphorylated on Ser-16. Phosphorylation of stathmin on Ser-16 is induced upon activation of the transmembrane tyrosine kinase epidermal growth factor receptor (EGFR) and these authors were able to show that a specific EGFR inhibitor blocked both signalling pathways, indicating a recruitment and transactivation of EGFR upon antibody-mediated PrPC clustering Citation[71]. This suggests that EGFR could be a functional transmembrane partner of PrPC. Interestingly, prior to ligand binding to EGFR in quiescent fibroblasts, a significant portion (65%) of the receptor is in the low density plasma membrane fractions that contain caveolae and non-caveolae rafts Citation[72]. After ligand binding, activated receptors rapidly move from this membrane fraction to non-raft regions of the plasma membrane where they are internalized by clathrin-coated pits Citation[73]. Other candidates as signal transduction transmembrane adaptors linking PrPC to the cytosolic signalling proteins are the transmembrane isoforms of NCAM Citation[70] (see above).
Although binding of stress-inducible protein 1 to PrPC induced neuroprotective signals Citation[69] and PrPC activates phosphatidylinositol 3-kinase that plays a pivotal role in cell survival Citation[74], another study reported that cross-linking of PrPCin vivo with specific monoclonal antibodies triggered rapid and extensive apoptosis in hippocampal and cerebellar neurons Citation[75]. From these and other studies it is not clear whether different ligands, possibly in different cell types, could promote alternative signalling pathways, or whether the localization of PrPC in different rafts or in raft and non-raft domains of the membrane could trigger different cellular responses. As yet no data have been presented to show whether the localization of PrPC in rafts is critical for these two opposing responses.
One mechanism that could account for these apparently contradictory results would be if PrPC resides in multiple subtypes of raft that differ in their protein (and lipid) compositions or if under basal conditions PrPC in one type of raft is segregated from particular signalling molecules which reside in another raft. Upon ligand binding or antibody cross-linking the PrPC containing rafts fuse with the rafts containing a particular subset of signalling molecules and the appropriate signal transduction cascade and cellular response is triggered. In relation to PrPC, evidence for the existence of different subtypes of rafts comes from the observation that in cerebellar granule cells DRMs containing most of PrPC, GAP-43 and protein kinase C can be separated from those DRMs containing Fyn and MARCKS Citation[76] and that PrPC and Thy-1 are present in different DRMs Citation[30] (see above). Moreover, in resting human T cells PrPC was predominantly localized to non-raft regions of the plasma membrane but upon antibody cross-linking was recruited into rafts where it activated the Src tyrosine kinase Citation[77]. This mechanism would be consistent with the emerging view that rafts may normally be relatively small, containing one or only a few protein molecules preferentially surrounded by a small group of lipids, so-called ‘lipid shells’ Citation[78]. Upon cell stimulation or antibody cross-linking these small domains fuse together to form larger platforms Citation[79]. The protein composition, and hence biological properties, of these larger, induced platforms will depend on which particular domains fuse in response to a particular stimulus.
Role of lipid rafts in the conversion of PrPC to PrPSc
Lipid rafts appear to play a critical role in the conformational conversion of PrPC to PrPScCitation[80]. Depletion of cellular cholesterol with the HMG CoA reductase inhibitor lovastatin, the polyene antibiotic filipin or the squalene synthase inhibitor squalestatin, diminished the formation of PrPScCitation[17], Citation[43], Citation[81] and removing PrPC from rafts by replacing its GPI anchor addition signal with the transmembrane and cytosolic domains from non-raft proteins, prevented the formation of PrPScCitation[17], Citation[39]. PrPSc is present in DRMs, although it did not cofractionate with PrPC on a Nycodenz density gradient suggesting that the two forms of the protein may be located in distinct DRMs of different densities and composition Citation[18], Citation[19].
The conversion of PrPC-like proteinase K-sensitive PrP (PrP-sen) to PrPSc-like proteinase K-resistant PrP (PrP-res) by exogenous PrP-res in a cell-free system has provided further insight into the role of rafts and the GPI anchor on PrPC in the conversion process. In this cell-free conversion assay, raft-bound PrP-sen resisted conversion to PrP-res unless the PrP-sen was released from rafts by phospholipase C digestion or the PrP-sen was inserted into contiguous membranes with the source PrP-res by polyethylene glycol fusion Citation[26], Citation[82]. Somewhat surprisingly, removal of the GPI anchor from the PrP-sen led to its conversion to PrP-res without phospholipase or polyethylene glycol treatment. These observations led Caughey and coworkers Citation[82] to conclude that generation of new PrPSc during TSE infection requires: (i) removal of PrPC from target cells, (ii) an exchange of membranes between cells, or (iii) insertion of incoming PrPSc into the raft domains of recipient cells. A more recent study using a modified version of the protein misfolding cyclic amplification (PMCA) cell-free conversion assay also observed that membrane attachment is not required for PrP-sen to convert efficiently into PrP-res Citation[83]. However, incubation of N2a cells with filipin, which induces the shedding of PrPC, inhibited the formation of PrP-res raising the possibility that the release of PrPC from the plasma membrane may decrease the amount of PrPC available for conversion to PrPScCitation[43].
In vitro structural studies in model raft membranes using recombinant Syrian hamster PrP (residues 90–231) showed that when refolded into ‘α-PrP’, a conformation containing predominantly α-helical secondary structure, the α-helix content increased on binding of the protein to raft-like membranes and that this form of the protein was protected from aggregation and fibrillization Citation[84]. In contrast, refolded ‘β-PrP’, which has predominantly β-sheet secondary structure, was converted into amyloid fibrils on binding to raft-like membranes Citation[85]. α-PrP was found to bind, with decreasing affinity, to palmitoyloleoylphosphatidylglycerol, dipalmitoylphosphatidylcholine and raft-like membranes, suggesting that the majority of PrP may exist preferentially outside of lipid rafts in the steady state Citation[86]. However, caution should be taken when interpreting the results of studies using recombinant bacterially-expressed truncated forms of PrP that do not contain any of the mammalian cell post-tanslational modifications, including the GPI anchor.
Evidence that PrPC and PrPSc need to be membrane-bound in order for conversion to occur came from a study in which infected Scrapie mouse brain (SMB) cells were co-cultured with uninfected target cells Citation[87]. This study showed that cell contact was required for efficient conversion of PrPC on the target cells. Both PrPC and PrPSc are released into the extracellular environment in association with exosomes, membranous vesicles that are secreted upon fusion of multivesicular endosomes with the plasma membrane Citation[88]. As these exosomes bearing PrPSc are infectious, they may represent a mechanism by which PrPSc is exchanged between membranes and enable the spread of PrPSc throughout the organism Citation[88]. As shown for other GPI-anchored proteins, PrPC can also be transferred efficiently between cells, a process that is dependent on an intact GPI anchor Citation[89].
A recent study using scrapie-infected transgenic mice expressing PrP lacking the GPI anchor, reported that abnormal PrP-res was deposited as amyloid plaques, rather then the usual nonamyloid form of PrP-res, which, although able to induce brain damage reminiscent of Alzheimer's disease, had minimal clinical manifestations Citation[90]. In addition, combined expression of anchorless and wild-type PrP produced accelerated clinical scrapie Citation[90]. These observations imply that the GPI anchor plays a critical role in the presentation and progression of prion disease, and that there is a link between the cell surface topology of PrPSc and prion disease pathogenesis. By disengaging PrPC from the cell surface, Chesebro and colleagues effectively uncoupled clinical disease from PrPSc formation Citation[91]. The available data can be accommodated in the following model (). For conversion and disease progression, the incoming PrPSc has to be inserted into a contiguous membrane with PrPC. Rafts provide a favourable environment for conformational conversion of PrPC to PrPSc, by concentrating the proteins within confined regions of the membrane, by aligning them in a way that promotes their interaction or by providing accessory molecules that are required for formation of PrPScCitation[20], Citation[80]. The conversion to PrPSc may affect signalling events involving PrPC, leading to the removal of neuroprotective signals and/or to the initiation of neurotoxic signals Citation[91] (). Soluble PrPC devoid of its membrane attachment can be converted into PrPSc, possibly more efficiently than membrane-bound PrPC, but lack of membrane-anchorage prevents disease progression as it fails to disrupt signal transduction processes.
Figure 3. A model for the role of lipid rafts in the conversion of PrPC to PrPSc and subsequent disease progression. PrPC is attached to the membrane via its GPI anchor and upon clustering in lipid rafts transduces neuroprotective signals into the cell. Infectious PrPSc inserts into the target cell membrane alongside the PrPC in the rafts. Conversion of the PrPC to PrPSc may affect signalling events involving PrPC, leading to neurotoxicity and cell death. Reproduced with permission from Citation[91]. This figure is reproduced in colour in Molecular Membrane Biology online.
![Figure 3. A model for the role of lipid rafts in the conversion of PrPC to PrPSc and subsequent disease progression. PrPC is attached to the membrane via its GPI anchor and upon clustering in lipid rafts transduces neuroprotective signals into the cell. Infectious PrPSc inserts into the target cell membrane alongside the PrPC in the rafts. Conversion of the PrPC to PrPSc may affect signalling events involving PrPC, leading to neurotoxicity and cell death. Reproduced with permission from Citation[91]. This figure is reproduced in colour in Molecular Membrane Biology online.](/cms/asset/20aff8b2-4bdf-4aad-855f-5012f8fc3fbe/imbc_a_144982_f0003_b.jpg)
Interestingly, during scrapie infection, although PrP-res accumulated in DRMs from retinas and optic nerves of mice, the PrPC interacting proteins caveolin-1 and synaptophysin were redistributed to detergent-soluble fractions Citation[92]. This alteration of the distribution of caveolin-1, synaptophysin and possibly other cytoplasmic signalling proteins upon prion replication could provide a mechanism by which signal transduction processes emanating from PrP on the surface of infected cells could be altered. Such alterations in signal transduction from PrPC may in turn contribute to disease pathogenesis. However, it remains to be seen what effect prion infection and the conversion of PrPC to PrPSc has on serotoninergic functions, neurite outgrowth and other PrPC-dependent functions.
Conclusions
It is clear that lipid rafts play a key role in both the normal and the pathological functioning of PrP. The association of PrPC with rafts is not a static event but should be considered as a dynamic process, with the protein exiting and entering rafts, and with the rafts containing PrPC fusing with other domains. Although PrPC probably interacts with rafts primarily via its GPI anchor, a region in its N-terminus is also important for regulating its raft association through interaction with an as yet unidentified raft component. Transmembrane adaptor proteins, both within rafts and in non-raft regions of the membrane, play important roles in the signal transduction and endocytosis of PrPC, respectively. Signal transduction from PrPC may be enhanced by the clustering of small raft domains containing PrPC, with other domains containing particular signalling proteins, although the precise molecular mechanisms by which clustering of PrPC triggers a variety of cellular responses requires further work. Finally, rafts are critically involved in the conformational conversion of PrPC to PrPSc, by providing a favourable environment for this process to occur and enabling disease progression. Whether conversion to PrPSc alters signal transduction processes emanating from PrPC in rafts remains to be clarified.
This paper was first published online on prEview on 27 January 2006.
DRT was in receipt of a studentship from the Medical Research Council of Great Britain. Work on PrP in our laboratory is supported by grants from the Medical Research Council.
References
- Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 2000; 97: 145–150
- Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95: 13363–13383
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73: 1339–1347
- Milhavet O, Lehmann S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Brain Res Rev 2002; 38: 328–339
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289: 1925–1928
- Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem 1998; 273: 33107–33110
- Watt NT, Hooper NM. The prion protein and neuronal zinc homeostasis. Trends Biochem Sci 2003; 28: 406–410
- Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG. Prion protein is necessary for normal synaptic function. Nature 1994; 370: 295–297
- Lichtenberg D, Goni FM, Heerklotz H. Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem Sci 2005; 30: 430–436
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987; 51: 229–240
- Stahl N, Baldwin MA, Hecker R, Pan K-M, Burlingame AL, Prusiner SB. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 1992; 31: 5043–5053
- Brewis IA, Ferguson MAJ, Mehlert A, Turner AJ, Hooper NM. Structures of the glycosyl-phosphatidylinositol anchors of porcine and human membrane dipeptidase. Interspecies comparison of the glycan core structures and further structural studies on the porcine anchor. J Biol Chem 1995; 270: 22946–22956
- Meri S, Lehto T, Sutton CW, Tyynela J, Baumann M. Structural composition and functional characterization of soluble CD59: heterogeneity of the oligosaccharide and glycophosphoinositol (GPI) anchor revealed by laser-desorption mass spectrometric analysis. Biochem J 1996; 316: 923–935
- Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol 1998; 14: 111–136
- Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem 2000; 275: 17221–17224
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992; 68: 533–544
- Taraboulos A, Scott M, Semenov A, Avraham D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 1995; 129: 121–132
- Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, Anderson RGW, Taraboulos A, Prusiner SB. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci USA 1996; 93: 14945–14949
- Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem 1997; 272: 6324–6331
- Sarnataro D, Campana V, Paladino S, Stornaiuolo M, Nitsch L, Zurzolo C. PrP(C) association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol Biol Cell 2004; 15: 4031–4042
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature 1997; 387: 569–572
- Pang S, Urquhart P, Hooper NM. N-Glycans, not the GPI anchor, mediate the apical targeting of a naturally glycosylated, GPI-anchored protein in polarised epithelial cells. J Cell Sci 2004; 117: 5079–5086
- Sarnataro D, Paladino S, Campana V, Grassi J, Nitsch L, Zurzolo C. PrPC is sorted to the basolateral membrane of epithelial cells independently of its association with rafts. Traffic 2002; 3: 810–821
- Uelhoff A, Tatzelt J, Aguzzi A, Winklhofer KF, Haass C. A Pathogenic PrP Mutation and Doppel Interfere with Polarized Sorting of the Prion Protein. J Biol Chem 2005; 280: 5137–5140
- Walmsley AR, Zeng F, Hooper NM. The N-terminal region of the prion protein ectodomain contains a lipid raft targeting determinant. J Biol Chem 2003; 278: 37241–37248
- Baron GS, Caughey B. Effect of glycosylphosphatidylinositol anchor-dependent and -independent prion protein association with model raft membranes on conversion to the protease-resistant isoform. J Biol Chem 2003; 278: 14883–14892
- Lee KS, Linden R, Prado MA, Brentani RR, Martins VR. Towards cellular receptors for prions. Rev Med Virol 2003; 13: 399–408
- Mahfoud R, Garmy N, Maresca M, Yahi N, Puigserver A, Fantini J. Identification of a common sphingolipid-binding domain in Alzheimer, prion, and HIV-1 proteins. J Biol Chem 2002; 277: 11292–11296
- Rosenmann H, Talmor G, Halimi M, Yanai A, Gabizon R, Meiner Z. Prion protein with an E200K mutation displays properties similar to those of the cellular isoform PrP(C). J Neurochem 2001; 76: 1654–1662
- Madore N, Smith KL, Graham CH, Jen A, Brady K, Hall S, Morris R. Functionally different GPI proteins are organised in different domains on the neuronal surface. EMBO J 1999; 19: 6917–6926
- Brugger B, Graham C, Leibrecht I, Mombelli E, Jen A, Wieland F, Morris R. The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J Biol Chem 2004; 279: 7530–7536
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AF, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE, Westaway D. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 1999; 292: 797–817
- Shaked Y, Hijazi N, Gabizon R. Doppel and PrP(C) do not share the same membrane microenvironment. FEBS Lett 2002; 530: 85–88
- Massimino ML, Ballarin C, Bertoli A, Casonato S, Genovesi S, Negro A, Sorgato MC. Human Doppel and prion protein share common membrane microdomains and internalization pathways. Int J Biochem Cell Biol 2004; 36: 2016–2031
- Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochem J 2004; 378: 281–292
- Shyng S-L, Huber MT, Harris DA. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J Biol Chem 1993; 268: 15922–15928
- Shyng SL, Heuser JE, Harris DA. A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J Cell Biol 1994; 125: 1239–1250
- Harmey JH, Doyle D, Brown V, Rogers MS. The cellular isoform of the prion protein, PrPc, is associated with caveolae in mouse neuroblastoma (N2a) cells. Biochem Biophys Res Commun 1995; 210: 753–759
- Kaneko K, Vey M, Scott M, Pilkuhn S, Cohen FE, Prusiner SB. COOH-terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scapie isoform. Proc Natl Acad Sci USA 1997; 94: 2333–2338
- Anderson RGW. The caveolae membrane system. Annu Rev Biochem 1998; 67: 199–225
- Parpal S, Karlsson M, Thorn H, Stralfors P. Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J Biol Chem 2001; 276: 9670–9678
- Peters PJ, Mironov A, Jr, Peretz D, van Donselaar E, Leclerc E, Erpel S, DeArmond SJ, Burton DR, Williamson RA, Vey M, Prusiner SB. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J Cell Biol 2003; 162: 703–717
- Marella M, Lehmann S, Grassi J, Chabry J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J Biol Chem 2002; 277: 25457–25464
- Parkin ET, Watt NT, Turner AJ, Hooper NM. Dual mechanisms for shedding of the cellular prion protein. J Biol Chem 2004; 279: 11170–11178
- Gorodinsky A, Harris DA. Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J Cell Biol 1995; 129: 619–627
- Parkin ET, Hussain I, Turner AJ, Hooper NM. The amyloid precursor protein is not enriched in caveolae-like, detergent-insoluble membrane microdomains. J Neurochem 1997; 69: 2179–2188
- Kirchhausen T. Clathrin. Annu Rev Biochem 2000; 69: 699–727
- Harris DA. Cellular biology of prion diseases. Clin Microbiol Rev 1999; 12: 429–444
- Conese M, Nykjaer A, Petersen CM, Cremona O, Pardi R, Andreasen PA, Gliemann J, Christensen EI, Blasi F. Alpha-2 Macroglobulin receptor/Ldl receptor-related protein(Lrp)-dependent internalization of the urokinase receptor. J Cell Biol 1995; 131: 1609–1622
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J 2003; 22: 3591–3601
- Nichols B. Caveosomes and endocytosis of lipid rafts. J Cell Sci 2003; 116: 4707–4714
- Magalhaes AC, Silva JA, Lee KS, Martins VR, Prado VF, Ferguson SS, Gomez MV, Brentani RR, Prado MA. Endocytic intermediates involved with the intracellular trafficking of a fluorescent cellular prion protein. J Biol Chem 2002; 277: 33311–33318
- Stockel J, Safar J, Wallacen AC, Cohen FE, Prusiner SB. Prion protein selectively binds copper(II) ions. Biochemistry 1998; 37: 7185–7193
- Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, Dyson HJ. Copper binding to the prion protein: structural implications of four identical cooperative binding sites. Proc Natl Acad Sci USA 1999; 96: 2042–2047
- Perera WSS, Hooper NM. Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr Biol 2001; 11: 519–523
- Taylor, DR, Watt, NT, Perera, WSS, Hooper, NM. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J Cell Sci 2005;118:5141–5153.
- Shyng S-L, Moulder KL, Lesko A, Harris DA. The N-terminal domain of a glycolipid-anchored prion protein is essential for its endocytosis via clathrin-coated pits. J Biol Chem 1995; 270: 14793–14800
- Kiachopoulos S, Heske J, Tatzelt J, Winklhofer KF. Misfolding of the prion protein at the plasma membrane induces endocytosis, intracellular retention and degradation. Traffic 2004; 5: 426–436
- Nunziante M, Gilch S, Schatzl HM. Essential role of the prion protein N terminus in subcellular trafficking and half-life of cellular prion protein. J Biol Chem 2003; 278: 3726–3734
- Zahn R. The octapeptide repeats in mammalian prion protein constitute a pH-dependent folding and aggregation site. J Mol Biol 2003; 334: 477–488
- Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM, Jr. Lipid rafts in neuronal signaling and function. Trends Neurosci 2002; 25: 412–417
- Horejsi V, Drbal K, Cebecauer M, Cerny J, Brdicka T, Angelisova P, Stockinger H. GPI-microdomains: a role in signalling via immunoreceptors. Immunology Today 1999; 20: 356–361
- Mouillet-Richard S, Pietri M, Schneider B, Vidal C, Mutel V, Launay J-M, Kellermann O. Modulation of serotonergic receptor signaling and cross-talk by prion protein. J Biol Chem 2005; 280: 4592–4601
- Spielhaupter C, Schatzl HM. PrPC Directly interacts with proteins involved in signaling pathways. J Biol Chem 2001; 276: 44604–44612
- Mattei V, Garofalo T, Misasi R, Gizzi C, Mascellino MT, Dolo V, Pontieri GM, Sorice M, Pavan A. Association of cellular prion protein with gangliosides in plasma membrane microdomains of neural and lymphocytic cells. Neurochem Res 2002; 27: 743–749
- Mattei V, Garofalo T, Misasi R, Circella A, Manganelli V, Lucania G, Pavan A, Sorice M. Prion protein is a component of the multimolecular signaling complex involved in T cell activation. FEBS Lett 2004; 560: 14–18
- Stuermer CA, Langhorst MF, Wiechers MF, Legler DF, Von Hanwehr SH, Guse AH, Plattner H. PrPc capping in T cells promotes its association with the lipid raft proteins reggie-1 and reggie-2 and leads to signal transduction. FASEB J 2004; 18: 1731–1733
- Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J 2002; 21: 3307–3316
- Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J 2002; 21: 3317–3326
- Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol 2005; 169: 341–354
- Monnet C, Gavard J, Mege RM, Sobel A. Clustering of cellular prion protein induces ERK1/2 and stathmin phosphorylation in GT1-7 neuronal cells. FEBS Lett 2004; 576: 114–118
- Mineo C, James GL, Smart EJ, Anderson RGW. Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J Biol Chem 1996; 271: 11930–11935
- Mineo C, Gill GN, Anderson RG. Regulated migration of epidermal growth factor receptor from caveolae. J Biol Chem 1999; 274: 30636–30643
- Vassallo N, Herms J, Behrens C, Krebs B, Saeki K, Onodera T, Windl O, Kretzschmar HA. Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem Biophys Res Commun 2005; 332: 75–82
- Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, Williamson RA. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004; 303: 1514–1516
- Botto L, Masserini M, Cassetti A, Palestini P. Immunoseparation of prion protein-enriched domains from other detergent-resistant membrane fractions, isolated from neuronal cells. FEBS Lett 2004; 557: 143–147
- Hugel B, Martinez MC, Kunzelmann C, Blattler T, Aguzzi A, Freyssinet JM. Modulation of signal transduction through the cellular prion protein is linked to its incorporation in lipid rafts. Cell Mol Life Sci 2004; 61: 2998–3007
- Anderson RG, Jacobson K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science 2002; 296: 1821–1825
- Mayor S, Rao M. Rafts: scale-dependent, active lipid organization at the cell surface. Traffic 2004; 5: 231–240
- Campana V, Sarnataro D, Zurzolo C. The highways and byways of prion protein trafficking. Trends Cell Biol 2005; 15: 102–111
- Bate C, Salmona M, Diomede L, Williams A. Squalestatin cures prion-infected neurons and protects against prion neurotoxicity. J Biol Chem 2004; 279: 14983–14990
- Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 2002; 21: 1031–1040
- Nishina K, Deleault NR, Lucassen RW, Supattapone S. In vitro prion protein conversion in detergent-solubilized membranes. Biochemistry 2004; 43: 2613–2621
- Sanghera N, Pinheiro TJ. Binding of prion protein to lipid membranes and implications for prion conversion. J Mol Biol 2002; 315: 1241–1256
- Kazlauskaite J, Sanghera N, Sylvester I, Venien-Bryan C, Pinheiro TJ. Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry 2003; 42: 3295–3304
- Critchley P, Kazlauskaite J, Eason R, Pinheiro TJ. Binding of prion proteins to lipid membranes. Biochem Biophys Res Commun 2004; 313: 559–567
- Kanu N, Imokawa Y, Drechsel DN, Williamson RA, Birkett CR, Bostock CJ, Brockes JP. Transfer of scrapie prion infectivity by cell contact in culture. Curr Biol 2002; 12: 523–530
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G. Cells release prions in association with exosomes. Proc Natl Acad Sci USA 2004; 101: 9683–9688
- Liu T, Li R, Pan T, Liu D, Petersen RB, Wong BS, Gambetti P, Sy MS. Intercellular transfer of the cellular prion protein. J Biol Chem 2002; 277: 47671–47678
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308: 1435–1439
- Aguzzi A. Cell biology. Prion toxicity: all sail and no anchor. Science 2005; 308: 1420–1421
- Russelakis-Carneiro M, Hetz C, Maundrell K, Soto C. Prion replication alters the distribution of synaptophysin and caveolin 1 in neuronal lipid rafts. Am J Pathol 2004; 165: 1839–1848