
Abstract
Over the last decade, several theoretical models have been put forth to describe how animals respond to adverse environments and how this response changes under different physiological demands across life history stages. These models capture the context- and condition-dependent nature of stress responses. Yet, application of the models has been limited thus far in part because each model addresses different aspects of the problems facing the field of stress biology. Thus, there is a need for a unifying theoretical model that incorporates changes in physiological demand with life history stages and age, intricate relationships among physiological systems, and biphasic nature of stress responses. Here, I propose a new integrative framework, the Damage-Fitness Model. In this model, regulators, such as DNA repair mechanisms and glucocorticoids, work together as anti-damage mechanisms to minimize damage at both the cellular and organismal level. When the anti-damage regulators are insufficient or inappropriate, persistent damage accumulates. Previous studies indicate that these damage directly impact reproductive performance, disease risk, and survival. The types of regulators, the threshold at which they are initiated, and the magnitude of the responses are shaped by developmental and current environments. This model unites existing theoretical models by shifting our focus from physiological responses to downstream consequences of the stress responses, circumventing context specificity. Discussions include (1) how the proposed model relates to existing models, (2) steps to test the new model, and (3) how this model can be used to better assess the health of individuals and a population.
Lay summary
The field of stress physiology faces a challenge of characterizing dynamic cellular, physiological, and behavioral responses when animals encounter a stressor. This paper proposes a new theoretical model which links stress avoidance, damage repair and accumulation, and fitness components.
Introduction
Physiologists, conservation biologists, and epidemiologists have long sought reliable physiological indicators of health and fitness for both wild and captive animals as well as for humans. One of the most commonly used indicators of health is glucocorticoids (Bonier, Martin, Moore, & Wingfield, Citation2009; Walker, Boersma, & Wingfield, Citation2005a; Wikelski & Cooke, Citation2006). The use of glucocorticoids is based on the assumption that the hormone levels before a disturbance (baseline) or after a standardized stressor (stress induced) can predict survival, reproductive performance, or other components of fitness (Arlettaz et al., Citation2007; Breuner, Patterson, & Hahn, Citation2008; Creel et al., Citation2002; Marra & Holberton, Citation1998; Thiel, Jenni-Eiermann, Braunisch, Palme, & Jenni, Citation2007; Walker, Boersma, & Wingfield, Citation2005b). However, over the last decade, this key assumption has been questioned (Bonier, Martin, et al., Citation2009; Bonier, Moore, Martin, & Robertson, Citation2009; Breuner et al., Citation2008; Romero et al., Citation2015). Although there is support for a negative relationship between baseline glucocorticoids and reproductive success (Sorenson, Dey, Madliger, & Love, Citation2017), examination of multiple vertebrate taxa revealed that this relationship is more complex (Bonier, Martin, et al., Citation2009). It is also known to change across substages of breeding for a given species (Bonier, Moore, et al., Citation2009). These studies indicate that the relationship between glucocorticoids and fitness components depends on context, sex, and species (Bonier, Martin, et al., Citation2009; Breuner et al., Citation2008; Schoenle, Zimmer, & Vitousek, Citation2018; Sorenson et al., Citation2017). These findings caution against using glucocorticoid levels from a single time point to predict health or reproductive outcome of the animal (Bonier, Martin, et al., Citation2009; Sorenson et al., Citation2017). This is particularly true if no consideration is given to sex, food availability, life history, or residual reproductive value, which provide context and selection pressure on glucocorticoid hormone levels (Bonier, Martin, et al., Citation2009; Breuner et al., Citation2008; Schoenle et al., Citation2018).
The recognition that the way animals respond and adjust to an adverse environment changes with life-history stage, age, rearing condition, and past experience with the stressor has led to the development of several theoretical models (Korte, Koolhaas, Wingfield, & McEwen, Citation2005; McEwen & Wingfield, Citation2003a; McEwen & Wingfield, Citation2010; Romero, Dickens, & Cyr, Citation2009; Romero et al., Citation2015; Schoenle et al., Citation2018). In spite of the growing interests, discussion, and reference to these models in scientific papers and at meetings, application of these models in predicting future responses and reproductive or survival outcomes is still scarce (Romero et al., Citation2015). In addition, much of research and models have revolved around glucocorticoid levels. However, glucocorticoids are only present in vertebrate animals. If we are to advance our understanding of the evolution of stress responses and the mechanistic links among stress responses, stress resilience, and fitness, it is vital to develop an integrative model which allows quantitative evaluation and formulaic predictions about how both individuals and populations will respond to a changing environment across taxa (Romero et al., Citation2015; Schoenle et al., Citation2018). To this end, I propose the Damage-Fitness Model. This new model integrates stress responses at cellular and organismal levels, shifts our focus from physiological responses to damage which are downstream effects of stress responses, and proposes to reevaluate how we measure and interpret stress in wild and captive animals, as well as in humans. Beyond biological systems, materials also respond to a stress (or force) and even can heal in the case of self-healing materials such as polymer–matrix composites. By shifting our focus to damage, we can start to unify various models in biology, psychology, and engineering to predict the outcome of those adverse events that is applicable to all taxa. In the following sections, I will lay out components of the Damage-Fitness Model, give examples and predictions of the described relationships, and suggest avenues of future studies.
Damage-Fitness Model
Overall definition of the model
In this model, any mechanism at the cellular or organismal level that minimizes or avoids persistent damage to a cell or a tissue are broadly defined as anti-damage regulator. The Damage-Fitness Model proposes that persistent damage and dysregulation of physiological systems result when (1) anti-damage regulators are not sufficient to avoid damage, (2) there are excessive levels of anti-damage regulators, which themselves cause damage, (3) damage cannot be repaired or removed, as well as from (4) normal molecular and cellular activities (Figure 1(A)). Persistent damage accumulates and directly impacts fitness-related measures (). Therefore, the model predicts a negative relationship between the extent of persistent damage or dysregulation of physiological systems and reproductive success and survival (). The threshold for turning on the anti-damage regulators and the magnitude and efficacy of cellular action initiated by the regulators change with developmental environment (Del Giudice, Ellis, & Shirtcliff, Citation2011) and life history stage (). When there is a match between the types and levels of stressors in the current environment and threshold, magnitude, and types of the regulators initiated, persistent damage is kept at minimum and fitness is expected to be high (Schoenle et al., Citation2018) (Zone I in and ). If there is a mismatch between the current environment and animals’ anti-damage regulator phenotype, damage accumulation accelerates leading to reduced fitness (arrows in and ). With this framework in mind, a stressor is defined as a stimulus that has the potential to inflict measurable damage at a molecular, cellular, organ, or organismal level.
Figure 1. Core concepts of the Damage-Fitness Model. (A) Error processes of normal biological activities in the absence of a stressor induce damage in healthy cells, tissues, and organs. This accrues with chronological age. In addition, stressors of endogenous or exogenous origin inflict damage to cells and the body. Organisms minimize persistent damage by avoiding the source of the stressor, damage repair, and damage removal. When the stressor or induced damage exceeds the capacity of the organism to minimize damage, persistent damage and dysregulation of physiological systems occur. Accumulation of the damage reduces fitness of an individual. (B) Due to error processes of biological activities, persistent damage increases with age (gray line). With increasing intensity, duration, and frequency of a stressor, persistent damage also increases (black line). These two types of damage are predicted to be additive.
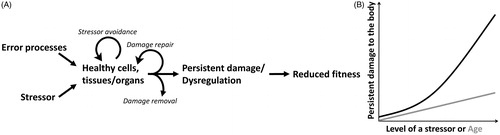
Figure 2. Relationships among anti-damage regulators, persistent damage and dysregulation of physiological systems, and fitness components/correlates. This figure may be considered a physiological explanation for an environmental change-fitness reaction norm presented in Monaghan (Citation2008). Anti-damage regulators are intricately connected to each other. Some of the connections are direct in that one physiological system directly influences another. Other connections are indirect through improving or reducing efficiency of another physiological system. Here, the networks of interconnected relationship between molecular/cellular and organismal anti-damage regulators are simplified as green vertical arrows between molecular/cellular and organismal anti-damage regulators (based on Physiological Regulatory Networks (Cohen, Martin, et al., Citation2012)). Anti-damage regulators are plastic and shaped by developmental environment and local and species adaptation through evolutoinary processes, while the strength of the stressor is in part determined by perception of the stressor. The purple arrow connecting organismal anti-damage regulators and fitness components depicts the relationship decribed in the Allostasis and Reactive Scope Models.
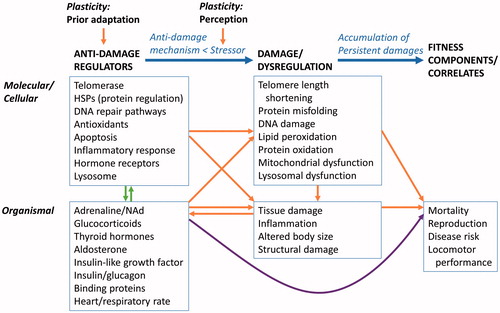
Figure 3. Predicted relationships between persistent damage and fitness-related measures (A) and the capacity of the anti-damage regulators and persistent damage (B). Persistent damage in these figures is a combination of damage caused by stressors as well as damage resulting from error processes of normal biological activities described in . (A) Vertical lines represent thresholds between low, moderate, and high cumulative damage with increasing impact of damage onto fitness-related measures. These thresholds depict stress tolerance. (B) Zone I refers to when a stressor level is lower than the capacity of the anti-damage regulators to evade damage. Zone II refers to when a level of the stressors exceeds the capacity of the regulators where persistent damage starts to accumulate. When a level of the stressors significantly exceeds the capacity of the regulators, persistent damage accumulates at a faster rate (Zone III). Past exposure to the stressor and appropriate regulators for the stressors present in the current environment likely expands the scope of stressor avoidance and damage repair, shifting the thresholds between zones toward the right. However, if the regulator phenotype does not match the occurring stressor, then the thresholds shift toward the left and faster accumulation of damage occurs.
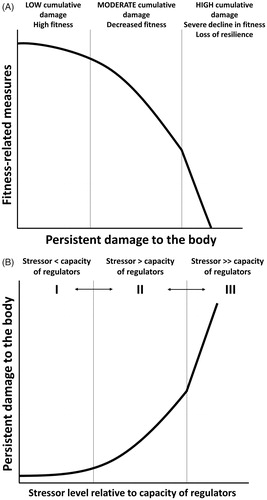
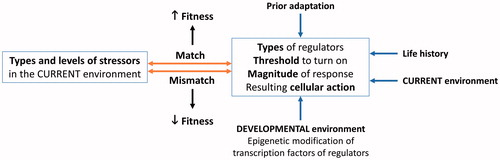
Figure 4. Predictors of fitness outcome based on an appropriateness of anti-damage regulators for the stressors present. Factors such as prior adaptation, life history stage, developmental environment, and current environment influence threshold, magnitude, and categories of anti-damage regulators as well as resulting cellular action (expanded in ). When the predetermined anti-damage regulator phenotype is appropriate for challenges present in the current environment, fitness is expected to increase. If the anti-damage regulator phenotype is not appropriate for the current environment, fitness is predicted to decline.
Anti-damage regulators
In this model, cellular and organismal stress responses, such as heat shock responses (cellular), DNA repair mechanisms (cellular), inflammatory responses (tissue), catecholamine responses (organismal), and adrenocortical responses (organismal), are all considered to be a part of the anti-damage regulators of the body (left column in ). In addition, proper degradation of nonfunctional cells or biomolecules through apoptosis and lysosomal degradation is critical to organ or cellular function (Boya, Citation2012; Folick et al., Citation2015); thus, it is also considered a part of the anti-damage mechanisms. These regulators protect cells, tissues, and animals from further exposures to the stressor, promote repair and recovery through repair proteins and molecules, and destroy removable damage in order to minimize persistent damage in the body. A stressor can be of endogenous or exogenous origin. In some instances, organisms acclimate and increase resistance to a stressor after exposure (Davies, Citation2000; Wiese, Pacifici, & Davies, Citation1995). Here, stress resistance, resilience, and tolerance are distinguished as follows: stress resistance is the animals’ ability to withstand a stressor through minimizing damage (Zone I in ), stress resilience is the animals’ ability to recover once damage occurs, while stress tolerance refers to the threshold at which damage negatively impacts fitness components (vertical lines in ). For instance, an animal is considered to have low resistance and high tolerance if a low level of challenge causes persistent damage, but accumulation of damage does not impact reproductive output, disease risk, or survival until the level of cumulative damage is high. Thus, the capacity of regulators to repair repairable damage and remove unrepairable damage contributes to stress resilience.
Persistent damage and fitness-related measures
When levels of stressors surpass the ability of the organism’s physiological systems or behavioral adjustment to avoid, repair, or remove damage, persistent damage to the body occurs. Damage also accumulates even in the absence of a stressor as a result of normal molecular and cellular activities, which accrues with age (Gladyshev, Citation2013) (). This age- and stressor-associated damage accrual includes telomere length shortening, protein misfolding, DNA damage, lipid peroxidation, protein oxidation, and chronic inflammation among others (Breuner, Delehanty, & Boonstra, Citation2013) (middle column in ). These types of damage can accumulate in the body, particularly when the damage occurs in nonreplaceable or nondividing postmitotic cells, such as neurons and myocytes (Iyama & Wilson, Citation2013). Lipid peroxidation and DNA damage, particularly to mitochondrial DNA (mtDNA), can lead to loss of cell and intracellular membrane integrity, resulting in necrosis (Vanlangenakker, Berghe, Krysko, Festjens, & Vandenabeele, Citation2008). At the tissue level, inadequate oxygen supply can lead to necrosis of the tissue (Vanlangenakker et al., Citation2008). As cell and tissue damage accumulates, organisms lose the ability to recover from damage (i.e. loss of resilience) and leads to increased disease risk and decreased reproductive and locomotor performance, and mortality (right column in ).
Persistent damage and dysregulation of physiological systems lead to disease and mortality risk in several ways. One way is through induced chronic inflammation. During necrotic cell death, cellular content with pro-inflammatory functions leaks out and initiates an inflammatory response (Rock & Kono, Citation2008; Scaffidi, Misteli, & Bianchi, Citation2002). Normally, apoptotic cells are removed by phagocytes before they release their cellular contents (Rock & Kono, Citation2008); however, when apoptotic cells cannot be removed by phagocytes, secondary necrosis occurs (Rock & Kono, Citation2008). This, too, triggers an inflammatory response. Similarly, misfolded proteins and other cellular injuries and abnormalities are sensed by pattern recognition receptors, which also induces an inflammatory response (Lotze et al., Citation2007). Although inflammatory responses are vital for wound healing and fight against pathogens, they can also kill neighboring normal cells and may lead to disease (Rock & Kono, Citation2008). Interestingly, over-nutrition also induces acute and chronic inflammation with an increase in the number of proinflammatory M1 macrophages and cytokines (Hotamisligil, Shargill, & Spiegelman, Citation1993; Lackey & Olefsky, Citation2016; Lumeng, Bodzin, & Saltiel, Citation2007; Mathis, Citation2013) where chronic insulin resistance is linked to macrophage- and cytokine-mediated inflammation (Hotamisligil et al., Citation1993; Lee et al., Citation2011). This could cause systemic insulin resistance and development of type-2 diabetes (Lackey & Olefsky, Citation2016; Lumeng et al., Citation2007). Although the trigger of chronic inflammation may differ, physiological burden of chronic inflammation is similar for wild and laboratory animals and humans.
A stressor can also induce dysregulation of the immune or reproductive systems, genomic instability, or alter relationships among physiological systems. For example, chronic psychological stress is shown to disrupt the inhibitory effect of glucocorticoids on immune function (Cohen, Martin, Wingfield, McWilliams, & Dunne, Citation2012). Non-stressed individuals showed a strong relationship between glucocorticoid levels and neutrophil/lymphocyte ratios, whereas no such relationship was found among stressed individuals, suggesting that immune cells become less sensitive to glucocorticoids with long-term stress (Cohen et al., Citation2012). Furthermore, this insensitivity to glucocorticoids was correlated with higher production of pro-inflammatory cytokines in response to a viral challenge. When acute inflammation persists or there is a failure to remove the source of inflammation, chronic inflammation results, damaging normal tissues and potentially leading to autoimmune disease, Alzheimer’s disease, and atherosclerosis (Khansari, Shakiba, & Mahmoudi, Citation2009; Rock & Kono, Citation2008). Another possible mechanism for loss of resilience is through transposons and retrotransposons. Transposons are genes that move within the genome (McClintock, Citation1984). High levels of transposons cause genomic instability and mutations, including DNA damage (Kemp & Longworth, Citation2015; Schmitt, Prindle, & Loeb, Citation2012). Thus, a level of transposons is regulated in healthy cells by either silencing transposon expression or degrading transposon products. Exposure to a stressor is suggested to change the activity of the transposable elements, although whether it actives or represses depends on the context (Horvath, Merenciano, & Gonzalez, Citation2017). Resulting high levels of transposons are associated with risks for diseases, aging, and infertility (Kazazian & Moran, Citation2017; Natt & Thorsell, Citation2016; Yang & Wang, Citation2016).
Many effects of whole-organism stress responses are double-edged swords. Stress-induced elevation in stress hormone levels is often protective at a low dose and over a short period of time; however, high-dose and long-term elevation is harmful. The latter can cause damage to and dysregulation of physiological systems. This damage and dysregulation, in turn, can lead to chronic inflammation, decreased body size, weakened cardiovascular system due to hypertension, and bone and muscle atrophy due to prolonged calcium mobilization and protein breakdown for glucose (de Vries et al., Citation2007; Kuo, Harris, & Wang, Citation2013; Saag et al., Citation1994; Sarnes et al., Citation2011; Zonana-Nacach, Barr, Magder, & Petri, Citation2000) (). Meta-analyses of clinical studies found that glucocorticoid treatment increased risks for bone fracture, peptic ulcer, cardiovascular disorders, and infection in a dose-dependent manner (Sarnes et al., Citation2011). In particular, the cumulative dose of glucocorticoids was positively correlated with risks for bone fracture, cardiovascular disease, and serious infections (de Vries et al., Citation2007; Saag et al., Citation1994; Sarnes et al., Citation2011; Zonana-Nacach et al., Citation2000). Another epidemiological cohort study revealed that exposure to childhood adversity (e.g. household dysfunction and physical neglect) increased risk for premature mortality (<50 years of age) (Kelly-Irving et al., Citation2013). Furthermore, women’s risk for premature mortality increased with a number of adversities in a dose-dependent manner (Kelly-Irving et al., Citation2013). This is in part due to the fact that individuals with exposure to childhood adversity are more prone to high-risk behaviors that leads to mortality and disability in a cumulative fashion (Campbell, Walker, & Egede, Citation2016). Childhood adversities are linked to altered whole-organism stress responses and elevated inflammatory signals, and when considered in conjunction with adulthood adversity, have an additive effect on inflammatory markers (Hostinar, Lachman, Mroczek, Seeman, & Miller, Citation2015). The cumulative effects of protective and harmful events through one’s lifetime are described in the Stress Accumulation Model and the Lifecourse Health Development Model in social sciences (Halfon, Larson, Lu, Tullis, & Russ, Citation2014; Hostinar et al., Citation2015). In parallel to the proposed model, these studies and theoretical models support the notion that stressful experiences have a cumulative impact on physiology, high-risk health behaviors, and disease risk (Campbell et al., Citation2016; Evans & Kim, Citation2010; Evans, Li, & Whipple, Citation2013; Hostinar et al., Citation2015).
Table 1. Roles of adrenaline, noradrenaline, corticotropin releasing hormone (CRH), glucocorticoids, and aldosterone in avoiding, repairing/recovering from damage, or when these mechanisms have failed and persistent damage and dysregulation occur.
Examples of anti-damage regulators, damage, and reduced fitness components
A cellular example of mtDNA damage and repair mechanisms illustrates how anti-damage regulators minimize persistent damage. mtDNA encodes proteins that are essential for electrical transport chain (Kazak, Reyes, & Holt, Citation2012). Cellular stressors, such as reactive oxygen species (ROS), radiation, and heavy metals can induce mtDNA damage (Karouna-Renier, White, Perkins, Schmerfeld, & Yates, Citation2014; Partridge, Huang, Hernandez-Rosa, Davidson, & Hei, Citation2007; Shokolenko, Venediktova, Bochkareva, Wilson, & Alexeyev, Citation2009). In vivo studies show that smoking and prenatal stress also increase mtDNA damage (Lee et al., Citation1999; Song et al., Citation2009). There are several mechanisms in a cell to protect DNA against oxidative stressors. To avoid cellular damage, antioxidants and related enzymes directly defuse oxidizing agents to mitigate damage to DNA (Zhang, Pi, Woods, & Andersen, Citation2010) (; antioxidants in ). When oxidizing stressors exceed anti-oxidizing capacity of the cell, lesions to the DNA occur (Avery, Citation2011). These lesions are repaired mainly by the base excision repair pathway (Alexeyev, Shokolenko, Wilson, & LeDoux, Citation2013; Kazak et al., Citation2012). It is interesting to note that mtDNA is more susceptible to damage compared to nuclear DNA (nDNA), shown by the fact that mtDNA damage and loss of function were observed at a lower concentration of hydrogen peroxide treatment compared to nDNA (Szczesny et al., Citation2018; Yakes, Chen, & Van Houten, Citation1997). Accumulation of single-strand break mtDNA damage leads to mitochondrial dysregulation and triggers apoptosis of the cell through activation of caspase-3 (Tann et al., Citation2011). mtDNA damage also serves as a pro-inflammatory signal, inducing sterile inflammation (i.e. no introduced pathogen) in vitro and in vivo (Nakayama & Otsu, Citation2018; Szczesny et al., Citation2018; West & Shadel, Citation2017). In vitro, induced mtDNA damage elevates pro-inflammatory mediators, such as interleukin (IL)-1α, IL-1β, IL-6, IL-8, TNFα, and VGFA, in lung epithelial cells (Szczesny et al., Citation2018). Moreover, damaged mtDNA is released from a stressed cell and is capable of inducing pro-inflammatory mediator release in non-stressed cells (Szczesny et al., Citation2018).
Table 2. Anti-damage mechanisms minimizing mitochondrial DNA (mtDNA) damage and its consequences when damage could not be avoided, repaired, or removed.
Similar pattern is observed for mtDNA mutation, including deletion and duplication. mtDNA mutation can occur from replication error (Kauppila & Stewart, Citation2015) which can transmit to the next generation (Ross, Coppotelli, Hoffer, & Olson, Citation2014). When there is enough nonmutated mtDNA, mitochondrial function is not impaired (Rossignol et al., Citation2003). However, when healthy mtDNA cannot compensate for the amount of mutation in mtDNA, deleterious outcome occurs at the level of transcription, translation, mitochondrial function, and phenotype (Rossignol et al., Citation2003). Interestingly, deleterious effects due to mtDNA mutation are thought to precipitate when past a certain threshold. At the translational level, mutation greater than 50 to 60% is shown to impair or arrest translation of mitochondrial proteins (Boulet, Karpati, & Shoubridge, Citation1992; Hayashi et al., Citation1991). At the phenotypic level, deletion greater than 60% and mutation greater than 90% result in phenotypic alterations (Shoffner & Wallace, Citation1995). Experimental elevation of mtDNA mutations through impairing proof-reading polymerase in mice led to loss of bone mineral density, premature decline in fertility in both sexes, and shorter lifespan (Trifunovic et al., Citation2004), indicating that accumulation of mtDNA mutation severely and negatively impacts fitness correlates and components.
Cellular events are intricately connected to whole-organism physiology and behavior as stress responses at cellular and organismal levels are linked. For instance, glucocorticoids play an important role in regulating energy availability via their interactions with metabolic pathways. Glucocorticoid levels increase when metabolic demand of reproduction is high, with a positive relationship between baseline glucocorticoids and reproductive investment (Bonier, Moore, et al., Citation2009; Casagrande et al., Citation2018). At low levels, glucocorticoids also enhance immune function and mitochondrial function (Dhabhar & McEwen, Citation1999; Du et al., Citation2009; Frank, Miguel, Watkins, & Maier, Citation2010; Lim, Müller, Herold, Van Den Brandt, & Reichardt, Citation2007). During times of environmental challenges, glucocorticoids mobilize glucose together with catecholamines, growth hormone, and glucagon and promote foraging and escape behavior (Sapolsky, Romero, & Munck, Citation2000) (). Catecholamines (e.g. adrenaline and noradrenaline) and corticotropin-releasing hormone facilitate the selective delivery of oxygen and glucose to brain and skeletal muscles (Charmandari, Tsigos, & Chrousos, Citation2005; Romero & Butler, Citation2007) (; ). These hormones together allow animals to avoid further exposure to the stressor. Glucocorticoids and aldosterone also coordinate physiological systems in aiding in the recovery from any damage through suppressing reproductive and immune functions and growth, as well as maintaining high blood glucose levels and blood pressure (; ) (Kubzansky & Adler, Citation2010; Sapolsky et al., Citation2000). When a rise in glucocorticoid level precedes an immune challenge, it enhances immune function (Dhabhar & McEwen, Citation1999; Frank et al., Citation2010; Lim et al., Citation2007), indicating that glucocorticoid responses can help the immune system to fight future infections. Together, these physiological changes make energy available to essential organs, promote escape behavior to minimize animal’s exposure to a damage-inducing stressor and help recover and prepare for future stressors (Charmandari et al., Citation2005; Romero & Butler, Citation2007). At the same time, prolonged elevation of blood glucose and blood pressure due to glucocorticoid and aldosterone can turn harmful. Chronic hyperglycemia causes the morphology of mitochondria to change and increases ROS production, leading to mitochondrial dysregulation, DNA damage, and apoptosis (Medikayala, Piteo, Zhao, & Edwards, Citation2011; Savu et al., Citation2011; Suzuki et al., Citation1999; Yu, Robotham, & Yoon, Citation2006). Similarly, in vitro treatment of high-dose glucocorticoids disrupted mitochondrial function, exaggerated kainic acid-induced apoptosis, and induced mtDNA damage in cortical neurons (Du et al., Citation2009). These studies indicate that anti-damage regulators can inflict damage when the levels are too high for a prolonged period of time.
Above examples show several key aspects of the Damage-Fitness Model. First, there is a multitude of regulators at the cellular and organismal level that act together to minimize persistent damage from accumulating in cells and organs. These anti-damage regulators are interconnected, creating a web of coordinated response to a stressor. Second, many regulators are protective at low dose but harmful at high dose where they can cause damage to cells and organs when they are dysfunctional or exceed a certain dose. Third, when the capacity of the regulators to minimize, repair, or remove damage is exceeded, persistent damage results (range II and III in ). Lastly, damage due to error processes accumulate with age, contributing to persistent damage and dysregulation of physiological systems over time (). Considering persistent damage is the outcome of insufficient, excessive, or inappropriate stress responses, the model takes into account biphasic consequences of stressor intensity as well as the responses' age- and season-specificity by focusing on persistent damage.
How developmental environment shapes anti-damage regulator phenotype and fitness outcome
Both developmental and current environments set the threshold for turning on the anti-damage regulators, prioritize which types of regulators are turned on, and determine the magnitude of cellular action activated by the regulators (). This is likely established through epigenetic modification of transcription factors for anti-damage regulators, such as hormone receptors (Bagot & Meaney, Citation2010; Kilvitis, Hanson, Schrey, & Martin, Citation2017; Weaver et al., Citation2004). When there is a mismatch between stressors present in the current environment and the pre-established threshold and capacity of anti-damage mechanisms, responses to a stressor are unbalanced with the cost of turning on the regulators at inappropriate times. This imbalance moves the threshold between the zone where persistent damage is kept to a minimum and the zone where damage starts to accumulate (Zone I and II in ) to the left. It also moves the threshold between zones II and III to the left. This leads to a faster accumulation of damage and a decline in fitness-related measures (; the mismatch to fitness arrow in ). On the other hand, when there is a match between stressor types and magnitude in the current environment and the anti-damage regulator phenotype (threshold, magnitude, and categories of regulator responses), the well-tuned regulator phenotype increases fitness-related measures (the match to fitness arrow in ). The emphasis here is that the mismatch is not strictly between predicted and actual environment (e.g. early- and late-life environments). Rather, it is the match or mismatch between the regulator phenotype and stressors present in the environment. This underscores the importance of choosing an appropriate environment to test whether developmental stressor negatively impacts fitness components. In other words, the context is critical if we want to know whether a certain environment or situation poses a threat to an organism (Wada & Sewall, Citation2014). For instance, it is likely that a developmental stressor enhances the effectiveness of the anti-damage mechanisms until developmental environment is too poor to support adequate growth and development. This will result in an inverted U-shaped dose-response curve when the adult environment is stressful (). Here, an organism gains stress resilience when its developmental environment is moderately poor. At the same time, when the adult environment is benign, individuals who experienced little to no developmental stressor may do better than individuals whose anti-damage mechanisms are prepared to deal with adverse environments (). This pattern likely results due to a cost associated with altered regulator pathways (mismatched). The cost of inappropriate regulator responses includes initiating the response prematurely or excessively, resulting in faster damage accumulation compared to when appropriate responses were initiated. If reproductive output of developmentally stressed and control animals were measured under a benign environment (2 arrows in ), one would conclude that the developmental stressor lowered the reproductive performance of that individual. However, the conclusion may vastly differ if reproductive output of the same individual was measured in a stressful environment (2 arrows in ).
Figure 5. Predicted relationship between developmental stressor intensity and fitness-related measures when the adult environment is stressful (A) and when the adult environment is benign (B). Arrows represent treatment groups in an experiment, control and developmentally stressed groups, in relation to fitness-related measures.
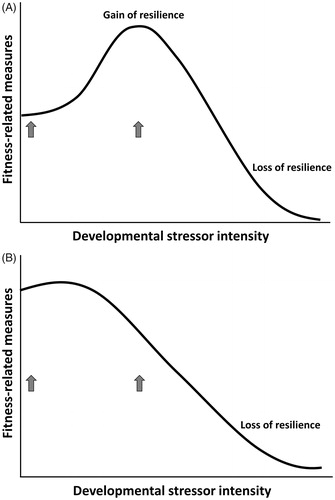
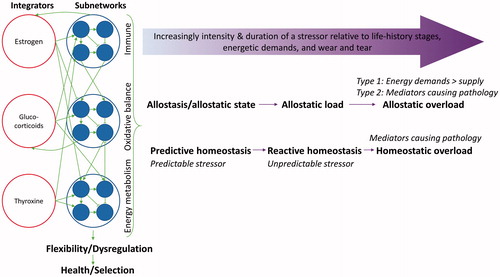
Figure 6. Schematic representation of Allostasis and Reactive Scope Models (horizontal purple arrows) and Physiological Regulatory Networks (green arrows among integrators, subnetworks, flexibility/dysregulation, and health/selection; redrawn from Cohen et al., Citation2012). Allostasis and Reactive Scope Models describe the temporal change of a particular physiological mediator, while Physiological Regulatory Networks describes integration of physiological systems within an individual that response together to a stressor. Wear and tear move the threshold of homeostatic overload closer to Reactive Homeostasis.
How the proposed model relates to existing models
The relationships described in the Damage-Fitness Model are built upon existing models of stress responses, namely the Allostasis, Reactive Scope Model, Physiological Regulatory Networks, and Control Theory Models (Supplementary Appendix). The Allostasis and Reactive Scope Models describe how animals’ energetic and physiological needs change with life-history stages and age and how internal stability is maintained through modifying physiology and behavior (McEwen & Wingfield, Citation2003a, Citation2003b; McEwen & Wingfield, Citation2010; Romero et al., Citation2009). The Physiological Regulatory Network model focuses on physiological interactions within an organism, emphasizing the importance of integrating various physiological systems, which together give rise to an organismal phenotype (Cohen, Martin, et al., Citation2012). Control theory, as applied to stress physiology, uses an engineering approach to illustrate changes in physiological reactions and anticipatory responses to a stressor (Bechhoefer, Citation2005; Stear, Citation1975; Zhang & Andersen, Citation2007; Zhang, Pi, Woods, & Andersen, Citation2009; Zhang et al., Citation2010). In essence, the Allostasis and Reactive Scope Models describe the temporal change of a particular physiological mediator (the purple arrow in and ), while the Physiological Regulatory Networks describe the integration of physiological systems within an individual that respond together to a stressor (green arrows in and ). Control Theory has been used to develop a mathematical model to simulate responses to a stressor, typically for a particular functional unit, such as redox balance (Zhang & Andersen, Citation2007; Zhang et al., Citation2009; Zhang et al., Citation2010). Despite their differences in approach, all these models aim to characterize the dynamic relationships among external environment, internal state, and internal demands associated with life history and environmental change and how internal stability of vital parameters are maintained.
In the Damage-Fitness Model, anti-damage mechanisms in relation to pathology are based on the Allostasis and Reactive Scope Models (McEwen & Wingfield, Citation2003a; Romero et al., Citation2009), and the intertwined relationships among molecular/cellular and organismal anti-damage regulators parallel the core ideas of the Physiological Regulatory Networks model (Cohen, Martin, et al., Citation2012; Martin & Cohen, Citation2014). As mentioned in the existing models, anti-damage regulator levels are plastic and can change due to developmental environment, prior experience to the stressor, population- and species-level adaptation, and age. When environmental conditions change within a lifetime, new transcription factors are activated and repair-and-prepare anti-damage mechanisms kick in to establish a new baseline for the regulators through feedforward and feedback loops, as described in Control Theory (Bechhoefer, Citation2005; Stear, Citation1975; Zhang & Andersen, Citation2007; Zhang et al., Citation2009; Zhang et al., Citation2010). This allows animals to adjust to the new environment. The feedforward loop senses the stressor and directly activates regulator genes which can increase stress resistance (Zhang & Andersen, Citation2007). With an evolutionary adaptation, the reactivity and capacity of the anti-damage mechanism also change and are passed down to the next generation (the adaptation to regulator arrow in ). Even with feedforward and feedback control to maintain low levels of damage, the more times a stressor level exceeds the ability of the animal to negate or repair stressor-induced damage, more damage the body accumulates. Consequently, it is more likely for dysregulation of the anti-damage mechanisms to occur. Perception of a stressor (including predictability and controllability) also plays an important role as it changes the intensity of the stressor for animals with cognition (Koolhaas et al., Citation2011) (the perception-to-stressor arrow in ). The feedback control of the anti-damage mechanism can decline with age (Sapolsky & Altmann, Citation1991; Sapolsky, Krey, & McEwen, Citation1983) due to natural accumulation of damage and dysregulation of the anti-damage mechanisms.
The proposed model regarding match and mismatch between regulator phenotype and the environment parallels the programing sensitivity model proposed by Nederhof and Schmidt (Nederhof & Schmidt, Citation2012). The authors integrated Mismatch and Cumulative Stress Hypotheses in psychopathology, stating that an individual’s sensitivity to physiological programing at an early life determines which hypothesis is applicable. For individuals with high programing sensitivity, developmental stressor exposure prepares organism’s physiology and behavior to better adjust to the stressful environment later on. However, this comes at a cost when the environment turns out to be benign (Mismatch Hypothesis) and the benefit is constrained to when the developmental challenge is at a moderate level. For individuals with low programing sensitivity, damage to cells and organs accumulates as developmental or adult environment becomes poor (Cumulative Stress Hypothesis), decreasing fitness. This means that physiological systems of individuals or species with high programing sensitivity are plastic and are shaped by an early-life environment. Organisms with short lifespans are likely to experience similar environments as young and adults. Thus, it is beneficial for them to tune in to early-life environmental signals. On the other hand, organisms with long lifespans or ones living in a fluctuating environment benefit less from being sensitive to early-life programing. Regardless of the programing sensitivity, an appropriate cellular and physiological response to the current challenge to neutralize, repair, and remove damage will abate negative impacts on fitness components. Thus, focusing on a match between anti-damage regulators and the current challenge, and resulting persistent damage inherently integrates hypotheses and models surrounding early-life and late-life environments.
Importantly, levels of the regulators change constantly with fluctuations in activity levels within a day and physiological demands associated with life-history stages, including growth, reproduction, and migration (Romero et al., Citation2009). As the Allostasis and Reactive Scope Models suggest, a short-term stressor or mild stressor is not likely to cause damage to the body. Similarly, the Damage-Fitness Model argues that the only set of parameters directly influencing fitness-related measures is the persistent damage to cells/tissues and dysregulation of regulators. The relationship between levels of persistent damage and fitness-related measures is predicted to be negative, for example, a negative asymptotic curve (). On the other hand, the relationship between levels of stressors and damage is highly context-specific and depends on the capacity of the anti-damage mechanism to negate the stressor or repair/remove damage caused by the stressor. Applying the concept from the field of engineering describing the damage progression after repeated stress (Diel & Huber, Citation2017), one possibility is that unrepairable, nonremovable damage accumulates in the body in non-linear fashion as intensity, duration, and frequency of the stressor increases (). It is likely that damage is not completely reversible in a sense that cells or tissue with repaired damage is more likely to endure damage again. If damage accumulation depends on prevailing damage, then it would potentially accumulate slowly initially, followed by an exponential increase in damage (Diel & Huber, Citation2017). As the damage occurs, regulator levels increase until the amount of damage exceeds the capacity of the anti-damage mechanisms to repair and remove them, accruing persistent damage (Zones II and III in ). Damage resulted from normal biological activities (Gladyshev, Citation2013) and one caused by stressors, including damage inflicted by dysregulation of the regulators, are predicted to be additive. As the levels of regulators likely change with stressor intensity, duration, and frequency, timing of sample collection influences whether a change in regulator levels is observed and the direction of the change. Thus, how well regulators can minimize persistent damage likely influences the relationships between (1) levels of stressors and fitness-related measures and (2) levels of anti-damage regulators and fitness-related measures. I believe that this is one of the reasons why literature reviews find an equivocal relationship between glucocorticoid levels or oxidative stress and reproductive performance or survival (Bonier, Martin, et al., Citation2009; Breuner et al., Citation2008; Zhang & Hood, Citation2016). Similarly, constant shifts in proportion of healthy and unhealthy cells based on a balance between anti-damage mechanisms and newly inflicted damage determines stress resilience at the whole-organism level. At the same time, persistent damage will only accumulate throughout one’s lifetime whose rate of accumulation vary among individuals.
Testing the Damage-Fitness Model
Rigorous testing of a theoretical model is critical. The proposed model involves multiple stages and dimensions, thus requiring a multi-step approach to test it. In this section, I will outline possible steps for testing the proposed model and predictions for each step.
Causality between persistent damage and fitness-related measures
The first step in testing the Damage-Fitness Model is to determine the relationship and causality between persistent damage and fitness-related measures (). Rather than nondiscriminatory relationships among damage parameters and fitness-related measures, it is likely that each damage parameter is linked to a set of particular fitness-related measures. Several studies connected telomere length or rate of shortening to lifespan, oxidative damage and body size to reproductive output, and protein misfolding, mitochondrial dysfunction, and inflammation to disease risks. Thus far, the vast majority of studies that examined these links have been correlative. Thus, one of the first steps should be to manipulate the level of persistent damage or experimentally induce dysregulation of the anti-damage mechanisms to determine the causal relationship between persistent damage and fitness-related measures. As the level of persistent damage to the cells or organs increases, fitness components and correlates such as reproductive output, protection from diseases, or survival are predicted to decline linearly or follow an inverse sigmoid function curve () (Damage-Fitness Hypothesis). More specifically, the model predicts that (1) as telomere shortening decelerates, lifespan increases, (2) as oxidative damage accumulates in the body, reproductive output declines, and (3) chronic organ-level or systemic inflammation increases disease risks. Using an experimental manipulation of telomere length, oxidative damage, and inflammation mentioned above, we can evaluate these predictions.
Capacity of the anti-damage mechanisms and persistent damage
The second step is to understand the relationship between capacity of the anti-damage regulators and levels of persistent damage () and identify mechanisms behind the relationship. Effectiveness of anti-damage mechanisms in averting damage may be difficult to determine. Identifying the pattern of regulator response that leads to more or less persistent damage will help distinguish a protective response versus an excessive or inappropriate response to a stressor. Similarly, classifying a stressor by whether it results in persistent damage, rather than duration of a stressor for example, may prove fruitful in future studies (instead of acute and chronic stress). As mentioned above, the context under which the regulators are measured is critical. The model predicts that when the intensity, frequency, or duration of a stressor is moderately low, anti-damage regulator levels, such as heat shock proteins, antioxidants, and DNA repair enzymes, are elevated correspondingly and effectively minimize damage (Regulator Capacity Hypothesis), hence persistent damage is predicted to remain low (Zone I in ). When the stressor level is moderate, the capacity of the anti-damage regulators to avoid persistent damage may be overwhelmed, and persistent damage increases accordingly (Zone II). When the stressor level is too high for the anti-damage regulators’ capacity, persistent damage will accumulate at an increased rate (Zone III).
Plasticity in anti-damage mechanisms
The third step is to examine how the developmental environment and prior exposure to a stressor alter the anti-damage regulators and their match with stressors in the current environment. This step tests a version of the Environmental Matching Hypothesis (Monaghan, Citation2008; Santarelli et al., Citation2014) and the Cumulative Stress Hypothesis (Choy, de Visser, Nichols, & van den Buuse, Citation2008), and a mechanistic link among developmental environment, the regulator phenotype of an individual, and resulting fitness components. Here, the proposed model predicts that when the environment during the developmental period is moderately poor, an epigenetic modification of transcription factors of the regulators leads to earlier onset of the anti-damage regulators, enhanced negative feedback on the anti-damage regulators, and higher cellular action for a given signal compared to when the developmental environment is benign or severely poor (Phenotype-Environment Match Hypothesis). This leads to an increase in lifetime reproductive success when the late-life environment is also moderately poor (). At the same time, if the adult environment is benign, having highly responsive anti-damage mechanisms that turn on the regulators early with more potency may decrease fitness (). Alternatively, poor developmental environments may shift the priority of the life history strategy from self-maintenance to reproductive output. In that case, reproductive output may plateau or even increase with a magnitude of developmental stressor before it declines (). With such shift in the life history strategy, survival will decline with a magnitude of developmental stressor.
Benefits of the proposed model
The Damage-Fitness Model and arising hypotheses and predictions will help select reliable indices of health in wild and captive animals as well as in humans. This would be particularly beneficial for studies on wild populations of animals where it is challenging to measure multiple parameters from the same individual. It also provides foci to determine mechanisms and constraint underlying adaptation to a new environment. Additionally, it offers additional tools to test quantitatively why some individuals suffer fitness consequences when exposed to a stressor while others do not. Once the strength of relationship between a particular damage parameter and reproductive output, disease risk, and/or survival is determined for a given population of animals, this model can be used as a predictive model.
The proposed model also sheds a light on a tradeoff between current and future reproduction. The core idea in the life history theory is that reproduction is costly and reduces likelihood of future reproduction (Stearns, Citation1989; Williams, Citation1966). However, many studies have failed to support this assumption (Beaulieu & Costantini, Citation2014; Zhang & Hood, Citation2016). In the case of oxidative stress, it has been argued that a rate of aging accelerates only when ROS levels exceeds antioxidant capacities or repair mechanisms for DNA, lipids, and proteins (Gladyshev, Citation2013; Monaghan, Metcalfe, & Torres, Citation2009). This parallels the core principle of the Damage-Fitness Model, and together they postulate that there is a tradeoff between current and future reproduction only when current reproductive event results in persistent damage in cells or organs. Thus, persistent damage may be the missing piece in our current framework of integrative stress physiology, and the proposed model offers a renewed approach to testing the tradeoffs between current reproduction and survival.
Recommendation for future studies
It has become clear that organisms’ response to a stressor is multifaceted and integrative (Romero et al., Citation2015). This makes it difficult to use a snapshot of one bioindicator level to assess health and condition of an animal. As previously suggested by other researchers, measuring multiple indicators of stress at multiple times will be insightful and needed. Equally important, damage and anti-damage regulators should be measured together whenever possible. This includes oxidative damage with antioxidant levels, as well as telomere length with telomerase activity. Of course, measuring mortality and reproductive success within and across generations is most informative. However, it is sometimes not feasible to measure reproductive success and mortality, sample animals repeatedly, or collect enough samples to quantify multiple physiological parameters. In those cases, the recommendation from the Damage-Fitness Model is to measure indicators of persistent damage or physiological dysregulation instead of anti-damage regulators because those regulators are often transient and improves fitness components only when capacity of the regulators to minimize damage exceeds the level of the stressor.
In various fields of biology, we aspire to predict population-level responses to an environmental change. However, no universal, single biomarker can assess individual and population health and predict the robustness of the population to an environmental change. It is likely that a predictive value of a damage parameter on relative fitness depends on species (Costantini & Dell'Omo, Citation2015). For each species of interest, we must first identify multiple parameters that correlate with fitness components (Beaulieu & Costantini, Citation2014). If a species is endangered or protected, it may be evaluated based on data of closely related species. Importantly, when we move from within-generation, within-individual physiological changes to across-generational, between-individual changes, we must determine the degree of heritability and whether the physiological responses to a stressor is beneficial (Wada & Sewall, Citation2014). If the traits only vary with the environment with low heritability, then it will neither be selected for nor against. Moreover, testing of the model should be done in healthy animals rather than animal models for certain disease, which may have altered relationships among anti-damage mechanisms, damage, and fitness components (Romero et al., Citation2015). Lastly, the list of parameters in this paper is not exhaustive; more controlled studies and studies on wild populations are needed to explore other types of damage and parameters indicative of physiological dysregulation that are directly linked to fitness-related measures and refine our understanding of those relationships.
Conclusion
Previous models of stress have incorporated wear and tear, phenotypic plasticity of stress responses across life history stages, and integrated various cellular and physiological parameters. The Damage-Fitness Model takes a performance-centered approach focusing on fitness-related measures and what is most directly linked to them. This underscores the importance of measuring components of fitness or performance metrics clearly related to reproduction and survival whenever possible. That is, it is paramount to demonstrate how an experimental treatment alters fitness-related measures and characterize changes in anti-damage mechanisms that led to the consequences. Connecting the three elements – fitness components, damage that directly impacts the fitness component, and regulator responses that led to the damage – will allow evaluation of beneficial and harmful regulator responses. For instance, when the goal of a study is to determine whether constructing a road is stressful to a toad population, then we need to first document any change in reproductive success, disease risk, or survival due to the road construction. We then can measure physiological damage that lowered the particular fitness component. If reproductive performance was reduced but survival was not affected, oxidative damage would be a better candidate than telomere length shortening. Once types of damage have been identified, it is imperative to determine the responsiveness of regulators such as antioxidant capacities and adrenocortical responses that permitted an accumulation of damage. This is different from a traditional approach of identifying an impact of road construction on adrenocortical responses then measuring possible consequences, such as immune function or oxidative damage. By depicting fitness-related measures, cellular and tissue damage that differ between the treatment groups, and various anti-damage mechanisms that allowed the damage to occur, we can begin to recognize beneficial and detrimental anti-damage mechanisms for that context. Most importantly, measuring damage and fitness components should lend itself to the creation of mathematical models, as they are likely directly related. Once that is established, more complex relationships among anti-damage mechanisms and damage as well as anticipatory and developmental effect may be incorporated. With RNAseq and other “-omics” technologies, there are millions of networks that are potential target of future studies. This model can help prioritize networks to focus on our studies. Finally, this model offers potential indicators of health and condition in animals and steps to be taken before they can be used to predict population-level responses to an environmental change.
Supplemental Material
Download MS Word (55.1 KB)Acknowledgements
I thank Creagh Breuner, John Finger, Britt Heidinger, Chloe Josefson, Alex Hoffman, Wendy Hood, Marty Martin, Michael Romero, Alex Rubin, Guillaume Salze, Tonia Schwartz, Masaru Wada, as well as two anonymous reviewers and the journal editor James Herman for their input on the manuscript. I also want to thank organizers and participants of the Stress in Healthy Animals workshop and the Editor’s Challenge workshop at the 2018 Society for Integrative and Comparative Biology meeting, and graduate students in the Stress: From cell to organism course for valuable discussion on the topic of stress. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
Notes on contributors
Haruka Wada
Haruka Wada is an integrative stress physiologist at Auburn University. For the last ∼20 years, she has been studying development of the hypothalamic-pituitary-adrenal axis and the effects of glucocorticoids and methylmercury on endocrine axes, immune function, cognition, and neurobiology of birds, amphibians, and mammals. Her laboratory is currently working on physiological underpinnings of stress resilience with heat conditioning in zebra finches.
References
- Alexeyev, M., Shokolenko, I., Wilson, G., & LeDoux, S. (2013). The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harbor Perspectives in Biology, 5, a012641. doi:10.1101/cshperspect.a012641
- Arlettaz, R., Patthey, P., Baltic, M., Leu, T., Schaub, M., Palme, R., & Jenni-Eiermann, S. (2007). Spreading free-riding snow sports represent a novel serious threat for wildlife. Proceedings of the Royal Society B, 274, 1219–1224. doi:10.1098/rspb.2006.0434
- Avery, S.V. (2011). Molecular targets of oxidative stress. Biochemical Journal, 434, 201–210. doi:10.1042/BJ20101695
- Bagot, R.C., & Meaney, M.J. (2010). Epigenetics and the biological basis of gene × environment interactions. Journal of the American Academy of Child and Adolescent Psychiatry, 49, 752–771. doi:10.1016/j.jaac.2010.06.001
- Beaulieu, M., & Costantini, D. (2014). Biomarkers of oxidative status: missing tools in conservation physiology. Conservation Physiology, 2. doi:10.1093/conphys/cou014
- Bechhoefer, J. (2005). Feedback for physicists: A tutorial essay on control. Reviews of Modern Physics, 77, 783–836. doi:10.1103/RevModPhys.77.783
- Bonier, F., Martin, P.R., Moore, I.T., & Wingfield, J.C. (2009). Do baseline glucocorticoids predict fitness? Trends in Ecology & Evolution, 24, 634–642. doi:10.1016/j.tree.2009.04.013
- Bonier, F., Moore, I.T., Martin, P.R., & Robertson, R.J. (2009). The relationship between fitness and baseline glucocorticoids in a passerine bird. General and Comparative Endocrinology, 163, 208–213. doi:10.1016/j.ygcen.2008.12.013
- Boulet, L., Karpati, G., & Shoubridge, E.A. (1992). Distribution and threshold expression of the tRNA(Lys) mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF). American Journal of Human Genetics, 51, 1187–1200.
- Boya, P. (2012). Lysosomal function and dysfunction: Mechanism and disease. Antioxidants and Redox Signaling, 17, 766–774. doi:10.1089/ars.2011.4405
- Breuner, C.W., Delehanty, B., & Boonstra, R. (2013). Evaluating stress in natural populations of vertebrates: Total CORT is not good enough. Functional Ecology, 27, 24–36. doi:10.1111/1365-2435.12016
- Breuner, C.W., Greenberg, A.L., & Wingfield, J.C. (1998). Noninvasive corticosterone treatment rapidly increases activity in Gambel's White-crowned Sparrows (Zonotrichia leucophrys gambelii). General and Comparative Endocrinology, 111, 386–394. doi:10.1006/gcen.1998.7128
- Breuner, C.W., Patterson, S.H., & Hahn, T.P. (2008). In search of relationships between the acute adrenocortical response and fitness. General and Comparative Endocrinology, 157, 288–295. doi:10.1016/j.ygcen.2008.05.017
- Briot, K., & Roux, C. (2015). Glucocorticoid-induced osteoporosis. RMD Open, 1, e000014. doi:10.1136/rmdopen-2014-000014
- Brown, M.R., & Fisher, L.A. (1983). Central nervous system effects of corticotropin releasing factor in the dog. Brain Research, 280, 75–79. doi:10.1016/0006-8993(83)91175-7
- Campbell, J.A., Walker, R.J., & Egede, L.E. (2016). Associations between adverse childhood experiences, high-risk behaviors, and morbidity in adulthood. American Journal of Preventive Medicine, 50, 344–352. doi:10.1016/j.amepre.2015.07.022
- Canalis, E., & Delany, A.M. (2002). Mechanisms of glucocorticoid action in bone. Annals of the New York Academy of Sciences, 966, 73–81. doi:10.1111/j.1749-6632.2002.tb04204.x
- Canalis, E., Mazziotti, G., Giustina, A., & Bilezikian, J.P. (2007). Glucocorticoid-induced osteoporosis: Pathophysiology and therapy. Osteoporosis International, 18, 1319–1328. doi:10.1007/s00198-007-0394-0
- Casagrande, S., Zsolt Garamszegi, L., Goymann, W., Donald, J., Francis, C.D., Fuxjager, M.J., … Hau, M. (2018). Do seasonal glucocorticoid changes depend on reproductive investment? A comparative approach in birds. Integrative and Comparative Biology, 58, 739–750.
- Charmandari, E., Tsigos, C., & Chrousos, G. (2005). Endocrinology of the stress response. Annual Review of Physiology, 67, 259–284. doi:10.1146/annurev.physiol.67.040403.120816
- Choy, K.H.C., de Visser, Y., Nichols, N.R., & van den Buuse, M. (2008). Combined neonatal stress and young-adult glucocorticoid stimulation in rats reduce BDNF expression in hippocampus: Effects on learning and memory. Hippocampus, 18, 655–667. doi:10.1002/hipo.20425
- Cohen, A.A., Martin, L.B., Wingfield, J.C., McWilliams, S.R., & Dunne, J.A. (2012). Physiological regulatory networks: Ecological roles and evolutionary constraints. Trends in Ecology and Evolution, 27, 428–435. doi:10.1016/j.tree.2012.04.008
- Cohen, S., Janicki-Deverts, D., Doyle, W.J., Miller, G.E., Frank, E., Rabin, B.S., & Turner, R.B. (2012). Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proceedings of the National Academy of Sciences of the United States of America, 109, 5995–5999. doi:10.1073/pnas.1118355109
- Costantini, D., & Dell'Omo, G. (2015). Oxidative stress predicts long-term resight probability and reproductive success in Scopoli's shearwater (Calonectris diomedea). Conservation Physiology, 3. doi:10.1093/conphys/cov024
- Creel, S., Fox, J.E., Hardy, A., Sands, J., Garrott, B., & Peterson, R.O. (2002). Snowmobile activity and glucocorticoid stress responses in wolves and elk. Conservation Biology, 16, 809–814. doi:10.1046/j.1523-1739.2002.00554.x
- Davies, K.J. (2000). Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life, 50, 279–289. doi:10.1080/15216540051081010
- de Vries, F., Bracke, M., Leufkens, H.G.M., Lammers, J.-W.J., Cooper, C., & van Staa, T.P. (2007). Fracture risk with intermittent high-dose oral glucocorticoid therapy. Arthritis & Rheumatism, 56, 208–214. doi:10.1002/art.22294
- Del Giudice, M., Ellis, B.J., & Shirtcliff, E.A. (2011). The adaptive calibration model of stress responsivity. Neuroscience and Biobehavioral Reviews, 35, 1562–1592. doi:10.1016/j.neubiorev.2010.11.007
- Dhabhar, F.S. (2008). Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection versus immunopathology. Allergy, Asthma & Clinical Immunology, 4, 2–11. doi:10.1186/1710-1492-4-1-2
- Dhabhar, F.S., & McEwen, B.S. (1999). Enhancing versus suppressive effects of stress hormones on skin immune function. Proceedings of the National Academy of Sciences of the United States of America, 96, 1059–1064. doi:10.1073/pnas.96.3.1059
- Diel, S., & Huber, O. (2017). A continuum damage mechanics model for the static and cyclic fatigue of cellular composites. Materials (Basel), 10, 951.
- Du, J., Wang, Y., Hunter, R., Wei, Y., Blumenthal, R., Falke, C., … Manji, H.K. (2009). Dynamic regulation of mitochondrial function by glucocorticoids. Proceedings of the National Academy of Sciences of the United States of America, 106, 3543–3548. doi:10.1073/pnas.0812671106
- Evans, G.W., & Kim, P. (2010). Multiple risk exposure as a potential explanatory mechanism for the socioeconomic status-health gradient. Annals of the New York Academy of Sciences, 1186, 174–189. doi:10.1111/j.1749-6632.2009.05336.x
- Evans, G.W., Li, D., & Whipple, S.S. (2013). Cumulative risk and child development. Psychological Bulletin, 139, 1342–1396. doi:10.1037/a0031808
- Fisher, L.A., Jessen, G., & Brown, M.R. (1983). Corticotropin-releasing factor (CRF): Mechanism to elevate mean arterial pressure and heart rate. Regulatory Peptides, 5, 153–161. doi:10.1016/0167-0115(83)90123-4
- Folick, A., Oakley, H.D., Yu, Y., Armstrong, E.H., Kumari, M., Sanor, L., … Wang, M.C. (2015). Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science, 347, 83–86. doi:10.1126/science.1258857
- Frank, M.G., Miguel, Z.D., Watkins, L.R., & Maier, S.F. (2010). Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behavior, and Immunity, 24, 19–30. doi:10.1016/j.bbi.2009.07.008
- Gladyshev, V.N. (2013). The origin of aging: imperfectness-driven non-random damage defines the aging process and control of lifespan. Trends in Genetics, 29, 506–512. doi:10.1016/j.tig.2013.05.004
- Halfon, N., Larson, K., Lu, M., Tullis, E., & Russ, S. (2014). Lifecourse health development: Past, present and future. Maternal and Child Health Journal, 18, 344–365. doi:10.1007/s10995-013-1346-2
- Hayashi, J., Ohta, S., Kikuchi, A., Takemitsu, M., Goto, Y., & Nonaka, I. (1991). Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proceedings of the National Academy of Sciences of the United States of America, 88, 10614–10618. doi:10.1073/pnas.88.23.10614
- Horvath, V., Merenciano, M., & Gonzalez, J. (2017). Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends in Genetics, 33, 832–841. doi:10.1016/j.tig.2017.08.007
- Hostinar, C.E., Lachman, M.E., Mroczek, D.K., Seeman, T.E., & Miller, G.E. (2015). Additive contributions of childhood adversity and recent stressors to inflammation at midlife: Findings from the MIDUS study. Developmental Psychology, 51, 1630–1644. doi:10.1037/dev0000049
- Hotamisligil, G.S., Shargill, N.S., & Spiegelman, B.M. (1993). Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science, 259, 87. doi:10.1126/science.7678183
- Iyama, T., & Wilson, D.M. (2013). DNA repair mechanisms in dividing and non-dividing cells. DNA Repair, 12, 620–636. doi:10.1016/j.dnarep.2013.04.015
- Karouna-Renier, N.K., White, C., Perkins, C.R., Schmerfeld, J.J., & Yates, D. (2014). Assessment of mitochondrial DNA damage in little brown bats (Myotis lucifugus) collected near a mercury-contaminated river. Ecotoxicology, 23, 1419–1429. doi:10.1007/s10646-014-1284-9
- Kauppila, J.H.K., & Stewart, J.B. (2015). Mitochondrial DNA: Radically free of free-radical driven mutations. Biochimica et Biophysica Acta Bioenergetics, 1847, 1354–1361. doi:10.1016/j.bbabio.2015.06.001
- Kazak, L., Reyes, A., & Holt, I.J. (2012). Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nature Reviews, Molecular Cell Biology, 13, 659. doi:10.1038/nrm3439
- Kazazian, H.H., Jr., & Moran, J.V. (2017). Mobile DNA in health and disease. The New England Journal of Medicine, 377, 361–370. doi:10.1056/NEJMra1510092
- Kelly-Irving, M., Lepage, B., Dedieu, D., Bartley, M., Blane, D., Grosclaude, P., … Delpierre, C. (2013). Adverse childhood experiences and premature all-cause mortality. European Journal of Epidemiology, 28, 721–734. doi:10.1007/s10654-013-9832-9
- Kemp, J.R., & Longworth, M.S. (2015). Crossing the LINE toward genomic instability: LINE-1 retrotransposition in cancer. Frontiers in Chemistry, 3, 68.
- Khansari, N., Shakiba, Y., & Mahmoudi, M. (2009). Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Patents on Inflammation & Allergy Drug Discovery, 3, 73–80. doi:10.2174/187221309787158371
- Kilvitis, H.J., Hanson, H., Schrey, A.W., & Martin, L.B. (2017). Epigenetic potential as a mechanism of phenotypic plasticity in vertebrate range expansions. Integrative and Comparative Biology, 57, 385–395. doi:10.1093/icb/icx082
- Klein, G.L. (2015). The effect of glucocorticoids on bone and muscle. Osteoporosis and Sarcopenia, 1, 39–45. doi:10.1016/j.afos.2015.07.008
- Koolhaas, J.M., Bartolomucci, A., Buwalda, B., de Boer, S.F., Flügge, G., Korte, S.M., … Fuchs, E. (2011). Stress revisited: A critical evaluation of the stress concept. Neuroscience and Biobehavioral Reviews, 35, 1291–1301. doi:10.1016/j.neubiorev.2011.02.003
- Korte, S.M., Koolhaas, J.M., Wingfield, J.C., & McEwen, B.S. (2005). The Darwinian concept of stress: benefits of allostasis and costs of allostatic load and the trade-offs in health and disease. Neuroscience and Biobehavioral Reviews, 29, 3–38. doi:10.1016/j.neubiorev.2004.08.009
- Kralj-Fišer, S., Scheiber, I.B.R., Kotrschal, K., Weiß, B.M., & Wascher, C.A.F. (2010). Glucocorticoids enhance and suppress heart rate and behaviour in time dependent manner in greylag geese (Anser anser). Physiology & Behavior, 100, 394–400. doi:10.1016/j.physbeh.2010.04.005
- Kubzansky, L.D., & Adler, G.K. (2010). Aldosterone: A forgotten mediator of the relationship between psychological stress and heart disease. Neuroscience and Biobehavioral Reviews, 34, 80–86. doi:10.1016/j.neubiorev.2009.07.005
- Kuo, T., Harris, C.A., & Wang, J.-C. (2013). Metabolic functions of glucocorticoid receptor in skeletal muscle. Molecular and Cellular Endocrinology, 380, 79–88. doi:10.1016/j.mce.2013.03.003
- Lackey, D.E., & Olefsky, J.M. (2016). Regulation of metabolism by the innate immune system. Nature Reviews. Endocrinology, 12, 15. doi:10.1038/nrendo.2015.189
- Lee, H.C., Lim, M.L.R., Lu, C.Y., Liu, V.W.S., Fahn, H.-J., Zhang, C., … Wei, Y.-H. (1999). Concurrent increase of oxidative DNA damage and lipid peroxidation together with mitochondrial DNA mutation in human lung tissues during aging—Smoking enhances oxidative stress on the aged tissues. Archives of Biochemistry and Biophysics, 362, 309–316. doi:10.1006/abbi.1998.1036
- Lee, Y.S., Li, P., Huh, J.Y., Hwang, I.J., Lu, M., Kim, J.I., … Kim, J.B. (2011). Inflammation is necessary for long-term but not short-term high-fat diet–induced insulin resistance. Diabetes, 60, 2474. doi:10.2337/db11-0194
- Lim, H.Y., Müller, N., Herold, M.J., Van Den Brandt, J., & Reichardt, H.M. (2007). Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology, 122, 47–53. doi:10.1111/j.1365-2567.2007.02611.x
- Limón-Pacheco, J., & Gonsebatt, M.E. (2009). The role of antioxidants and antioxidant-related enzymes in protective responses to environmentally induced oxidative stress. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, 674, 137–147. doi:10.1016/j.mrgentox.2008.09.015
- Lotze, M.T., Zeh, H.J., Rubartelli, A., Sparvero, L.J., Amoscato, A.A., Washburn, N.R., … Billiar, T. (2007). The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunological Reviews, 220, 60–81. doi:10.1111/j.1600-065X.2007.00579.x
- Lumeng, C.N., Bodzin, J.L., & Saltiel, A.R. (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. Journal of Clinical Investigation, 117, 175–184. doi:10.1172/JCI29881
- Manson, S.C., Brown, R.E., Cerulli, A., & Vidaurre, C.F. (2009). The cumulative burden of oral corticosteroid side effects and the economic implications of steroid use. Respiratory Medicine, 103, 975–994. doi:10.1016/j.rmed.2009.01.003
- Marra, P.P., & Holberton, R.L. (1998). Corticosterone levels as indicators of habitat quality: Effects of habitat segregation in a migratory bird during the non-breeding season. Oecologia, 116, 284–292. doi:10.1007/s004420050590
- Martin, L.B., & Cohen, A.A. (2014). Physiological regulatory networks: The orchestra of life? In L. B. Martin, C. K. Ghalambor & H. A. Woods, (Eds.), Integrative Organismal Biology (p. 137–152).
- Mathis, D. (2013). Immunological goings-on in visceral adipose tissue. Cell Metabolism, 17, 851–859. doi:10.1016/j.cmet.2013.05.008
- McClintock, B. (1984). The significance of responses of the genome to challenge. Science (New York, N.Y.), 226, 792. doi:10.1126/science.15739260
- McEwen, B.S., & Wingfield, J.C. (2003a). The concept of allostasis in biology and biomedicine. Hormones and Behavior, 43, 2–15. doi:10.1016/S0018-506X(02)00024-7
- McEwen, B.S., & Wingfield, J.C. (2003b). Response to commentaries on the concept of allostasis. Hormones and Behavior, 43, 28–30. doi:10.1016/S0018-506X(02)00039-9
- McEwen, B.S., & Wingfield, J.C. (2010). What is in a name? Integrating homeostasis, allostasis and stress. Hormones and Behavior, 57, 105–111. doi:10.1016/j.yhbeh.2009.09.011
- Medikayala, S., Piteo, B., Zhao, X., & Edwards, J.G. (2011). Chronically elevated glucose compromises myocardial mitochondrial DNA integrity by alteration of mitochondrial topoisomerase function. American Journal of Physiology: Cell Physiology, 300, C338–C348. doi:10.1152/ajpcell.00248.2010
- Monaghan, P. (2008). Early growth conditions, phenotypic development and environmental change. Philosophical Transactions of the Royal Society B: Biological Sciences, 363, 1635–1645. doi:10.1098/rstb.2007.0011
- Monaghan, P., Metcalfe, N.B., & Torres, R. (2009). Oxidative stress as a mediator of life history trade-offs: Mechanisms, measurements and interpretation. Ecology Letters, 12, 75–92. doi:10.1111/j.1461-0248.2008.01258.x
- Nakayama, H., & Otsu, K. (2018). Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochemical Journal, 475, 839–852. doi:10.1042/BCJ20170714
- Natt, D., & Thorsell, A. (2016). Stress-induced transposon reactivation: A mediator or an estimator of allostatic load? Environmental Epigenetics, 2, dvw015. doi:10.1093/eep/dvw015
- Nederhof, E., & Schmidt, M.V. (2012). Mismatch or cumulative stress: Toward an integrated hypothesis of programming effects. Physiology and Behavior, 106, 691–700. doi:10.1016/j.physbeh.2011.12.008
- Partridge, M.A., Huang, S.X., Hernandez-Rosa, E., Davidson, M.M., & Hei, T.K. (2007). Arsenic induced mitochondrial DNA damage and altered mitochondrial oxidative function: Implications for genotoxic mechanisms in mammalian cells. Cancer Research, 67, 5239–5247. doi:10.1158/0008-5472.CAN-07-0074
- Rock, K.L., & Kono, H. (2008). The inflammatory response to cell death. Annual Review of Pathology: Mechanisms of Disease, 3, 99–126. doi:10.1146/annurev.pathmechdis.3.121806.151456
- Romero, L.M., & Butler, L.K. (2007). Endocrinology of stress. International Journal of Comparative Psychology, 20, 89–95.
- Romero, L.M., Dickens, M.J., & Cyr, N.E. (2009). The reactive scope model – A new model integrating homeostasis, allostasis, and stress. Hormones and Behavior, 55, 375–389. doi:10.1016/j.yhbeh.2008.12.009
- Romero, L.M., Platts, S.H., Schoech, S.J., Wada, H., Crespi, E., Martin, L.B., & Buck, C.L. (2015). Understanding stress in the healthy animal – potential paths for progress. Stress, 18, 491–497. doi:10.3109/10253890.2015.1073255
- Ross, J.M., Coppotelli, G., Hoffer, B.J., & Olson, L. (2014). Maternally transmitted mitochondrial DNA mutations can reduce lifespan. Scientific Report, 4, 6569.
- Rossignol, R., Faustin, B., Rocher, C., Malgat, M., Mazat, J. P., & Letellier, T. (2003). Mitochondrial threshold effects. Biochemical Journal, 370, 751. doi:10.1042/bj20021594
- Saag, K.G., Koehnke, R., Caldwell, J.R., Brasington, R., Burmeister, L.F., Zimmerman, B., … Furst, D.E. (1994). Low dose long-term corticosteroid therapy in rheumatoid arthritis: An analysis of serious adverse events. The American Journal of Medicine, 96, 115–123. doi:10.1016/0002-9343(94)90131-7
- Santarelli, S., Lesuis, S.L., Wang, X.-D., Wagner, K.V., Hartmann, J., Labermaier, C., … Schmidt, M.V. (2014). Evidence supporting the match/mismatch hypothesis of psychiatric disorders. European Neuropsychopharmacology, 24, 907–918. doi:10.1016/j.euroneuro.2014.02.002
- Sapolsky, R.M. (1996). Stress, glucocorticoids, and damage to the nervous system: The current state of confusion. Stress, 1, 1–19. doi:10.3109/10253899609001092
- Sapolsky, R.M., & Altmann, J. (1991). Incidence of hypercortisolism and dexamethasone resistance increases with age among wild baboons. Biological Psychiatry, 30, 1008–1016. doi:10.1016/0006-3223(91)90121-2
- Sapolsky, R.M., Krey, L.C., & McEwen, B.S. (1983). Corticosterone receptors decline in a site-specific manner in the aged rat brain. Brain Research, 289, 235–240. doi:10.1016/0006-8993(83)90024-0
- Sapolsky, R.M., Romero, L.M., & Munck, A.U. (2000). How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocrine Reviews, 21, 55–89. doi:10.1210/er.21.1.55
- Sarnes, E., Crofford, L., Watson, M., Dennis, G., Kan, H., & Bass, D. (2011). Incidence and US costs of corticosteroid-associated adverse events: A systematic literature review. Clinical Therapeutics, 33, 1413–1432. doi:10.1016/j.clinthera.2011.09.009
- Savu, O., Sunkari, V.G., Botusan, I.R., Grunler, J., Nikoshkov, A., & Catrina, S.B. (2011). Stability of mitochondrial DNA against reactive oxygen species (ROS) generated in diabetes. Diabetes/Metabolism Research and Reviews, 27, 470–479. doi:10.1002/dmrr.1203
- Scaffidi, P., Misteli, T., & Bianchi, M.E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature, 418, 191. doi:10.1038/nature00858
- Schmitt, M.W., Prindle, M.J., & Loeb, L.A. (2012). Implications of genetic heterogeneity in cancer. Annals of the New York Academy of Sciences, 1267, 110–116. doi:10.1111/j.1749-6632.2012.06590.x
- Schoenle, L.A., Zimmer, C., & Vitousek, M.N. (2018). Understanding context dependence in glucocorticoid-fitness relationships: The role of the nature of the challenge, the intensity and frequency of stressors, and life history. Integrative and Comparative Biology, 58, 777–789. doi:10.1093/icb/icy046
- Shoffner, J., & Wallace, D. (1995). Oxidative phosphorylation diseases. In C. R. Scriver, A. L. Beaudet, W. S. Sly & D. Valle (Eds.), The metabolic basis of inherited disease (pp. 1535–1609). New York: McGraw Hill.
- Shokolenko, I., Venediktova, N., Bochkareva, A., Wilson, G.L., & Alexeyev, M.F. (2009). Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Research, 37, 2539–2548. doi:10.1093/nar/gkp100
- Song, L., Zheng, J., Li, H., Jia, N., Suo, Z., Cai, Q., … Zhu, Z. (2009). Prenatal stress causes oxidative damage to mitochondrial DNA in hippocampus of offspring rats. Neurochemical Research, 34, 739–745. doi:10.1007/s11064-008-9838-y
- Sorenson, G.H., Dey, C.J., Madliger, C.L., & Love, O.P. (2017). Effectiveness of baseline corticosterone as a monitoring tool for fitness: A meta-analysis in seabirds. Oecologia, 183, 353–365. doi:10.1007/s00442-016-3774-3
- Stear, E.B. (1975). Application of control theory to endocrine regulation and control. Annals of Biomedical Engineering, 3, 439–455. doi:10.1007/BF02409328
- Stearns, S.C. (1989). Trade-offs in life-history evolution. Functional Ecology, 3, 259–268. doi:10.2307/2389364
- Suzuki, S., Hinokio, Y., Komatu, K., Ohtomo, M., Onoda, M., Hirai, S., … Toyota, T. (1999). Oxidative damage to mitochondrial DNA and its relationship to diabetic complications. Diabetes Research and Clinical Practice, 45, 161–168. doi:10.1016/S0168-8227(99)00046-7
- Szczesny, B., Marcatti, M., Ahmad, A., Montalbano, M., Brunyanszki, A., Bibli, S.I., … Szabo, C. (2018). Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Scientific Reports, 8, 914. doi:10.1038/s41598-018-19216-1
- Tann, A.W., Boldogh, I., Meiss, G., Qian, W., Van Houten, B., Mitra, S., & Szczesny, B. (2011). Apoptosis induced by persistent single-strand breaks in mitochondrial genome: Critical role of EXOG (5'-EXO/endonuclease) in their repair. Journal of Biological Chemistry, 286, 31975–31983. doi:10.1074/jbc.M110.215715
- Thiel, D., Jenni-Eiermann, S., Braunisch, V., Palme, R., & Jenni, L. (2007). Ski tourism affects habitat use and evokes a physiological stress response in capercaillie Tetrao urogallus: A new methodological approach. Journal of Applied Ecology, 45, 845–853. doi:10.1111/j.1365-2664.2008.01465.x
- Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J.N., Rovio, A.T., Bruder, C.E., … Larsson, N.-G. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature, 429, 417. doi:10.1038/nature02517
- Vanlangenakker, N., Berghe, T.V., Krysko, D.V., Festjens, N., & Vandenabeele, P. (2008). Molecular mechanisms and pathophysiology of necrotic cell death. Current Molecular Medicine, 8, 207–220. doi:10.2174/156652408784221306
- Vyas, S., Rodrigues, A.J., Silva, J.M., Tronche, F., Almeida, O.F.X., Sousa, N., & Sotiropoulos, I. (2016). Chronic stress and glucocorticoids: From neuronal plasticity to neurodegeneration. Neural Plasticity, 2016, 1. doi:10.1155/2016/6391686
- Wada, H., & Sewall, K.B. (2014). Introduction to the symposium-uniting evolutionary and physiological approaches to understanding phenotypic plasticity. Integrative and Comparative Biology, 54, 774–782. doi:10.1093/icb/icu097
- Walker, B.G., Boersma, P.D., & Wingfield, J.C. (2005a). Field endocrinology and conservation biology. Integrative and Comparative Biology, 45, 12–18. doi:10.1093/icb/45.1.12
- Walker, B.G., Boersma, P.D., & Wingfield, J.C. (2005b). Physiological and behavioral differences in magellanic penguin chicks in undisturbed and tourist-visited locations of a colony. Conservation Biology, 19, 1571–1577. doi:10.1111/j.1523-1739.2005.00104.x
- Weaver, I.C.G., Cervoni, N., Champagne, F.A., D'Alessio, A.C., Sharma, S., Seckl, J.R., … Meaney, M.J. (2004). Epigenetic programming by maternal behavior. Nature Neuroscience, 7, 847–854. doi:10.1038/nn1276
- West, A.P., & Shadel, G.S. (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nature Reviews Immunology, 17, 363–375. doi:10.1038/nri.2017.21
- Wiese, A.G., Pacifici, R.E., & Davies, K.J.A. (1995). Transient adaptation to oxidative stress in mammalian cells. Archives of Biochemistry and Biophysics, 318, 231–240. doi:10.1006/abbi.1995.1225
- Wikelski, M., & Cooke, S.J. (2006). Conservation physiology. Trends in Ecology and Evolution, 21, 38–46. doi:10.1016/j.tree.2005.10.018
- Williams, G.C. (1966). Natural selection, the cost of reproduction and a refinement of Lack’s principle. American Naturalist, 100, 687–690. doi:10.1086/282461
- Yakes, F.M., Chen Y., Van Houten, B. (1997). Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proceedings of the National Academy of Sciences of the United States of America, 94, 514–519. doi:10.1073/pnas.94.2.514
- Yang, F., & Wang, P.J. (2016). Multiple LINEs of retrotransposon silencing mechanisms in the mammalian germline. Seminars in Cell and Developmental Biology, 59, 118–125. doi:10.1016/j.semcdb.2016.03.001
- Yu, T., Robotham, J.L., & Yoon, Y. (2006). Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proceedings of the National Academy of Sciences of the United States of America, 103, 2653. doi:10.1073/pnas.0511154103
- Zhang, Q., & Andersen, M.E. (2007). Dose response relationship in anti-stress gene regulatory networks. PLoS Computational Biology, 3, e24. doi:10.1371/journal.pcbi.0030024
- Zhang, Q., Pi, J., Woods, C.G., & Andersen, M.E. (2009). Phase I to II cross-induction of xenobiotic metabolizing enzymes: A feedforward control mechanism for potential hormetic responses. Toxicology and Applied Pharmacology, 237, 345–356. doi:10.1016/j.taap.2009.04.005
- Zhang, Q., Pi, J., Woods, C.G., & Andersen, M.E. (2010). A systems biology perspective on Nrf2-mediated antioxidant response. Toxicology and Applied Pharmacology, 244, 84–97. doi:10.1016/j.taap.2009.08.018
- Zhang, Y., & Hood, W.R. (2016). Current versus future reproduction and longevity: A re-evaluation of predictions and mechanisms. The Journal of Experimental Biology, 219, 3177–3189. doi:10.1242/jeb.132183
- Zonana-Nacach, A., Barr, S.G., Magder, L.S., & Petri, M. (2000). Damage in systemic lupus erythematosus and its association with corticosteroids. Arthritis & Rheumatism, 43, 1801–1808. doi:10.1002/1529-0131(200008)43:8<1801::AID-ANR16>3.0.CO;2-O