
Abstract
The biologic effects of inorganic arsenic predominantly involve reaction of the trivalent forms with sulfhydryl groups in critical proteins in target cells, potentially leading to various toxicologic events including cancer. This mode of action is a threshold process, requiring sufficient concentrations of trivalent arsenic to disrupt normal cellular function. Nevertheless, cancer risk assessments for inorganic arsenic have traditionally utilized various dose-response models that extrapolate risks from high doses assuming low-dose linearity without a threshold. We present here an approach for a cancer risk assessment for inorganic arsenic in drinking water that involves considerations of this threshold process. Extensive investigations in mode of action analysis, in vitro studies (>0.1 µM), and in animal studies (>2 mg/L in drinking water or 2 mg/kg of diet), collectively indicate a threshold basis for inorganic arsenic-related cancers. These studies support a threshold for the effects of arsenic in humans of 50–100 µg/L in drinking water (about 65 µg/L). We then evaluate the epidemiology of cancers of the urinary bladder, lung, and skin and non-cancer skin changes for consistency with this calculated value, focusing on studies involving low-level exposures to inorganic arsenic primarily in drinking water (approximately <150 µg/L). Based on the relevant epidemiological studies with individual-level data, a threshold level for inorganic arsenic in the drinking water for these cancers is estimated to be around 100 µg/L, with strong evidence that it is between 50 and 150 µg/L, consistent with the value calculated based on mechanistic, in vitro and in vivo investigations. This evaluation provides an alternative mode of action-based approach for assessing health-protective levels for oral arsenic exposure based on the collective in vitro, in vivo, and human evidence rather than the use of a linear low-dose extrapolation based on default assumptions and theories.
1. Introduction
Inorganic arsenic is a naturally occurring element present in the earth's crust and occurs in soil and water by various processes. Ultimately, some inorganic arsenic enters into the food chain in both plants and animals (Cullen Citation2008; Cullen and Reimer Citation2017). The inorganic form is metabolized by various organisms to a variety of organic metabolites, and thiolation can occur by chemical reaction with hydrogen sulfide. Inorganic arsenic has long been known to be associated with a variety of toxicities, both acute and chronic. Among the chronic effects that have been noted, the earliest and most well-known are the effects on skin, including hyperkeratosis and hypo- and hyperpigmentation, most notably arising on the palms and soles in contrast to similar lesions occurring in sun-exposed areas of the body secondary to ultraviolet radiation (Cullen Citation2008; Cullen and Reimer Citation2017). These skin changes are known as either arsenicosis or arseniasis. These benign lesions occurred following ingestion of a variety of solutions that had been developed over time as possible therapies (Fowler's solution, Gay's solution), but an association of these lesions with the evolution of skin cancer was first noted by Jonathan Hutchinson in 1887 (Cullen Citation2008). He reported five cases of cancer arising from these skin keratoses. These skin changes and skin cancer have been documented following a variety of arsenical therapeutic exposures, including the anti-syphilitic arsenical drugs developed by Ehrlich in the early 20th century.
Subsequently, the relationship of these skin changes, including cancer, to high levels of inorganic arsenic in the drinking water was documented in a variety of studies. The relationship was particularly well-documented in populations in Taiwan (Tseng et al. Citation1968; Cheng et al. Citation1988). However, it was not until 1985 that detailed epidemiologic evaluations of these populations showed that internal cancers were possibly related to inorganic arsenic exposure in the drinking water (Chen et al. Citation1985, Citation1986, Citation1988; Chen et al. Citation2016). Most notable was the relationship with urinary bladder cancer, which has subsequently been documented in a wide variety of populations, particularly in Asia and in Latin America (NRC Citation1999, Citation2001). Exposure to inorganic arsenic (mostly as arsenic trioxide) in various occupational settings, particularly in the mining industry, was first associated with an increased incidence of lung cancer in 1974 (Ott et al. Citation1974), although previous studies had suggested a relationship (IARC Citation1973). An increased risk of lung cancer related to a high level of inorganic arsenic in the drinking water has also been documented beginning with the studies in southwest Taiwan (Chen et al. Citation1985). Thus, it is now well-accepted that exposure to high levels of inorganic arsenic in the drinking water, and high levels of arsenicals in various therapeutic modalities and in various occupations, are associated with an increased risk of various types of cancers, most notably, skin, urinary bladder, and lung (IARC Citation1980, Citation2009).
Some studies suggest a relationship between inorganic arsenic exposure and cancers of the kidney, liver, and prostate, but these associations, if any, are much less well documented, and may be confounded by other factors (e.g. hepatitis for liver cancer). However, a study by Ferreccio et al. (Citation2013) in a Chilean population demonstrated that the increased risk of kidney cancer was related to an increased risk of kidney pelvis urothelial carcinomas, not renal cell carcinomas. These urothelial tumors arise from the same epithelium that extends from the kidney pelvis to the ureters to the urinary bladder, and they are likely the result of similar processes as those leading to the increased incidence of urinary bladder tumors in populations exposed to high levels of inorganic arsenic in the drinking water.
The relationship of inorganic arsenic in the drinking water to various cancers is well-documented at high exposure levels, generally >200 µg/L (ppb), but the risk at lower exposures is less clear. Nevertheless, the epidemiologic evidence has strongly suggested a threshold for the relationship between inorganic arsenic exposure and cancer (Abernathy et al. Citation1996; Mink et al. Citation2008; Cohen et al. Citation2013; Tsuji, Alexander et al. Citation2014; Lamm et al. Citation2015; Zhou and Xi Citation2018), and the various chemical and biological effects of inorganic arsenic in biological systems support the possibility of a threshold (Snow et al. Citation2005; Gentry et al. Citation2010, Gentry, Clewell et al. Citation2014; Gentry, Yager et al. Citation2014; Cohen et al. Citation2013). A threshold for the epidemiologic findings for lung cancer and inhalation exposure to inorganic arsenic has also been described by Lewis et al. Citation2015, and supported by a mode of action evaluation. Despite these observations and mechanistic underpinnings, the cancer risk assessment for inorganic arsenic in the drinking water has usually involved a linear, non-threshold dose-response, indicating risks in excess of regulatory guidelines even for drinking water exposures of 10 µg/L and lower (USEPA Citation1995, Citation2006, Citation2008, Citation2010; NRC Citation2001; JECFA Citation2011; FDA Citation2016)
In 1973, the International Agency for Research on Cancer Monograph Program evaluated inorganic arsenic and classified it as a known human carcinogen based mostly on reports of epidemiology studies related to skin cancer and a few reports suggesting a relationship occupationally to lung cancer (IARC Citation1973). At that time, there was little in the literature regarding animal studies, and very little was known about the mechanisms involved in inorganic arsenic carcinogenesis, although numerous hypotheses had been suggested.
In the 1980s, the United States Environmental Protection Agency (USEPA) reviewed the carcinogenicity of inorganic arsenic and developed a cancer slope factor for risk assessment of ingested arsenic based on linear extrapolation of skin cancer risk at high doses (mid-points of three dose groups: 170, 470, 800 µg/L) in an area of southwestern Taiwan with elevated arsenic levels in well water (USEPA Citation1984, Citation1988). USEPA subsequently reevaluated the 1975 inorganic arsenic drinking water standard, and in 2001 made a determination of a Maximum Contaminant Level (MCL) of 10 µg/L in the drinking water (66 CRF 6975) using a low-dose linear, non-threshold extrapolation model based primarily on the studies from southwestern Taiwan (NRC Citation1999, Citation2001). In 2010, a revised evaluation was presented by the USEPA suggesting an even steeper dose-response slope for cancer risk, but this assessment was subsequently withdrawn and has not been finalized (USEPA Citation2010). At present, the MCL for inorganic arsenic in the drinking water in the United States (U.S.) remains at 10 µg/L. A recent evaluation by Lynch et al. (Citation2017a, Citation2017b) calculated a somewhat lower risk assessment than proposed by USEPA in 2010, based on meta-regression analysis of epidemiological studies of lung and bladder cancer. Although the analysis performed by Lynch et al. (Citation2017a, Citation2017b) was ultimately based on a low-dose linear, non-threshold regression model, they noted that the association at low doses was largely determined by the relationship at high doses and detailed the evidence for a possible non-linear threshold approach to the risk assessment for inorganic arsenic (Cohen Citation2018a).
The intent of our study is to examine the mode of action basis for a threshold for oral exposure to inorganic arsenic leading to an increased risk of cancer, followed by the evidence from in vitro and in vivo animal studies regarding potential tissue concentration and dose around which such a threshold might occur. An estimated value for a threshold for inorganic arsenic in the drinking water is calculated based on extensive mechanistic, in vitro and in vivo investigations. We then evaluate the consistency of this threshold with the epidemiology literature by focusing on low-level arsenic exposure studies, e.g. drinking water exposures less than approximately 150 µg/L, and the risk of cancers of the lung, urinary bladder, and skin (with some consideration of evidence from skin arsenicosis).
2. Mode of action
The underlying basis for any assessment of the shape of the dose-response for cancer risk at low doses must first consider the mode of action for carcinogenesis. A number of publications have previously reviewed the mode of action of inorganic arsenic (Kitchin and Wallace Citation2008a; Gentry et al. Citation2010; Gentry, Yager et al. Citation2014; Gentry, Clewell et al. Citation2014; Kitchin and Conolly Citation2010; Cohen et al. Citation2013; Lynch et al. Citation2017a). These sources and the literature cited were reviewed, along with subsequently published literature.
2.1. Metabolism and kinetics
The understanding of mode of action is of fundamental importance for any biological effect of inorganic arsenic and first requires an evaluation of its chemistry, metabolism, and kinetics. Inorganic arsenic exists in the environment predominantly as the +5 (arsenate) and +3 (arsenite) oxidation states. In addition, a variety of organic arsenicals exist in the environment, including methylated arsenicals (monomethyl arsonic acid, MMAV, dimethyl arsinic acid, DMAV), a variety of arsenosugars and arsenolipids, and arsenobetaine and arsenocholine (Cullen Citation2008; Thomas Citation2015). Arsenobetaine and arsenocholine are considered biologically inactive and are minimally metabolized to DMAV (Thomas Citation2015). Arsenocholine can be metabolically converted to arsenobetaine (Marafante et al. Citation1984; Thomas Citation2015). Evidence for arsenobetaine being metabolized to a limited extent to DMAV and other metabolites has been reported in some species, but the evidence for humans is limited and contradictory (Thomas Citation2015). Furthermore, the metabolism in mammals appears to occur via gastrointestinal flora, not mammalian enzymes (Thomas Citation2015). The other forms of arsenic have the potential to be metabolized when exposure occurs in mammalian systems (Thomas Citation2015).
In oxygen-rich environments, inorganic arsenic is present primarily as arsenate (+5), which is the most common form present in drinking water (Cullen Citation2008; El-Masri and Kenyon Citation2008; Thomas Citation2015). Arsenite (+3) can also be present in the environment, particularly under anaerobic conditions. After exposure through drinking water or diet, arsenate is rapidly transported across the gastrointestinal tract, although some can be reduced in the gastrointestinal tract before absorption (Thomas Citation2010, Citation2015). Once absorbed, much of the arsenate is reduced to arsenite by several enzymes present in liver and other tissues, including the blood. It then undergoes oxidative methylation () to the monomethyl form, MMAV, which is then reduced to monomethylarsonous acid (MMAIII) and again oxidatively methylated to the dimethyl form (DMAV). DMAV can also be reduced to dimethylarsonic acid (DMAIII) and then further methylated to the trimethyl arsenic oxide (TMAVO) (Le et al. Citation2000; Cullen Citation2008; Thomas Citation2015). Methylation is primarily performed in mammalian systems by the enzyme arsenic (+3 oxidation state) methyltransferase (As3mt) (Chen, Arnold et al. Citation2011; Thomas Citation2015). The methylated forms and non-methylated inorganic arsenic are excreted predominantly in the urine (El-Masri and Kenyon Citation2008; Kenyon et al. Citation2008; El-Masri et al. Citation2018).
Figure 1. Metabolism of inorganic arsenic through progressive reductions and then oxidative methylations. The trivalent forms can react with sulfhydryl groups producing biologic effects. Although formation of TMAVO readily occurs in rodents, its formation is limited in humans unless exposed to very high (toxic) levels of inorganic arsenic.
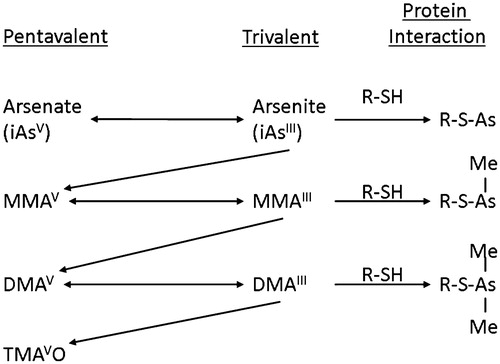
There are species differences in arsenic metabolism (Drobna, Walton, Paul et al. Citation2010; Thomas Citation2015) and interindividual differences (Drobna et al. Citation2004). DMAV is metabolized to TMAVO in rodents to an appreciable extent, particularly in rats. In contrast, DMAV is a poor substrate for the human enzyme As3mt, consequently, little TMAVO is present in human urine unless one is exposed to very high levels of inorganic arsenic. Under most circumstances, TMAVO is not detectable in human urine (Lu et al. Citation2003; Thomas Citation2015).
Transport of arsenicals is also dependent on oxidative state and whether they are in inorganic or organic form (Drobna, Walton, Harmon et al. Citation2010; Cohen et al. Citation2013; El-Masri et al. Citation2018). Arsenate can be transported by a number of transporters in various tissues, primarily by those involved in phosphate transport. In contrast, arsenite is readily transported by a variety of transporters in various cell systems, as are the trivalent methylated forms of arsenic. The pentavalent methylated forms of arsenic are poorly transported across cell membranes.
During the past decade, thiolated arsenicals have been identified, with one or more of the oxygens present in the various arsenicals replaced by a sulfur atom (Pinyayev et al. Citation2011). Thiolated arsenicals predominantly form non-enzymatically (chemically) by reaction of arsenicals with hydrogen sulfide (H2S) in the gastrointestinal tract (Pinyayev et al. Citation2011; Rehman and Naranmandura Citation2012; Thomas Citation2015). The microbiome of the gastrointestinal tract can influence arsenic metabolism, quite possibly by affecting sulfur-forming bacterial composition. Furthermore, arsenicals can influence the microbiome (Lu et al. Citation2013, Citation2014; Coryell et al. Citation2018). For reasons that are unclear thiolated arsenicals whether pentavalent or trivalent are readily transported across cell membranes. However, once inside the cell, it appears that the thiolated forms are rapidly reduced to the trivalent oxygenated forms, which are toxic (Suzuki et al. Citation2010; Rehman and Naranmandura Citation2012; see below).
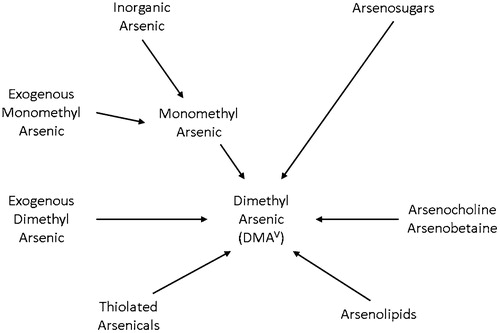
Arsenosugars from ingested foods appear to be metabolized primarily to dimethylarsinic acid (DMAV) and excreted in the urine (Thomas Citation2015; Thomas and Bradham Citation2016) (). Arsenolipids can also be metabolized to DMAV and excreted in the urine (Thomas and Bradham Citation2016). This can add significant confounding to assessments of human exposures of inorganic arsenic based on urinary levels of DMA1. This could be the explanation as to why DMA in the urine has been found to be predominantly from inorganic arsenic in the drinking water in populations exposed to high levels of inorganic arsenic (the levels are unclear, but clearly above 10 µg/L), whereas much of the urinary DMA at lower exposures of inorganic arsenic in the drinking water is derived from other sources of DMA, primarily dietary, such as DMAV, and especially arsenosugars and arsenolipids (Aylward et al. Citation2014; Thomas Citation2015; El-Masri et al. Citation2018).
Figure 2. Dimethylarsinic acid (DMAV) can be derived from numerous starting arsenicals. The DMAV is excreted predominantly in the urine. At high exposures of inorganic arsenic in the drinking water, most of the urinary DMAV is derived from the inorganic arsenic. However, at low exposure levels in the drinking water, these other sources of DMAV, primarily from food sources, will predominate.
Several variables can alter the metabolism and kinetics of inorganic arsenic and arsenicals in general, which could readily influence potential susceptibility to toxicity of exposure to arsenicals. A major factor is nucleotide polymorphisms in As3mt, which can influence the ability of the enzyme to metabolize inorganic arsenic to methylated forms (Fu et al. Citation2014; Li et al. Citation2017). This effect in its extreme occurs in the As3mt knockout mouse model developed by David Thomas and his colleagues at the USEPA (Drobna et al. Citation2009). In the knockout mice, inorganic arsenic is not methylated so the inorganic arsenic is retained longer in the knockout mouse than in the wild type mouse, leading to greater toxicity (Drobna et al. Citation2009; Hughes et al. Citation2010; Chen, Arnold et al. Citation2011; Yokohira et al. Citation2011). This is strong evidence that overall, methylation is a detoxifying process for inorganic arsenic. However, as described below, the intermediate trivalent methylated metabolites are also highly toxic, more than the inorganic trivalent arsenic itself (Petrick et al. Citation2000; Styblo et al. Citation2000; Cohen et al. Citation2002), such that less efficient metabolism results in more exposure to more toxic intermediates.
Other variables that contribute to differences in susceptibility to toxicity of inorganic arsenic include the available methylation capacity of an individual. This is predominantly influenced by dietary folate levels, but other aspects also can contribute (Gamble et al. Citation2005, Citation2006). Under certain rare circumstances, disorders of folate metabolism can influence this. However, such uncommon disorders have serious health consequences by themselves and are not considered in assessing risks associated with arsenic exposure in drinking water, given that they are not representative of the general population (Fraser et al. Citation2016).
2.2. Reaction with sulfhydryl moieties
The biologic effects of inorganic arsenic result predominantly from the chemical interaction of trivalent arsenicals with sulfhydryl groups, mainly in critical proteins in tissues (Kitchin and Wallace Citation2005, Citation2006, Citation2008b; Cohen et al. Citation2013) (). Reaction with smaller thiolated chemicals, such as glutathione, also can lead to toxicity, although there is an ample reserve of glutathione in most cells, considerably higher than the amounts that interact with trivalent arsenicals, even with high exposures. Glutathione is present in most cells at levels of 1–2 mM, and up to 10 mM in some cells such as hepatocytes (Forman et al. Citation2009). This contrasts with trivalent arsenical concentrations of less than 10 µM, even with significant toxicity. Arsenate itself, when the concentration is high, also can have effects on cells related to its substitution for phosphate, particularly in the process of oxidative phosphorylation (Aposhian Citation1997; Cullen Citation2008). This may be a major contributor to the acute toxicity of exposure to inorganic arsenic, although its relevance to the chronic effects of inorganic arsenic, such as cancer, is less likely.
Figure 3. Inorganic arsenic can be metabolized to thioarsenicals and methylated arsenicals. Ultimately trivalent arsenicals are generated inside the cell that can react with sulfhydryl groups. Reaction with critical cellular proteins will produce biologic effects. These effects are all non-cancerous. However, the cytotoxicity produced in epithelial cells, such as skin, lung, and urinary bladder, will lead to regenerative cell proliferation. If prolonged, this ultimately leads to an increased risk of these cancers.
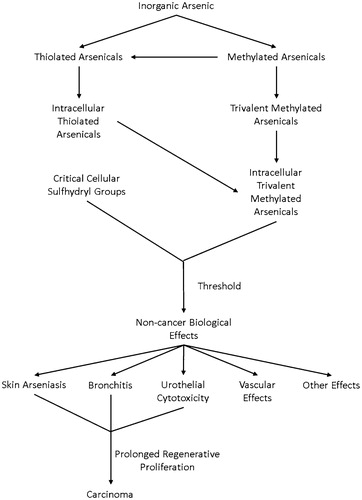
The reaction of trivalent arsenicals with sulfhydryl groups produces a carbon-arsenic bond that is generally thermodynamically stable. Thus, the reaction with many proteins produces essentially an irreversible binding to that sulfhydryl group (Lu et al. Citation2004, Citation2007; Kitchin and Wallace Citation2006, Citation2008b).
To examine the biologic effects of the reaction between trivalent arsenicals and sulfhydryl groups, extensive investigation of the reaction of trivalent arsenicals with a cysteine present in the alpha chain of rat hemoglobin has been conducted (Lu et al. Citation2007). There is a free cysteine in rat hemoglobin that is not present in the hemoglobin of most other species, including humans. In the rat, this interaction with hemoglobin results in accumulation of arsenic in red blood cells, which essentially has the half-life of the red blood cells themselves (Aposhian Citation1997). Interestingly all trivalent arsenicals whether arsenite, MMAIII, or DMAIII, reacted in vitro with this cysteine in purified rat hemoglobin. In contrast, when the rat was orally exposed to inorganic arsenic, MMAV, or DMAV, the only form of arsenic detected bound to rat hemoglobin was DMAIII (Lu et al. Citation2004, Citation2007). This is but one example of the precaution that must be exercised in extrapolating from in vitro to in vivo systems.
Nevertheless, the species difference in binding to hemoglobin exemplifies an important issue in extrapolating toxicities between species. Available sulfhydryl groups are present in a variety of proteins, especially zinc finger proteins and others that are important in the regulation of various cell processes, and the available free sulfhydryl groups present in these proteins can vary significantly between species (Kitchin and Wallace Citation2005, Citation2006, Citation2008b). Furthermore, which proteins are available will differ within tissues, likely serving as the basis for differences in tissue specificity following exposure to high levels of inorganic arsenic and other arsenicals.
To understand the biologic effects of inorganic arsenic and other arsenicals, it is essential to take into account these differences in metabolic capacity and kinetics, especially the availability of sulfhydryl groups in critical proteins in specific tissues, which can vary between species.
2.3. Trivalent arsenicals and toxicity
As indicated above, inorganic arsenic is methylated to the mono and dimethyl forms (and trimethyl in rodents), that serves primarily as a detoxifying process leading to excretion of the substances predominantly in the urine. However, as part of this process, trivalent forms of arsenic, including MMAIII and DMAIII, are formed (Le et al. Citation2000; Cullen Citation2008). The actual enzymatic process remains unclear, but increasingly it appears to involve a non-oxidative model with methylation steps occurring with arsenic bound to the enzyme, without the involvement of glutathione (Packianathan et al. Citation2018). This is also supported by the observations by Thomas (Citation2015) that the process is not influenced by glutathione concentrations. In various in vitro systems, these methylated trivalent species are more toxic than inorganic trivalent arsenic (Petrick et al. Citation2000; Styblo et al. Citation2000; Thomas et al. Citation2001; Cohen et al. Citation2002). Trivalent inorganic arsenic itself is highly toxic to cells, however, the trivalent methylated forms are usually 2 to 5 times more toxic than inorganic arsenic depending on the cell system. To estimate the overall toxicity of arsenicals, the amount of total trivalent arsenic available in cells is critical, along with which proteins they are able to interact. Differences that might occur between in vitro and in vivo exposures need to be addressed. A small part of this pool of trivalent arsenicals could be derived from the intracellular uptake of thiolated arsenicals, whether trivalent or pentavalent, since they are converted rapidly to the trivalent oxygenated forms (Suzuki et al. Citation2010; Pinyayev et al. Citation2011). The concentration for cytotoxicity of the trivalent arsenicals in various in vitro systems varies from 0.1 to 10 µM (Gentry et al. Citation2010). In contrast, the pentavalent methylated arsenicals (MMAV, DMAV, TMAVO) are cytotoxic at millimolar concentrations in vitro (Cohen et al. Citation2002; Dodmane, Arnold, Muirhead et al. Citation2014), concentrations that are not attainable in vivo.
A key to understanding the basis for a threshold for inorganic arsenic carcinogenicity on the biologic effects of arsenicals is the reaction with sulfhydryl groups. Small molecules, such as glutathione, are present in ample reserve in cells, so it is unlikely that they will be depleted to a significant extent, even with relatively high exposures to inorganic arsenic. To maintain cellular and tissue homeostasis, proteins are continuously turning over but vary in their half-lives. Structural proteins such as collagen tend to turn over slowly, with a half-life of years. In contrast, regulatory proteins such as transcription factors, enzymes related to DNA synthesis and repair, and proteins involved in numerous cell functions, have half-lives of minutes to hours (Doherty et al. Citation2009; Bojkowska et al. Citation2011; Hinkson and Elias Citation2011). Since regulatory proteins are of particular interest regarding carcinogenic mechanisms, the rapid turnover will assure the presence of functional protein until adequate amounts of trivalent arsenicals have reacted with them to actually deplete their levels below critical functional levels. Once these levels have been superseded, a biologic response such as the various arsenic associated toxicities will occur.
An indication of the capacity of proteins to handle arsenic binding is illustrated by the reaction of trivalent arsenicals to rat hemoglobin ( Lu et al. Citation2004, Citation2007). Rat hemoglobin has an unique cysteine in the alpha chain to which trivalent arsenicals can bind. The bound arsenic on the hemoglobin is retained for the life of the red blood cells in which it occurs (30–90 days). Even though a relatively large amount of arsenic can be bound to the hemoglobin, there is no effect on the overall functioning of the red cells since there are many times more hemoglobin molecules available that will not have the arsenic bound and new hemoglobin is constantly being synthesized.
Based on the extensive reviews of Gentry et al. (Citation2010); Gentry, Clewell et al. (Citation2014) and Gentry, Yager et al. (Citation2014), the critical concentration for trivalent arsenicals to cause biological effects based on in vitro studies is >0.1 µM. This is a highly conservative estimate since in vivo it is likely that with various protective mechanisms, such as metabolism and cell protection processes such as mucin, glycosaminoglycans, cell junctions, etc., a higher concentration is required to produce a biologic effect. Nevertheless, >0.1 µM is a reasonable estimate for the cellular concentration required for obtaining a biologic effect such as the various toxicities.
It has become apparent that the exogenous exposure to inorganic arsenic leads to biologic effects in humans at exposure levels lower than that appears to be necessary for animal systems, particularly with respect to carcinogenicity. Thus, increased incidences of cancer are detectable at drinking water levels above ≈100 µg/L, whereas in the animals a biologic response is generally obtained only at exposure levels of mg/L. This appears predominantly to be due to differences in toxicokinetics between the animal species, rather than being related to the susceptibility of human cells versus animal cells (Clewell et al. Citation2007; El-Masri and Kenyon Citation2008; Kenyon et al. Citation2008; Dodmane et al. Citation2013; El-Masri et al. Citation2018). The cytotoxicity of various trivalent arsenicals in rat and/or mouse cells is similar to human cells of the same tissues. In these in vitro systems, the biologic responses occur at similar concentrations of the trivalent arsenicals, usually within two- to four-fold differences, but always less than ten-fold among species (Dodmane et al. Citation2013). If anything, the human cells tend to be less susceptible to the biologic effects of the trivalent arsenicals in these in vitro systems when they are compared to rat or mouse cells. This has been true for established cell lines and for primary human cells (Gentry et al. Citation2010; Yager et al. Citation2013).
2.4. Mode of action for arsenical carcinogenesis
Several modes of action have been suggested to explain the carcinogenicity of arsenicals, including direct genotoxicity (i.e. DNA reactivity), indirect genotoxicity (i.e. non-DNA reactivity), oxidative stress, epigenetic changes, and cytotoxicity with regenerative proliferation.
2.4.1. Reaction with DNA
The only mode of action that is compatible with a linear extrapolation without a threshold is direct genotoxicity involving direct interaction of the chemical with DNA, leading to adduct formation, mutation, and ultimately carcinogenicity (Cohen and Arnold Citation2011). Chemicals of this type include the polycyclic aromatic hydrocarbons, aromatic amines, N-nitrosamines, aflatoxins, and others. However, Nesnow et al. (Citation2002) clearly demonstrated that arsenicals do not react directly with DNA, and based on chemical principles, such a reaction is highly unlikely. Some investigators have suggested that there is free radical formation by arsenicals, but this occurs only under extreme conditions with high concentrations and specific biologic circumstances (Nesnow et al. Citation2002; Lantz and Hays Citation2006), none of which are likely to occur in viable mammalian organisms.
2.4.2. Indirect genotoxicity
It is well accepted that indirect mechanisms of damage to DNA that occur by interactions with targets or processes other than DNA are expected to show nonlinear, threshold dose responses (Kirkland and Müller Citation2000; MacGregor et al. Citation2015). There is evidence that indirect genotoxicity can occur by a variety of means, and there is evidence that such effects could occur in mammalian cellular systems exposed to trivalent arsenicals. However, these involve interaction with proteins and will have a threshold effect. The two most plausible indirect processes that could lead to indirect genotoxicity are inhibition of DNA repair and interaction with the proteins involved in the formation of the mitotic spindle, i.e. tubulin (Cohen et al. Citation2013). Interaction with a variety of DNA repair enzymes has been demonstrated (Kitchin and Wallace Citation2008b; Andrew et al. Citation2009; Ebert et al. Citation2011; Zhou et al. Citation2011; Faita et al. Citation2013), although these studies involved predominantly in vitro systems and especially involved purified enzymes. The role that DNA repair and mitotic-spindle interference play as causal events in arsenical carcinogenesis are not established. Nevertheless, given that these effects involve interactions with proteins, a threshold effect is expected, as described above. Furthermore, it is unclear if these effects would occur at concentrations attained in vivo that would not be overtly toxic to humans.
Likewise, interactions with tubulin will certainly have a threshold effect (Kitchin and Wallace Citation2008b). If this actually occurs at high enough exposure levels, it is likely to result in cell death. However, in whole tissues and in intact organisms there is no evidence that such a reaction with tubulin with consequent inhibition of the formation of the mitotic spindle actually occurs. A number of substances are known to interact with the mitotic spindle, such as colchicine, but the result of such an interaction is metaphase arrest (Garland Citation1978). Colchicine is the substance used in cytogenetic evaluations and in the assessment of various indirect genotoxicity parameters. There is no evidence in animal studies or in humans that this actually leads to a carcinogenic effect. Colchicine is widely used in human medicine in the treatment of gout (Chen and Schumacher Citation2008), with no evidence of a carcinogenic effect.
Thus, although there is a possibility for indirect genotoxicity, if it does occur it will only be at very high concentrations. Such concentrations are higher than those that would be attained systemically in animals or humans. Therefore, it is unlikely that this is the basis for the carcinogenicity of arsenicals either in animals or in humans, particularly at lower doses.
A variety of in vitro and in vivo studies have demonstrated genotoxicity using primarily assays involving micronucleus formation or chromosomal aberrations (Kligerman et al. Citation2003; Kligerman and Tennant Citation2007). Studies evaluating direct mutagenesis such as the Ames assay, have generally been negative, particularly those done under guidelines currently in place (Nesnow et al. Citation2002; Kligerman and Tennant Citation2007; OECD 2015; EFSA Scientific Committee Citation2017). A recent in vivo assay in gpt delta transgenic mice showed a suggestion of a positive response in liver to arsenite in the drinking water (Takumi et al. Citation2014). This isolated finding is inconsistent with the lack of mutagenicity observed in a variety of assays and with the lack of DNA reactivity of arsenicals. The dose used was 85 ppm arsenite which can produce liver toxicity in some mouse strains (Yokohira et al. Citation2011). No evaluation for liver toxicity was performed in this study. A major difficulty in interpreting these genotoxicity assays, particularly those in vitro are the high concentrations that are needed to obtain a positive result. In general, these are well above the 10 µM concentration that is lethal to cells in vitro. A difficulty in interpreting the in vitro assays is that the assessment of cytotoxicity may be performed only after a few hours of exposure whereas cell death may not be evident for 3 to 5 days of exposure. Nevertheless, the cytotoxic process has already begun and is likely the basis for the genotoxicity (Styblo et al. Citation2000; Dodmane et al. Citation2013). These high concentrations also suggest the potential for interaction with critical proteins, such as DNA repair enzymes and tubulin, that could produce indirect genotoxic effects, but that has not been adequately evaluated.
2.4.3. Evaluation of genotoxicity in humans
In vivo assessment of indirect genotoxicity has also been evaluated, both in animal models and in humans. In animal models, the exposure levels are always high (Kligerman and Tennant, Citation2007). In humans, the major endpoint that has been utilized to evaluate genotoxicity has been micronucleus formation, particularly evaluating urothelial cells that have been exfoliated in the urine. These studies in humans have numerous shortcomings which have been previously described (Cohen, Chowdhury et al. Citation2016).
A major shortcoming of these human studies has been the lack of a clear dose response, despite large increases in exposure. As an example, in a study by Basu et al. (Citation2004) in which they evaluated exfoliated urothelial cells, buccal mucosa squamous cells, and blood lymphocytes from populations exposed to various drinking water levels of inorganic arsenic, there was little or no variability in the number of micronuclei per 1000 cells at different exposure levels. Three groups with exposure levels of 50–150, 151–250, or >250 µg/L inorganic arsenic in the drinking water were evaluated. In urothelial cells, the number of micronuclei per 1000 cells was 6.30, 6.48, and 6.98, respectively; in buccal cells, it was 5.75, 5.78, and 5.90; and in blood lymphocytes, it was 9.01, 9.39, and 9.42. Standard errors were not presented in the publication and no group with low drinking water exposures (<10 µg/L) was evaluated. None of these changes were statistically significant.
Furthermore, the assessment of inorganic arsenic exposure has been limited. Either drinking water levels or measurement of urinary arsenic was used, with all of the shortcomings described below for the epidemiologic studies. Frequently, total arsenic rather than speciated arsenic in urine has been measured, which is particularly difficult to interpret and can be greatly influenced by organic arsenic forms from the diet. Even speciated urinary arsenic measures may also be influenced by DMA or by DMA precursors in the diet. Furthermore, the evaluation of urinary arsenic levels has been performed at a single point in time, with no control for time of day or any follow-up. Although some have suggested that urinary arsenic levels are reasonably stable, other studies have shown that they are highly variable, not only from day to day but even during the same day. Variation over spans of months and years is likely to be considerable (Wang et al. Citation2016). In addition, the populations that are being compared are at different locations with a large number of confounding factors, including wide variation in the nutritional status of the individuals. This is particularly important regarding folate and other influences on methylation status (Gamble et al. Citation2005, Citation2006).
A further confounding factor is exposure to tobacco products. This is generally evaluated by self or third-party reporting, without verification of nicotine exposure, such as by measurement of cotinine in the urine. Furthermore, tobacco exposure may only be an assessment of cigarette smoking whereas many of the populations, such as in West Bengal and Bangladesh, have considerable oral tobacco exposure through betel quid, which could further confound the findings. Betel quid contains substantial amounts of nicotine. Recent studies have shown that nicotine orally administered to mice and rats produces a similar cytotoxic and regenerative proliferative effect on the urinary bladder urothelium as inorganic arsenic (Dodmane, Arnold, Pennington et al. Citation2014; Suzuki et al. Citation2018).
Other variables that are not adequately taken into account in the assessment of micronuclei in urothelial cells include the fact that the cells that are exfoliated are dead and have undergone considerable autolysis in the urine. In addition, some of the micronuclei that have been counted in individuals exposed to high levels of inorganic arsenic could be intracytoplasmic inclusions. These inclusions have been demonstrated in mice exposed to inorganic arsenic and in exfoliated human urothelial cells from patients that had been treated with extremely high doses of arsenic trioxide for the treatment of promyelocytic leukemia (PML) (Wedel et al. Citation2013; Dodmane, Arnold, Muirhead et al. Citation2014). In mice and in humans, it appears that these inclusions contain inorganic arsenic and serve as a reservoir for binding inorganic arsenic so that it cannot induce toxicity in the cells, a means of handling excess inorganic arsenic by these cells. This was particularly well demonstrated in the As3mt knockout mice administered inorganic arsenic (Dodmane, Arnold, Muirhead et al. Citation2014). Furthermore, these inclusions are not found in rat urothelium (Dodmane, Arnold, Muirhead et al. Citation2014), and rats do not have evidence of DNA damage or micronuclei in the urothelium following oral administration of arsenate or DMAV in drinking water (Wang et al. Citation2009). Overall, given the significant limitations of the studies in humans, it is unlikely that the inorganic arsenic is acting as a clastogen.
2.4.4. Epigenetics and oxidative stress
Epigenetics has also been suggested as a mode of action for arsenic carcinogenicity (Smeester et al. Citation2011; Rager et al. Citation2017). However, the causal linkage and relationship of epigenetic mechanisms with arsenical carcinogenicity is poorly defined, and does not take into account that exposures to other environmental toxicants or dietary intake of food will lead to changes in the methylation pattern of DNA or the histone acetylation pattern, as protein expressions will be turned on and off to handle even normal metabolic and cellular processes (Goodman et al. Citation2010). To be related to carcinogenicity, it would have to be demonstrated that the epigenetic changes were irreversible, and this has not been adequately addressed. For the most part, epigenetics serves as a marker for biologic processes happening at a given time and does not provide an explanation for long term effects such as carcinogenicity.
Another mode of action that has been suggested is DNA damage secondary to oxidative stress (Kitchin and Wallace Citation2008a; Kitchin and Conolly Citation2010). This should be distinguished from activation of oxidation pathways detected by genomic assays, which is not necessarily indicative of DNA damage. Although oxidative stress and toxicity have been demonstrated repeatedly in various in vitro systems, these have generally not been validated in vivo exposures. In various in vitro studies, the exposures can readily produce oxidative stress, particularly at concentrations that are cytotoxic (Wei et al. Citation2005; Kitchin and Wallace Citation2008a; Kitchin and Conolly Citation2010). Most of the in vitro studies reporting oxidative stress as a factor have been performed at concentrations of arsenic above 10 µM, which, as described below, is lethal to cells. It is also higher than systemic or urinary concentrations attainable in animals or humans. Any oxidative damage likely would be a consequence of the toxicity rather than the cause. Co-administration of arsenic with various antioxidants in vitro inhibits the process, although this could be simply due to chemical interaction with the arsenical directly in some cases. Administration of such antioxidants has not blocked various biologic effects when evaluated in the in vivo setting (Wei et al. Citation2005; Suzuki et al. Citation2009). In the review of DMA by the USEPA Office of Pesticides Programs (OPP), it was concluded that the mode of action was sustained cytotoxicity and regenerative cell proliferation rather than reactive oxygen species (ROS)-induced DNA damage (USEPA Citation2006), consistent with the conclusions of the USEPA Science Advisory Board (USEPA Citation2007).
2.5. Cytotoxicity and regenerative proliferation
The most likely mode of action based on considerable evidence available is cytotoxicity with consequent regenerative proliferation (Cohen et al. Citation2013). This has been demonstrated specifically in the DMAV rat bladder cancer model, and it was accepted by the USEPA in its review of the DMAV pesticide registration (USEPA Citation2006) and by the USEPA SAB in its review (USEPA Citation2007). Evidence supporting this mode of action has been demonstrated for inorganic arsenic in a variety of animal models, in vitro systems, and in humans.
2.5.1. DMA-Induced urinary bladder cancer in rats
The most extensively investigated model for cytotoxicity and regenerative proliferation regarding arsenical carcinogenesis is the DMAV bladder cancer model in rats. This has been reviewed extensively (Cohen et al. Citation2006) and was accepted by the USEPA in its review of the DMAV pesticide registration in 2006 (USEPA Citation2006). It has been used as a model chemical for international and governmental training programs on applications of the mode of action/human relevance framework originally developed by a committee of the International Life Sciences Institute (ILSI) Risk Science Institute sponsored by the USEPA and Health Canada (Sonich-Mullin et al. Citation2001; Meek et al. Citation2003; Seed et al. Citation2005). This framework was extended internationally by the International Programme on Chemical Safety (IPCS) (Boobis et al. Citation2006, Citation2008) and has been widely used for evaluation of mode of action analyses of animal models and extrapolation to human relevance.
In the DMAv model, as described below, an increased incidence of urinary bladder tumors occurs in rats when it is administered in the drinking water or in the diet, but does not produce an effect in mice in a two-year bioassay. Based on extensive investigations, the mode of action was determined to be cytotoxicity with regenerative proliferation (Cohen et al. Citation2006). The key events were reduction of DMAV to DMAIII with excretion and concentration in the urine. This led to superficial cytotoxicity of the urothelium with regenerative proliferation and ultimately an increased incidence of tumors. Cytotoxicity and proliferation were increased at dietary doses as low as 10 mg/kg (10 ppm) of diet, with negative results at 2 mg/kg of diet. Urinary concentrations of DMAIII were >1 µM after treatment for 1 day with 100 mg/kg DMAV in the diet and ≈5 µM after treatment for 10 weeks (Cohen et al. Citation2002). After treatment for 25 weeks, the urinary concentration of DMAIII decreased to around 1.0 µM. In vitro investigations demonstrated that DMAIII was cytotoxic to immortalized rat (MYP3) and human (1T1) urothelial cells (Cohen et al. Citation2002; Dodmane et al. Citation2013) at a concentration similar to that resulting in cytotoxicity and other alterations in biologic parameters investigated in vitro in a variety of cell systems (Gentry et al. Citation2010; Gentry, Clewell et al. Citation2014; Gentry, Yager et al. Citation2014). There was a clear threshold response by assessment of the morphologic endpoints and based on the urinary concentration of the reactive metabolite, DMAIII.
Cytotoxicity with regenerative proliferation has been identified as the mode of action for numerous chemicals involving numerous target tissues, including liver (Meek et al. Citation2003), kidney (Lock and Hard Citation2004), forestomach (Proctor et al. Citation2018), small intestine (Haney Citation2015; Thompson et al. Citation2017), urinary bladder (Cohen Citation2018b), and others. Cytotoxicity is a well-accepted mode of action of toxicity with a threshold response that can then be used for estimating risk to humans. In the case of treatment with DMAV, as in many others, the cytotoxicity if prolonged, can lead to an increased risk of cancer. However, if the exposure is below the level that produces the cytotoxicity, then no cytotoxicity will occur and no tumors (Andersen et al. Citation2000).
2.5.2. Inorganic arsenicals and cytotoxicity
For inorganic arsenic, whether arsenate or arsenite, a similar cytotoxic response, and consequent regenerative proliferation are seen in the urinary bladder of rats and mice (Simeonova et al. Citation2000; Cohen et al. Citation2013; Arnold et al. Citation2014). However, in contrast to DMAV, prolonged administration of inorganic arsenic to rats and mice does not appear to produce a statistically significant increased incidence of urothelial tumors (see Section 4). This may be related to the attenuation of the hyperplastic response as the animal's age; the hyperplasia is minimal to absent after the age of 26 weeks (Arnold et al. Citation2014). Why the attenuation occurs has not been determined, but is likely related to toxicokinetic considerations.
In vitro evaluations of exposure to inorganic arsenic in rat urothelial cells produced a cytotoxic and regenerative effect similar to that observed in vivo (Cohen et al. Citation2013). Furthermore, the evaluation of the urothelium in the animals exposed to inorganic arsenic utilizing genomic analyses showed an initial change corresponding to cytotoxicity (at 2 weeks of administration) with later genomic changes indicative of a proliferative response (12 weeks of exposure) (Clewell et al. Citation2011). These transcriptomic findings correspond to the morphologic changes and immunohistochemical evaluation of proliferation in the bladder urothelium (Arnold et al. Citation2014).
A similar response was also seen utilizing primary urothelial cells from patients, with a mixture of various arsenicals leading to similar transcriptomic changes as observed in vivo (Yager et al. Citation2013). Primary human urothelial cells had a variation in response of only approximately three-fold, which is potentially useful information in the extrapolation to a quantitative assessment in humans. Again, one has to keep in mind that the in vitro changes will be conservative compared to in vivo since the cells in vitro will not have the normal protective processes present in a fully differentiated tissue such as the urothelium in the urinary bladder. In vitro systems have several aspects that need to be considered when extrapolating to in vivo systems. One important consideration is the loss of chemical metabolizing capabilities in the in vitro systems. A second consideration is the lack of full differentiation of cells in vitro. The in vitro epithelial systems frequently lack the protective barriers of the fully differentiated tissues in vivo, such as cell junctions, membrane protections (e.g. uroplakins in the urothelium), blockage of cell transport, production of protective materials (e.g. mucins, proteoglycans, glycosaminoglycans, etc.) as well as interactions with other cell types and products (e.g. inflammatory cells, cytokines, growth factors and inhibitors). Furthermore, established cell lines have abnormalities that allow them to grow indefinitely that do not occur in vivo. Such changes include abnormalities in p53, which influences DNA repair and cell growth, as well as alterations in other proteins involved in cell growth and proliferation. Thus, the in vitro studies will likely overestimate the risk to humans.
Investigations in other cell types produced similar results as in urothelial cells (Petrick et al. Citation2000; Styblo et al. Citation2000; Thomas et al. Citation2001; Vega et al. Citation2001; Dodmane et al. Citation2013). However, in vivo models to evaluate the in vitro findings with these other cell types, including bronchial epithelial cells and skin keratinocytes, are not available. Nevertheless, the concentrations of the various trivalent arsenicals required to produce cytotoxicity in vitro were similar across the different tissue types, and the transcriptomic response was similar in all three cell types (Dodmane et al. Citation2013). These studies involved established cell lines for urothelium and keratinocytes and primary cells for bronchial epithelium.
2.5.3. Cytotoxicity and carcinogenesis in humans
As described above, there is evidence in humans that cytotoxicity with consequent regenerative proliferation is the mode of action involved with arsenic-related tumors. The reason for the urothelium being a target for inorganic arsenic carcinogenesis is apparently based on the concentration and excretion of the trivalent forms in the urine. The reason that the skin and lung are also targets is likely related to high concentrations of sulfhydryl-containing proteins in skin keratin and in lung surfactant (Kishore et al. Citation2006) for binding trivalent arsenicals in blood. Lung cancer that arises from oral exposure may also differ in other ways than airborne delivery to the lung with occupational exposure by inhalation. An evaluation of lung cancer cases in cancer registry data for the arseniasis-endemic area of southwest Taiwan revealed that squamous cell carcinoma (but not adenocarcinoma or small cell carcinoma) was associated with arsenic exposure in drinking water, most prevalently at concentrations in excess of 640 µg/L in contrast to the greater prevalence of adenocarcinomas in historical copper smelter workers (Kuo et al. Citation2017). Although animal models similar to that for bladder cancer are lacking for lung and skin cancer from oral exposure to arsenic, in vitro data indicate the similar sensitivity to arsenic of bronchial epithelial cells, keratinocytes, and bladder epithelial cells.
Cytotoxicity with consequent regenerative proliferation is most clearly demonstrated in the skin, with the precancerous changes defined as hyperkeratosis and increased epidermal proliferation as the response to toxicity of the epidermis (Cohen et al. Citation2013). This is associated with increased proliferation, and eventually decreased differentiation of the epidermis as the lesion evolves toward actinic keratosis with dysplasia, carcinoma in situ, and eventually squamous or basal cell carcinoma. Interestingly, melanoma, another skin malignancy, does not appear to be increased with arsenic exposure, perhaps because it arises from melanocytes rather than keratinocytes and has a different molecular basis than squamous and basal cell carcinomas (Shain and Bastian Citation2016).
Likewise, epidemiologic investigations suggest that inflammatory changes in the lung (indicative of cytotoxicity), such as bronchitis (Mazumder Citation2007) and bronchiectasis (Mazumder et al. Citation2005; Mazumder Citation2007) are increased in response to exposure to high levels of inorganic arsenic in the drinking water, but this has not been investigated to the same extent as the skin arseniasis changes (Cohen et al. Citation2013). For the urinary bladder, there are no noninvasive techniques that can evaluate the precursor changes in the bladder, so this cannot be directly investigated in the human urothelium. However, in an industrial accident with exposures to extremely high levels of inorganic arsenic, resulting in a high mortality rate, several of the individuals developed hematuria, a clear sign of urothelial toxicity (Xu et al. Citation2008). The superficial urothelial toxicity observed in rats and mice administered inorganic arsenic is not detectable clinically in humans.
An important consideration in this mode of action is that it is a clear threshold event, requires prolonged exposure with a long latency period, and has been well documented in several human populations and with all three tumor sites related to high exposures of arsenicals. Most importantly, inorganic arsenic is not acting as a carcinogen directly on the target tissue but is producing toxicity, which is a non-cancerous endpoint (Cohen et al. Citation2013). However, under these circumstances and in tissues with prolonged exposure, this cytotoxicity, and regenerative proliferation evolves into an increased risk of cancer (). Based on these considerations, the risk assessment for inorganic arsenic effects on cancer and non-cancer endpoints can be evaluated the same, both involving thresholds.
Identification of this threshold for cytotoxicity leading to carcinogenicity requires an estimate of the amount of exposure from exogenous sources that will yield concentrations greater than 0.1 µM of trivalent arsenicals in the target tissues. This is most readily measurable for the urinary bladder urothelium, since urine is the medium through which the exposure occurs that leads to the cytotoxicity, and several studies have measured urinary arsenic concentrations and arsenic drinking water concentrations. Based on calculations described in Cohen et al. (Citation2013) and below, the estimated ingestion of inorganic arsenic in the drinking water, that will produce trivalent arsenicals concentrated and excreted in the urine at levels >0.1 µM is ≈50–100 µg/L.
3. Evidence of dose-response from in vitro investigations
Studies conducted in vitro provide important information on the likely tissue concentrations for the effects of inorganic arsenic in humans at environmentally relevant drinking water concentrations. These in vitro dose response results can be used together with the results from epidemiology studies to provide evidence regarding concentrations that are likely to be below the threshold for inorganic arsenic carcinogenicity. The use of in vitro data for this purpose as supported by evidence in vivo is consistent with the recommendation of the National Research Council (NRC Citation2013) that mode of action data for inorganic arsenic should be used to inform the shape of the dose-response curve in the low-concentration region.
Gentry et al. (Citation2010) reported the results of a comprehensive literature review focused on the evaluation and integration of gene or protein expression changes following exposures to inorganic arsenic compounds over a range of concentrations. The in vivo and in vitro data identified for the Gentry et al. (Citation2010) review were organized by dose/concentrations, species, tissue and cell type (primary, immortalized, tumor-derived), with the results also organized by functional category (i.e. oxidative stress signaling, proteotoxicity signaling, inflammatory signaling, cell cycle checkpoint control, DNA repair activities, and cell survival or cell death signaling). For each gene or protein expression change, the lowest concentration associated with a significant change was identified, and then a comparison of the changes by functional category and dose was conducted. In reviewing the available data for the different cell types, Gentry et al. (Citation2010) noted that a striking observation was the consistency in response, not only across cell lines (primary and immortalized) but also across different tissue types and species.
The results from studies with primary cells reported by Gentry et al. (Citation2010), the combined concentration-response information (at concentrations ranging from 0.005 to up to 100 µM) provided clear evidence of concentration dependence of arsenic effects on the expression of various genes. Responses at concentrations ≤0.1 µM inorganic arsenic indicated adaptive responses, while those studies in which exposures were between 0.1 and 10 µM resulted in potentially toxic responses in cellular control pathways associated with response to stress (to be distinguished from oxidative damage to DNA), proteotoxicity, inflammation, and proliferative signaling (). The results also suggested that the mode of action was arsenic-induced included inhibition of DNA repair processes in the cell, as had been previously suggested (Snow et al. Citation2005). The authors concluded that the gene changes observed across different mammalian cells and cells from different organs were consistent with a transition from one of adaptation in response to arsenic exposure at low concentrations to gene expression changes that reflect frankly toxic effects at higher concentrations, supporting a dose-dependent transition or nonlinear dose-response relationship with increasing exposures to arsenic.
Table 1. Dose-response for the in vitro effects of arsenic in primary cells.
To update the Gentry et al. (Citation2010) review, a search was conducted for in vitro studies reported with inorganic arsenic or its metabolites published since the Gentry et al. (Citation2010) comprehensive review was conducted, in order to identify any new in vitro studies that evaluated gene or protein expression changes in human bladder, lung or skin primary cells as a function of arsenic concentration. Keywords considered included: in vitro, arsenic, arsenite, arsenate, dimethylarsinous acid, dimethylarsonic acid, DMA, monomethylarsonous acid, monomethylarsonic acid, and MMA. Over 1000 articles were identified using combinations of the keywords.
As differences in response between primary, immortalized and cancer-derived cells have been reported, the screen of the available titles and abstracts was focused on identifying concentration-response studies conducted in primary or immortalized primary cells from the human bladder, lung or skin, which would be most appropriate for in vitro to in vivo extrapolation to estimate a potential threshold or transition concentration. A review of the available titles and abstracts indicated ≈30 in vitro studies conducted in human primary cells or immortalized primary cell lines (Supplementary material).
Overall, the results from these newer studies were consistent with the conclusions reached previously by Gentry et al. (Citation2010). Unfortunately, all but three studies only investigated single concentrations within the range of those reviewed by Gentry et al. (Citation2010) (0.05 to 100 µM), so they were not informative for characterizing the dose-response for cellular responses. One study compared global gene expression profiles of normal human epithelial keratinocytes exposed to arsenite, MMAIII or DMAIII at four concentrations (0.1, 1.0, 1.5, and 5 µM) for 24 h (Bailey et al. Citation2010). Unfortunately, this study did not include dose-response modeling of the genomic responses, but no differentially expressed transcripts were observed following arsenite or DMAIII exposures at 0.1 µM and responses to MMAIII at 0.1 µM were limited to anti-apoptotic signaling, cell-cell communication, and cell morphology.
The search also identified two studies that conducted exposures at multiple concentrations in primary human bladder urothelial cells (Yager et al. Citation2013) and lung cells (Efremenko et al. Citation2015) and performed benchmark dose analysis to identify a No Observed Transcriptional Effect Level (NOTEL). The Yager et al. (Citation2013) study was conducted to assess the genomic response in human primary urothelial cells from multiple individuals (n = 15) in which the cells were treated in vitro with mixtures of arsenite and its methylated metabolites (trivalent or pentavalent, total arsenic concentrations ranging from 0.06 to 18 µM) that were based on the proportion of arsenic and its metabolites reported in the urine of humans ingesting arsenic in drinking water. This study is unique in that the cells were exposed to a mixture of inorganic arsenic and methylated metabolites comparable to the in vivo situation, making direct comparisons between in vitro and in vivo exposures easier. All other studies in the published literature focused on exposures to arsenite or single metabolites.
Following incubation of the cells for 24 h with the mixture of arsenite and its metabolites, the observed genomic responses were consistent with the integrated in vitro data reported previously by Gentry et al. (Citation2010). A number of genes were identified that demonstrated a similar dose-response across subjects, including genes related to oxidative stress signaling (heme oxygenase-1 (HMOX1), thioredoxin reductase (TXNRD1), thioredoxin (TXN), metallothionine regulation (MT1E), protein folding (FKBP5), DNA damage sensing (DDB2), cell adhesion and growth regulation (LGALS8) and immune response (THBD)). Benchmark dose analyses on these gene expressions result in primary human bladder cells identified benchmark dose lower confidence limits (BMDLs) for the changes in these genes in the range of 0.09–0.58 µM for total arsenic in the trivalent arsenical mixtures (). BMDLs for the mixtures of arsenite and its pentavalent metabolites ranged from 0.35–1.7 µM (not shown). Benchmark doses (BMDs) and BMDLs only varied by an approximate factor of three across individuals, suggesting that the default factor of 3 typically used in human health risk assessments for interhuman variability in pharmacodynamics (IPCS Citation2005; USEPA Citation1994) would be adequately protective.
Table 2. Benchmark dose ranges for genes with a statistically significant dose-response trend in primary urothelial cells from most subjects after treatment with arsenite, MMAIII, and DMAIII (trivalent) mixtures.
In addition to the benchmark dose analysis, Yager et al. (Citation2013) conducted an alternative statistical method for analysis, the no statistical significance of trend method (NOSTASOT), which confirmed that NOTELs ranged from 0.18 to 1.8 µM total arsenic concentration for these same genes. This study provides strong evidence for a dose-dependent transition in response in the range of 0.1–1.0 µM. This study was the first to examine gene expression changes in primary urothelial cells from multiple human subjects and provided evidence for no observed transcriptomic effect levels in normal human cells exposed to biologically plausible concentrations of arsenic mixtures.
The Efremenko et al. (Citation2015) study was conducted as a complementary experiment to the Yager et al. (Citation2013) study; the concentrations used in Efremenko et al. (Citation2015) were the same as those in the human urothelial study (Yager et al. Citation2013). In addition to the arsenical trivalent mixture exposures, exposures to arsenic trioxide were also performed to compare responses for exposures of lung epithelial cells at the apical membrane from inhalation and exposures at the basal membrane from oral exposure. Similar analyses of the gene expression results were conducted as those in the Yager et al. (Citation2013) study for urothelial cells, focusing on those genes expressed most in common among cells from three individuals. Benchmark dose analysis confirmed similarity in the concentration-response relationship between lung and bladder epithelial cells, with comparable benchmark dose estimates across tissue types for the trivalent mixtures, as well as comparable benchmark dose estimates following exposures of lung cells to either the trivalent mixture or arsenic trioxide. The consistency of the genomic responses in human primary cells from two different tissues (bladder and lung) and for two different trivalent arsenic exposures (arsenite and its trivalent metabolites vs. arsenite alone) supports the usefulness of this data to characterize the dose response for the cellular effects of trivalent inorganic arsenic.
Because the cells in both Efremenko et al. (Citation2015) and Yager et al. (Citation2013) studies were treated with arsenic mixtures for 24 h, whereas humans are exposed to arsenic in drinking water throughout their lifetime, it is reasonable to question whether the dose-response for gene expression changes observed following short-term exposure are indicative of the dose-response for potential responses following chronic exposure. Thomas, Philbert et al. (Citation2013) and Thomas, Wesselkamper et al. (Citation2013) conducted studies with a number of compounds to evaluate this question and concluded that the dose-response for genomic alterations in short-term studies provides a conservative predictor of both cancer and noncancer outcomes in lifetime bioassays. In the case of arsenite, a study conducted in which mice were exposed to concentrations of arsenate in drinking water ranging from 0.2 to 50 ppm for 1 or 12 weeks of exposure (Clewell et al. Citation2011) demonstrated that benchmark doses for cellular responses did not decrease between 1 and 12 weeks of exposure.
A study conducted by Dodmane et al. (Citation2013) investigated gene expression changes in three human cell types (urothelial (1T1), keratinocyte (HEK001) and bronchial epithelial (HBE) cells) corresponding to the target organs for inorganic arsenic-induced cancer following administration of arsenite, MMAIII and DMAIII at cytotoxic concentrations (1.6 to 10 µM) for 24 h. While the specific gene changes observed across the arsenicals differed, the changes appeared to be related to similar pathways (NRF2-mediated stress response, interferon, p53, cell cycle regulation, and lipid peroxidation). Overall, the results demonstrated that cytotoxicity from the trivalent arsenicals occurs at similar concentrations and provided evidence of similar responses across the different cell types corresponding to the target organs for inorganic arsenic-induced cancer.
3.1. In vitro to in vivo extrapolation
Gentry, Yager et al. (Citation2014) conducted an in vitro to in vivo extrapolation to estimate an acceptable drinking water concentration based on the BMDLs for genomic changes in human bladder cells reported by Yager et al. (Citation2013) (). They determined that since the mixture ratio used in the Yager et al. (Citation2013) study in vitro was selected to be equivalent to those present in the urine in vivo, only a simple conversion of the micromolar concentrations by the molecular weight of arsenic was needed to estimate an “equivalent” in vivo urine concentration. Based on the range of benchmark doses reported in vitro in Yager et al. (Citation2013) (0.09 to 0.58 µM – trivalent mixture; 0.37 to 1.7 µM – pentavalent mixture), Gentry, Yager et al. (Citation2014) calculated in vivo “equivalent” urine concentrations for the PODs of 6.5 to 43.5 µg/L for the trivalent mixture and 26.25 to 127.5 µg/L for the pentavalent mixture. Using data on the ratio of trivalent and pentavalent arsenic in urine (Mandal et al. Citation2001), they then combined these results to obtain an estimated range of BMDLs for inorganic arsenic and its methylated metabolites in urine of 21–104 µg/L.
The in vitro to in vivo extrapolation approach used in Gentry, Yager et al. (Citation2014) was based on the BMDLs for the most sensitive genomic responses in cells exposed in vitro. However, the lowest genomic responses, even in repeated dose in vivo studies, have been shown to occur at somewhat lower concentrations than those associated with chronic toxicity or carcinogenicity (Thomas, Philbert et al. Citation2013; Thomas, Wesselkamper et al. Citation2013). Moreover, cells in culture are likely to be more sensitive to stress than in their natural in vivo setting. In a critical review on the carcinogenicity of inorganic arsenic, Cohen et al. (Citation2013) noted there are limitations that should be considered when extrapolating from such in vitro data to predict in vivo responses. Focusing on the in vitro evidence for skin, urinary bladder and lung cancer, Cohen et al. (Citation2013) noted that cell systems such as urothelium, epidermis, and bronchus are likely to be more sensitive in vitro than in vivo, since the fully differentiated epithelium has numerous protective barriers that are not present in cell culture. Thus, the use of the BMDL for the most sensitive genomic response in cells exposed in vitro as a point of departure in a risk assessment would be overly conservative.
When interpreting genomic data it is also important to keep in mind that the cellular control network involves a high level of interaction and redundancy to respond robustly to stressors (Zhang, Bhattacharya et al. Citation2014). A limited cellular response is typically required to maintain homeostasis or adapt to the stress at low concentrations of chemical stress, but as the concentration increases, it becomes necessary for the cell to recruit additional network control elements and pathways to avoid toxicity. In the case of trivalent arsenic, it has been noted that at low concentrations on the order of 0.1 µM, the effects of arsenic appear to be adaptive, while concentrations above 1 µM are clearly cytotoxic (Snow et al. Citation2005, Gentry et al. Citation2010). Until recently it has not been possible to determine where in this range of concentrations the transition from adaptation to toxicity occurs; however, the genomic dose-response results reported in Yager et al. (Citation2013) now make it possible to estimate the location of this concentration-dependent transition. As shown in , the lowest BMDLs, around 0.1 µM, are for alterations in genes associated with oxidative stress signaling (HMOX1, MT1E, TXN) and cell adhesion/growth regulation (LGALS8). These low concentration effects of trivalent arsenic represent a protective response that allows the cell to prevent possible stress (Snow et al. Citation2005). Apart then from these protective responses, the lowest BMDLs for the genes in are above 0.2 µM.
Table 3. Studies of low-level arsenic exposureTable Footnote* included in the main evaluation of dose-response in the current study in comparison to Lynch et al. (Citation2017a) and non-ecological studies in Lamm et al. (Citation2015) and Tsuji, Alexander et al. (Citation2014).
Cohen et al. (Citation2013) determined that the trivalent arsenic species (arsenite + MMAIII+DMAIII) represent ≈23% of the total arsenic in urine based on the most conservative analytic method (i.e. the estimate of the highest percentage of trivalent forms), described by Styblo and colleagues (Currier et al. Citation2011). Adjusting the BMDL of 0.2 µM (15 µg/L) from the Yager et al. (Citation2013) study by this ratio results in a genomic BMDL of 15/0.23 = 65 µg/L total arsenic species (inorganic arsenic, MMA, DMA) in urine.
The literature search conducted as part of the Gentry, Yager et al. (Citation2014) study identified multiple studies conducted in various areas in Mexico and in Canada with a wide range of exposures to arsenic concentrations in drinking water (Del Razo et al. Citation1997, Citation2011; Valenzuela et al. Citation2005). The data from these studies suggested a range of drinking water to urine total arsenic species (inorganic arsenic, MMA, DMA) concentration ratios ranging from 0.33 to 2.1. A recent study conducted in Taiwan (Hsu et al. Citation2017) suggests similar ratios ranging from 0.68 to 2.25. These data support the use of an average factor of 1 for converting arsenic urinary concentrations into arsenic concentrations in drinking water. The range of variability in the observed drinking water:urine ratios also provides information on the interhuman variability in toxicokinetics (or dietary sources of arsenic): the range of approximately 6-fold in these studies (from a low of 0.33 to a high of 2.1, with an average of about 1.0) is consistent with the default factor of 3 typically used in human health risk assessments for interhuman variability (from an average individual to a sensitive individual) in toxicokinetics (USEPA Citation1994; IPCS Citation2005). That is, water:urine ratio of 0.33, which would suggest a sensitive individual from the viewpoint of toxicokinetics is 3-fold below the average value of 1.0.
In summary, the overall in vitro evidence for the cellular effects of trivalent arsenic demonstrates a change from adaptive to potentially adverse effects between 0.1 and 1.0 µM (Gentry et al. Citation2010). Based on the BMDLs calculated for cellular effects of trivalent arsenic mixtures representative of human internal exposures (Yager et al. Citation2013), the threshold for potentially adverse cellular effects from exposure to inorganic arsenic in drinking water is likely to occur at urinary concentrations of trivalent arsenic above 0.2 µM, which corresponds to drinking water total arsenic concentrations above 65 µg/L. Concentrations below this level are unlikely to result in adverse cellular effects, even after chronic exposure.
This conclusion is further supported by the results of a recent study on changes in genome-wide DNA methylation, microRNA expression, mRNA expression, and protein expression levels in cord blood from pregnant women exposed to varying levels of inorganic arsenic in drinking water (Rager et al. Citation2017). Benchmark dose modeling was conducted on a robust measure informed by multiple -omic profiles using weighted gene co-expression analysis. Benchmark dose modeling of this integrated measure resulted in a BMD(BMDL) of 58(45) μg/L speciated arsenic in urine.
4. Animal models of inorganic arsenic carcinogenesis
One of the major difficulties in furthering our understanding of the mode of action for inorganic carcinogenesis has been the lack of a reliable animal model for any of the target sites, including skin, urinary bladder, and lung. Part of this difficulty is the lack of a complete, standard 2-year bioassay in rats or mice conducted under Good Laboratory Practices (GLP). A summary of the evaluation of inorganic arsenic and other arsenicals in animal models was extensively reviewed by Tokar, Benbrahim-Tallaa et al. (Citation2010) and Cohen et al. (Citation2013). A search by PubMed and by Google for carcinogenicity studies since 2012 was performed using arsenic plus mice, arsenic plus rat, and arsenic plus carcinogenicity as search terms, and these yielded 200 publications. Most of these involved assays for genotoxicity, metabolism, metabolic effects, in vitro studies and reviews and included several redundancies. Seven studies were identified from these 200 publications that involved long-term bioassays on arsenicals alone or following DNA-reactive carcinogens and are included in the various studies described in the following sections.
4.1. Transplacental carcinogenesis model in mice
The closest to an animal model of inorganic arsenic carcinogenesis is the two-generation model originally developed by Waalkes and his colleagues (Waalkes et al. Citation2003). This model involves the administration of inorganic arsenic in the drinking water to mice in utero (gestation days 8 to 18) at concentrations of 42.5 and 85 mg/L (ppm) and then following the animals for their lifetime. Under these circumstances, there was an increased incidence of lung tumors and a slight increase of other tumors. However, using this model, subsequent studies by these investigators have only been able to confirm the increased incidence of lung tumors (Waalkes et al. Citation2004; Waalkes, Liu, Ward and Diwan Citation2006; Waalkes, Liu, Ward; Powell et al. 2006). In addition, studies by another laboratory using this model (Nohara et al. Citation2012; Takumi et al. Citation2015) have not confirmed the findings of lung tumors; instead, they observed increased incidences of liver tumors.
In more recent studies, Waalkes et al. (Citation2014) and Tokar et al. (Citation2011) administered inorganic arsenic as sodium arsenite in the drinking water before pregnancy, during pregnancy and lactation, and then continuing for the lifetime of the animals, up to two years. Under these circumstances, they again obtained an increased incidence of lung tumors (bronchiolo-alveolar tumors), although the dose-response was inconsistent. Increased lung tumor incidences were observed at 500 and 24 000 µg/L drinking water concentrations in males and at 50, 12 000 and 24 000 µg/L in females, with negative results at 5000, 6000 and 12 000 µg/L in males and at 500, 5000, and 6000 µg/L in females. These authors claimed that the incidence was increased even at low doses (50 µg/L).
However, such conclusions, including at the low dose, were based on statistical significance at p < 0.05, and are likely false positives (Cohen et al. Citation2014, Citation2015; Burgoon and Druwe Citation2016; Cohen, Arnold et al. Citation2016). As described by Haseman (Citation1983), p values of <0.01 instead of 0.05 should be used for common tumors (defined as tumors with a background incidence of >1%) because of the marked variability of incidences in controls. This variability was evident in the incidence of tumors in controls observed by Waalkes and colleagues in their various studies, which overlapped with the incidences in treated mice in the Tokar et al. (Citation2012) and Waalkes et al. (Citation2014) studies (Cohen et al. Citation2014, Citation2015; Cohen, Arnold et al. Citation2016). A similar rule of using p values of <0.01 instead of 0.05 has been adopted by the International Commission on Harmonization (FDA Citation2001) for regulation of pharmaceuticals and is also included in the Organization of Economic Co-operation and Development (OECD Citation2010) Guidelines. With consideration of p < 0.01 for significance, the only statistically significant incidences were present at the highest doses evaluated by Waalkes and his associates (e.g. 12 000 to 24 000 µg/L; Cohen et al. Citation2014, Citation2015, Cohen, Arnold et al. Citation2016 ). Overall, the experiments by Waalkes and his colleagues suggest that inorganic arsenic administered in utero leads to an increased incidence of lung tumors, but only at high concentrations. Extrapolation of these findings to humans is problematic not only because of the difficulties with respect to dose-response observed in these studies and the difficulties of replication in other laboratories, but also because of the differences between lung carcinogenesis and lung tumors in mice compared to humans (Nikitin et al. Citation2004; Strupp et al. Citation2016; Yamada et al. Citation2017). The various other tumors observed by Waalkes and colleagues in some of their studies, including benign ovarian tumors, liver tumors, and adrenal cortical adenomas, have not generally been replicated in other studies or the incidences are not statistically significant, particularly utilizing the Haseman rule. An earlier study by Waalkes et al. (Citation2000) involving intravenous administration of arsenate to dams produced increased proliferative lesions of the ovary and liver, but no significant increased incidences of neoplasms. An overall critique of this transplacental model is described in Garry et al. (Citation2015).
The various experiments involving transplacental studies in mice involved administration of sodium arsenite during pregnancy followed by observation for the rest of the animals’ life. Other studies have used this same model in mice involving transplacental exposure to sodium arsenite followed by administration of various compounds to the offspring, including 12-O-tetradecanoyl phorbol-13-acetate (TPA) (Waalkes et al. Citation2004, Citation2008), diethylstilbestrol (DES) (Waalkes, Liu, Ward and Diwan Citation2006, Waalkes, Liu, Ward, Powell et al. Citation2006), or tamoxifen (TAM) (Waalkes, Liu, Ward and Diwan Citation2006). The use of TPA was an attempt to enhance the production of skin tumors since TPA is considered a potent promotor of skin carcinogenesis in mice. The experiment with TPA after transplacental administration of arsenic produced a slight increase in liver tumor incidences and lung adenomas, but the dose response was not indicative of an effect by TPA. In contrast, administration with DES following transplacental administration of arsenic produced increased uterine and vaginal carcinomas, urinary bladder hyperplasia and urinary bladder proliferative lesions (hyperplasia plus tumors) in addition to liver tumors in female offspring. Although the incidences of some of these tumors, such as the urinary bladder lesions, were not statistically significant, their rarity and the presence of pre-neoplastic hyperplasia suggested that the tumors were actually a treatment-related effect. A similar response was seen with the other estrogen-related compound, TAM.
In summary, these various studies with this transplacental model supported the hypothesis that transplacental exposure to sodium arsenite increased susceptibility to tumorigenesis, but only at high dose levels (e.g. 12 000 to 24 000 µg/L). Nevertheless, a large number of epidemiological evaluations have been conducted on populations exposed to high concentrations of inorganic arsenic in drinking water throughout their lifetime for multiple generations. Therefore, to the extent there is early life sensitivity to carcinogenicity, this would be included in these studies as well as in studies that examine the highest risks for various exposure metrics including, average, cumulative, or highest exposures at various earlier life stages (e.g. various lag times).
4.2. Other animal models of inorganic arsenic carcinogenesis
To evaluate tumor promotion in skin carcinogenesis, administration of sodium arsenite to Tg.AC homozygous mice, which are considered genetically initiated, also was evaluated. Sodium arsenite by itself did not enhance the tumorigenicity in this model (Germolec et al. Citation1998). Likewise, administration to Crl:SKl-hrBR (hairless) mice in combination with UV radiation did not produce an effect by arsenite (Rossman et al. Citation2001) other than a slight increase in numbers of tumors, but not incidences. A recent study utilizing an initiation-promotion protocol administering arsenite in drinking water at 2, 20, or 200 mg AsIII/L to Sencar mice gave variable results on tumor number but not incidences, increasing or decreasing numbers depending on time of observation (Palmieri and Molinari, Citation2015). No studies have produced consistent changes in mouse skin models.
A lifelong study in rats using a protocol developed at the Ramazzini Institute yielded a variety of tumors, including a single urinary bladder tumor (Soffritti et al. Citation2006). However, these were not statistically significant. Furthermore, these studies have been questioned because all animals were infected and developed pneumonia, the pathology was not peer-reviewed, it was not a standard protocol, and was not conducted according to GLP (NTP Citation2011; Gift et al. Citation2013).
Administration of arsenate in drinking water to mice produced an overall increased incidence of tumors but no significant increase in any particular tumor (Ng et al. Citation1999). This type of analysis is generally not accepted as a valid assessment of carcinogenesis based on statistical and biologic principles.
A variety of other studies have evaluated the potential carcinogenicity of inorganic arsenic but used non-oral routes of administration, as well as other species besides mice. Intratracheal administration of calcium arsenate led to a slight increase in lung adenomas in hamsters (Yamamoto et al. Citation1987), but this experimental model has limitations with regard to its relevance to humans because of the marked inflammatory reaction that accompanies the intratracheal administration (Driscoll et al. Citation2000).
Administration of sodium arsenite after a known carcinogen has also been examined in various model systems such as administration of sodium arsenite in the drinking water after diethylnitrosamine (DEN) administration, but this did not lead to a significant increase of tumor incidences (Shirachi et al. Citation1983). Administration of arsenite or DMAV in the drinking water concurrently with BBN slightly increased urothelial dysplasia, but without statistical significance as the BBN treatment alone produced urothelial alterations in 100% of the mice (Dai et al. Citation2017). Furthermore, there were only ten mice per group. DMAV or arsenite alone produced no effects on the bladder urothelium.
In a colon cancer model in mice, arsenite administered after pretreatment with azoxymethane and dextran sodium sulfate (AOM/DDS) had no significant effect on tumor incidence but increased tumor number and size (Wang et al. Citation2012). The dose of arsenite was 58 mg/L in drinking water. This model has wide variability in response to AOM/DDS without other treatments, raising concerns about the relationship to arsenic treatment. Arsenical treatments of mice without pretreatment with AOM/DDS has not been reported in any studies to affect the intestines.
In summary, inorganic arsenic has not produced a significant incidence of tumors in a variety of models other than in the transplacental model developed by Waalkes and associates. However, in the Waalkes model, the tumor incidence is increased significantly only at high concentrations, and there is some question with respect to the reproducibility of this model and comparison of the tumor incidences with historical controls. In contrast, arsenite administered in drinking water or diet increased the incidence of urothelial hyperplasia of the urinary bladder in mice and rats, but this does not appear to evolve into tumors even in long term studies (Simeonova et al. Citation2000: Suzuki et al. Citation2008; Arnold et al. Citation2014).
4.3. Animal models of carcinogenesis by methylated and other arsenicals
Other arsenicals have also been evaluated in animal models. The most noteworthy of these is the administration of DMAV in either the diet (a GLP two-year study) or drinking water to rats leading to the induction of urinary bladder urothelial tumors, including invasive carcinomas (Wei et al. Citation1999; Arnold et al. Citation2006). In contrast, administration of DMAV to mice was without a tumorigenic effect (a GLP two year study) (Arnold et al. Citation2006). The dose-response with DMAV in rats was noteworthy for its positive results only at high doses. In the diet (Arnold et al. Citation2006), tumors were increased at concentrations of 40 and 100 ppm, whereas there was some evidence of hyperplasia at 10 ppm but was completely negative at 2 ppm. In the drinking water (Wei et al. Citation1999), bladder tumors were induced at concentrations of 50 and 200 ppm but was negative at 12 ppm in a 2-year bioassay. These studies are discussed in greater detail in the section mode of action.
Administration of DMAV after prior treatment with the urinary bladder DNA reactive carcinogen N-(4-hydroxybutyl)-N-butylnitrosamine (BBN) also produced an increased incidence of bladder tumors due to DMAV at concentrations of 10 ppm and higher (Wanibuchi et al. Citation1996). In a protocol involving administration of 5 DNA reactive carcinogens to "initiate" a number of organ systems, there was some evidence DMAV induced an increase not only in urinary bladder tumors (BBN was one of the 5 administered DNA reactive chemicals) but also increased tumor incidences in kidney, liver, and thyroid (Yamamoto et al. Citation1995). DMAV was administered in the drinking water at various concentrations, with increased tumor incidences at 400 ppm. However, this protocol produces a wide variation in tumor incidences with the 5 DNA reactive chemicals alone, so it is difficult to determine if subsequent treatment with DMAV increased tumor incidences above background. However, given these studies with DMA alone and after treatment with DNA reactive carcinogens, it is clear that DMAV is a urinary bladder carcinogen in the rat, although not in the mouse in full 2-year bioassays performed under GLP conditions. The tumors with DMA in rats were observed primarily in female rats and not in male rats.
In contrast, MMAV was negative in GLP studies in rats and in mice (Arnold et al. Citation2003). There is one report with regard to trimethylarsinic acid in rats with a suggestion of an increased incidence of liver adenomas, but the background incidence of these tumors suggests that this may have been within historical controls and not due to the treatment (Shen et al. Citation2003).
Some of these arsenicals have also been administered in specific mouse models to evaluate specific effects, but many of these suffer from difficulties in interpretation as described in Cohen et al. (Citation2006, Citation2013). These include studies of DMAV in K6/ODC mice in an attempt to evaluate skin tumor promotion (Morikawa et al. Citation2000). However, the results were equivocal and difficult to interpret and were considered negative by the authors. Another study involving administration of inorganic arsenate to two different strains of mice reported an increase in total numbers of tumors but not at any particular site (Ng et al. Citation1999), and the increase was not considered treatment-related. Administration of DMAV to Ogg1-/- mice (deficient in base excision repair of 8-oxoguanine, a mutagenic byproduct from oxidative stress) was purported to increase the lung tumor incidence (Kinoshita et al. Citation2007). However, the control mice in the study had an incidence considerably lower than historical controls reported in other publications, and it is likely that the effect was not due to the DMAV treatment but due to the reduced number of tumors in the controls (Cohen et al. Citation2006). This is particularly likely since DMA did not produce tumors by itself in mice in a 2-year bioassay. Administration of DMAV in drinking water at 0, 50, and 200 ppm to heterozygous p53 (±) knockout mice showed a slight increase in lymphoma incidence (Salim et al. Citation2003). However, this model has a very high spontaneous incidence of lymphoma with considerable variability, so it is unlikely that these results were treatment-related (Donehower et al. Citation1992; Storer et al. Citation2001). Administration of arsenate or DMAV in the drinking water to A/J male mice has produced a statistically significant increased number of lung tumors (Hayashi et al. Citation1998; Cui et al. Citation2006), but this is a strain of mice that has essentially a 100% incidence of lung tumors, and the number of tumors per mouse is highly variable, even in untreated mice. It is unlikely that this is actually a true treatment effect with either arsenate or DMAV, as there are numerous difficulties with this model (Nikitin et al. Citation2004). Likewise, administration of DMAV after pretreatment with 4-nitroquinoline 1-oxide to mice increased the number of lung tumors but not the incidence (Yamanaka et al. Citation1996). This model has many of the same issues as the A/J mouse model, with a high and variable background incidence of lung tumors. Application of DMAV orally to hairless mice resulted in an increased number of skin tumors, either in combination with UV radiation or after UV radiation or dimethylbenz(a)anthracene (DMBA) administered to the skin (Yamanaka et al. Citation2001). Findings regarding DMAV or inorganic arsenic in mouse skin tumors models have been inconsistent, but by itself, inorganic arsenic does not appear to induce skin tumors.
The administration of MMAIII to mice utilizing the transplacental protocol used for evaluation of inorganic arsenic showed an increase in ovarian and adrenal tumors in the females and an increase in liver, lung, and testicular tumors in males (Tokar et al. Citation2012). A difficulty in evaluating this study is the instability of MMAIII in aqueous solutions (the chemical was administered in the drinking water) without verification of the quantitative levels in the drinking water at various times during the experiment. Nevertheless, this was again at high concentrations, 12.5 or 25 ppm in the drinking water.
In a medium-term assay, administration of diphenylarsine, a chemical warfare neurotoxicant, to rats at 12.5, 25, or 50 ppm in the drinking water following pretreatment with diethylnitrosamine (DEN) showed a slight effect on liver foci numbers and size at the highest dose (Wei et al. Citation2013). Diphenylarsine without DEN pretreatment had no effect on liver foci numbers or area.
Lastly, administration of gallium arsenide produced an increased incidence of lung tumors in female rats in an inhalation study, but not in male rats or male or female mice (NTP 2000). However, this was likely due to the particulate matter of the gallium arsenide that produced a marked inflammatory reaction similar to that seen with other particulates and was not due to the chemical itself (Bomhard et al. Citation2013).
4.4. Summary of animal studies
In summary, inorganic arsenic has only been positive in animal assays when administered to pregnant mouse dams with long term follow-up of the pups, taking into consideration historical controls and statistical analyses (Cohen et al. Citation2014, Citation2015; Cohen, Arnold et al. Citation2016). Although there remain concerns about this model, it has produced positive results, albeit at high concentrations (at least 12,000 µg/L in the drinking water). DMAV has reproducibly induced urinary bladder urothelial tumors when administered at high concentrations in drinking water or diet. An increase in urothelial tumors was observed at 50 ppm in drinking water and 40 ppm in the diet. An increased incidence of urinary bladder tumors was produced when DMAV was administered at 10 ppm in drinking water after pretreatment with BBN, and increased hyperplasia was produced by DMAV at 10 ppm of the diet but not at 2 ppm. As discussed above, these thresholds for DMAV correspond to oral doses that produce a urinary concentration of >0.1 µM dimethylarsinous acid (DMAIII), the threshold concentration for biologic effects of trivalent arsenicals in vitro (Gentry et al. Citation2010). The threshold for inorganic arsenicals appears to be higher than the threshold for tumorigenic effects of DMAV.
5. Epidemiology
5.1. Study selection criteria
Our review and evaluation of epidemiological studies builds upon previous efforts by Lynch et al. (Citation2017a), who conducted literature searches and systematically reviewed studies of all cancer endpoints initially, followed by more refined assessments of the quality of epidemiological studies on lung and bladder, the endpoints with the most robust evidence for assessing dose-response associations. Our search also considered the literature reviews conducted as the basis for recent meta-analyses of low-level arsenic exposure in association with risk of bladder cancer (Tsuji, Alexander et al. Citation2014), the meta-regression analysis of arsenic exposure and risk of lung cancer (Lamm et al. Citation2015), and the review of skin lesions and skin cancer (Karagas et al. Citation2015).
In addition to including skin cancer studies, along with studies of bladder and lung cancer, we focused our efforts on studies of low-level arsenic exposure, defined initially as arsenic water concentrations (or equivalent dose) of less than approximately 150 µg/L, but recognizing that differences in water intake and nutrition among populations may affect the actual external and internal arsenic dose.
Inclusion/exclusion criteria for initial study consideration and subsequent selection of studies for dose-response evaluation were based in part on criteria of Lynch et al. (Citation2017a) and others (e.g. Tsuji, Alexander et al. Citation2014; Lamm et al. Citation2015). These criteria were also used for study identification and assessment of study quality.
The initial selection criteria were as follows:
Peer-reviewed original epidemiological studies of clinically verified bladder, lung, or non-melanoma skin cancer in relation to inorganic arsenic exposure;
Individual-level exposure and outcome data;
Quantitative estimates of relative risk (e.g. hazard ratios or odds ratios) and their variability (e.g. 95% confidence intervals, CI), with reported number of cancer cases and non-cases or person-time at risk for each exposure category; and
Assessment of low-level exposure (approximately <150 µg/L or equivalent dose).
Further study evaluation for relevance in dose-response assessment at low doses was based on the following eligibility criteria:
More than two dose groups in the low-level range, with reported numeric boundaries;
Incidence (not mortality) for bladder and skin cancers (lung cancer has a higher mortality rate than bladder and non-melanoma skin cancers, so risks based on mortality for lung cancer are comparable to those based on incidence);
Individual-level adjustment for smoking in bladder and lung cancer studies;
Longest follow-up (cohort) or largest sample size (case-control) if multiple studies were available from the same study population; and
Arsenic concentration in water or means of converting to water exposure concentration.
The last criterion was based on assessing whether additional studies might update the low-level studies considered in the primary analysis of Lynch et al. (Citation2017a), which modeled studies reporting cancer risks in association with exposure to arsenic in water, the most commonly used exposure metric in epidemiological studies.
As an additional line of evidence in examining the dose-response for low-level arsenic exposure in relation to lung and bladder cancer risks, we also evaluated studies that reported associations in never smokers, because of the high potential for effect modification and/or residual confounding from incomplete adjustment for intensity and duration of smoking. For the analysis of never smokers, we did not restrict the exposure metric to arsenic water concentration and also included cumulative dose (e.g. mg or mg/L-years) or arsenic biomarkers.
5.2. Literature search
A literature search was conducted in PubMed, the National Library of Medicine’s database of citations for biomedical literature, to identify articles published from 2016 (prior to the June 2016 cutoff of Lynch et al. Citation2017a, Citation2017b; with some overlap in case of missed studies) to May 2018 using the search terms “arsenic” or synonyms (e.g. 7440–38-2, 75–60-5, monomethylarsenic, dimethylarsenic, monomethylarsonic, monomethylarsonous, dimethylarsinic, dimethylarsinous, or arsenate) and “cancer” or synonyms (e.g. carcinoma, malignant, malignancy, neoplasm, neoplasia, neoplastic, nonmelanoma, non-melanoma, mortality, or cohort), with restriction of some keywords to titles and abstracts. A secondary search was also conducted using the search terms “arsenic” and “skin lesions” to identify publications on arsenic and nonmalignant skin lesions published since the July 2014 cutoff of Karagas et al. (Citation2015). Review papers were not considered for the purpose of dose-response evaluations but were considered for additional insights on studies and any publications that may have been missed by the literature search. We also examined the reference lists of identified articles to identify any additional relevant publications.
This literature search for arsenic and cancer resulted in 178 studies, and the search for arsenic and skin lesions resulted in 85 studies. The abstracts of all identified studies were initially reviewed for relevance. After exclusion of animal and in vitro studies, exposure assessments, risk assessments, case reports and case series, studies of occupational or therapeutic arsenic exposure, studies of other health endpoints not considered here, editorials or commentaries, and other irrelevant studies (but counting review articles that were examined but not considered eligible for inclusion), 25 studies for arsenic and cancer remained for full-text review for eligibility. A full-text review of 15 studies was conducted for information on arsenic exposure and skin lesions.
After the 25 studies for cancer were screened for the initial study selection criteria, 16 studies were excluded because they did not report original epidemiological study results, did not evaluate skin, bladder, or lung cancer, did not report quantitative estimates of relative risk between arsenic exposure and cancer, or were ecological in design (i.e. lacked individual-level exposure and outcome data). The remaining nine studies, which were not assessed by Lynch et al. (Citation2017a), met the initial study selection criteria, and are summarized in Supplemental Table 1 (design) and Supplemental Table 2 (results). All 25 full-text articles reviewed, along with reasons for exclusion or inclusion, are listed in Supplemental Table 3. We also considered whether epidemiological studies identified by Lynch et al. (Citation2017a) in their literature search, but not included in their dose-response assessment, could contribute relevant information to the dose-response evaluation.
5.3. Exposure metric
Epidemiological studies of environmental arsenic exposure and cancer have used various measures of exposure including arsenic exposure media concentration (e.g. in water, soil, or air); biomarkers in human fluids and tissues (e.g. in urine, hair, or nails); and arsenic dose based on reported drinking water consumption rates and water concentration. Each of these measures comes with various uncertainties that need to be considered in the evaluation of the evidence.
5.3.1. Arsenic concentration in exposure media
Water concentration is the most commonly used exposure metric in epidemiological studies, likely because of the relative ease of data collection, availability of historical records, and ability to associate water levels with individual residential and workplace locations. Water is also the primary source of elevated arsenic exposures for most human populations because of its widespread natural occurrence. A key uncertainty with this measure is the availability of historical data for past water exposures. Although untreated well water concentrations may be relatively stable over time (Steinmaus et al. Citation2005), participant changes in residential location or water source, or changes in concentration with installation of water treatment will affect exposures. Some studies attempted to obtain or estimate previous water exposure data (e.g. Meliker et al. Citation2010; Dauphine et al. Citation2013; Steinmaus et al. Citation2013; Baris et al. Citation2016), whereas others used only recent well water data (Chen et al. Citation2010a, Citation2010b). Mitigating factors in the case of Chen et al. (Citation2010a, Citation2010b), a study based in northeastern Taiwan, are that the water source was not treated and the population was relatively isolated and stable. More than half lived at the same location since birth and most of those who moved did so only once.
At lower exposure levels, inorganic arsenic in food becomes an important contributor to exposure, but it is more difficult to quantify because of the variable amounts in the diet, depending on the type of food, differences in growing conditions and food preparation methods (particularly if arsenic-contaminated water is also used for growing and cooking rice or other crops), and individual diet composition (Tsuji et al. Citation2007; Tsuji, Perez et al. Citation2014; Xue et al. Citation2010). Few data are available to characterize inorganic arsenic separately from organic forms in the diet, particularly for individuals. The contribution of inorganic arsenic in food for the U.S. population, even for upper bound exposures, would be less than exposure to drinking water at 2 L/day at 10 µg/L (Tsuji et al. Citation2007; Xue et al. Citation2010). Thus, arsenic from food is expected to be a lesser source of exposure than contaminated drinking water.
Arsenic in soil can also be a source of exposure, but greatly elevated soil concentrations are required to elevate exposure in excess of dietary arsenic (Tsuji et al. Citation2005, Citation2007), likely because of relatively low bioavailability (Diamond et al. Citation2016) and small amounts of daily soil intake (Stanek et al. Citation2001). Accordingly, biomonitoring in young children at a smelter site indicated no increases in inorganic arsenic exposure over background levels from the diet and water until residential soil arsenic concentrations were in excess of 300 ppm (Hwang et al. Citation1997a). Epidemiological studies of cancer and other health outcomes in relation to arsenic in soil in residential populations typically have poor assessment of individual-level exposures; therefore, these studies were not eligible for inclusion. The most rigorously conducted case-control study (Frost et al. Citation1987) and retrospective cohort study (Tollestrup et al. Citation2003) of residential populations with historical exposure to arsenic from both air emissions and elevated levels in soil and dust from a copper smelter (as high as >1000 ppm arsenic in soil) did not find significantly elevated risks of lung or bladder cancer mortality.
Arsenic in the air also does not contribute measurably to arsenic exposure except in the presence of unusually high industrial emissions sources (as for the population studied by Frost et al. Citation1987 and Tollestrup et al. Citation2003). Residential exposure scenarios involving airborne arsenic in resuspended dust from soil results in only a fraction of arsenic exposure from incidental soil ingestion (USEPA Citation2018). Inhalation exposure to arsenic is assessed separately from the oral route and is based on studies in historical smelter workers (Erraguntla et al. Citation2012; Lewis et al. Citation2015).
5.3.2. Arsenic concentration in biomarkers
Biomarkers such as arsenic in urine, blood, hair, or toenails have also been used to assess arsenic exposure (ATSDR Citation2007). Arsenic in urine, followed by toenails, has been the most frequently used biomarker in epidemiological studies. Urinary arsenic concentrations “speciated” for inorganic arsenic and its methylated metabolites are considered to best represent inorganic arsenic exposure, rather than total urinary arsenic, which may also include a number of less toxic dietary organic forms (ATSDR Citation2007). As noted above, ingested inorganic arsenic (the more toxic form) is metabolized in a series of oxidation/reduction reactions to MMA, followed by DMA, which is the primary form in urine with environmental exposures (Cohen et al. Citation2013). More rapid and extensive metabolism to DMAV and excretion in urine thereby reduces internal exposure to the more cytotoxic trivalent arsenic forms. Studies that report speciated (i.e. sum of inorganic arsenic MMA and DMA) arsenic levels rather than total blood or urinary arsenic levels are preferred for greater exposure specificity to inorganic arsenic. Exposures to less toxic DMA and organic precursor compounds of DMA in various foods, however, complicate assessment of speciated arsenic levels in biomarkers of inorganic arsenic exposure (Schoof et al. Citation1999; Choi et al. Citation2010; Cascio et al. Citation2011; Aylward et al. Citation2014; Nearing et al. Citation2014; Tsuji et al. Citation2015; Taylor et al. Citation2017).
Urinary arsenic is a short-term measure of exposure, with up to 85% of an inorganic arsenic dose excreted in 1–3 days (ATSDR Citation2007). Arsenic in blood reflects even more recent exposure, within hours of intake (ATSDR Citation2007). In epidemiological studies, both are typically sampled at only one point in time. In populations with consistent dietary and water sources and consistent consumption rates over time, these transient biomarkers may be reflective of longer-term exposure, particularly for first-morning void urine samples (Hwang et al. Citation1997b). Blood samples can vary depending on variation in arsenic intake over a day and are usually not speciated. Similarly, for convenience, particularly with large study populations, most urine samples are spot samples taken at one time over a day rather than a 24-h urine sample or standardized first-morning void sample. Spot urine samples are particularly uncertain because of greater variation in sample hydration state.
Creatinine in urine is typically used to adjust for hydration state of arsenic in spot urine samples; however, creatinine adjustment can introduce variation because of differences in creatinine excretion rates depending on gender, age, dietary intake of protein/malnutrition, or disease states (Barr et al. Citation2005). In a population in Bangladesh, low urinary creatinine was associated with malnutrition, which also increased susceptibility to adverse health effects, including those associated with arsenic (Pilsner et al. Citation2009). Low muscle mass and decreased creatinine excretion also occur in the early stages of chronic kidney disease, prompting caution regarding overestimation of urinary analyte concentrations if adjusted using urinary creatinine (Tynkevich et al. Citation2014). Chronic kidney disease has been associated with various other diseases such as hypothyroid, diabetes, cardiovascular disease and cancer, including urinary tract cancers (Wong et al. Citation2016), and possibly lung and non-melanoma skin cancer through associations with smoking in the former and from evidence in kidney transplant patients in the latter (Stengel Citation2010). Creatinine adjustment of urinary arsenic levels for individuals with low creatinine in urine could, therefore, accentuate an association between arsenic exposure and adverse health effects. Because of these issues, epidemiological studies using urinary arsenic were considered with caution in this assessment.
Arsenic in hair and toenails reflect exposure over the weeks of hair or nail growth and are reported to contain mostly inorganic arsenic and much less organic arsenic than urine (Button et al. Citation2009); however, these findings may be affected by external contamination of hair and nails by inorganic arsenic in soil/dust or water, even after sample cleaning (Hindmarsh and McCurdy Citation1986; Tsuji et al. Citation2005; Button et al. Citation2009). In addition, information to relate these measures to a daily dose or to other common dose measures, such as water arsenic exposure concentration, often are not provided.
Biomarker studies were not included in the modeling by Lynch et al. (Citation2017a) in their primary dose-response analysis because most otherwise eligible epidemiological studies used arsenic in drinking water as their dose metric. We additionally considered the bladder cancer results of Karagas et al. (Citation2004), because this research group published a quantitative relationship of arsenic in toenails with arsenic drinking water concentration that allowed conversion between exposure metrics for this population (Karagas et al. Citation2000).
5.3.3. Calculated arsenic dose
In addition to the concentration of arsenic in an exposure matrix (e.g. water, urine, toenails, food), some epidemiological studies of arsenic and cancer have examined associations with a calculated arsenic dose (reported water intake multiplied by water concentration) instead of an exposure matrix concentration. Although this metric may provide more accurate individual information on arsenic exposure, it is subject to uncertainty in recall for historical intake decades ago and potential bias in case-control studies, whereby cases may have biased recall of drinking more water. Incorporating survey information on drinking water intake may also introduce additional uncertainty due to the reliance on multiple self-reported variables or unverified assumptions.
Comparisons among studies from different countries may also be affected by differences in water intake rates. For example, populations with fewer sources of fluids other than the elevated-arsenic water source or with cooking practices that include more water (e.g. cooking rice in Taiwan or dishes with excess water in Bangladesh and West Bengal) will have greater water intake than for tap water consumption in the U.S. (Tsuji, Perez et al. Citation2014; Lynch et al. Citation2017a).
Studies have also examined associations with either arsenic dose or arsenic exposure matrix concentrations based on average daily exposure, highest lifetime daily exposure, highest 5-year daily exposure, cumulative exposure (e.g. total mg arsenic or µg/L multiplied by years of exposure), or exposure lagged for various time periods from diagnosis up to 40 years or more prior. However, biological guidance is uncertain as to which exposure metric is most plausibly linked to cancer development. The practice of testing multiple exposure metrics is prone to model selection bias, by which the metric with the strongest statistical association with cancer risk is emphasized as the “correct” one, resulting in positive findings due to multiple comparisons.
Dose calculations are also complicated by exposure to large changes in arsenic drinking water concentrations, either from moving between locations or water sources with changes in arsenic concentrations from installation of water treatment plants. Several towns in northern Chile had widely varying drinking water concentrations with high concentrations during historical periods of high arsenic exposure prior to 1971 (e.g. 860 µg/L in two towns, 250 µg/L in two towns) and between 1971 and 1977 (636 µg/L in two towns with 250 µg/L previously, 287 µg/L in another town) (Steinmaus et al. Citation2013). Except for one town with 600 µg/L continuously over time, water treatment led to a decrease in arsenic water concentrations over the following decades. Seven other towns have had stable arsenic concentrations of 1 to 60 µg/L since at least 1930. Relating average exposure metrics for studies of this population (Smith et al. Citation2009; Ferreccio et al. Citation2013; Steinmaus et al. Citation2013) to other populations is thus particularly difficult because of large variation in potential exposure for the study population. Some participants in the study of skin cancer in Hungary, Romania, and Slovakia (Leonardi et al. Citation2012) also had high historical exposures (up to 250 to 400 µg/L) before water treatment interventions in the mid-1980s to 1990s. Studies of populations in Hanford, California, and Fallon, Nevada had longer durations of exposure to elevated arsenic levels in public supplies (most around 100 µg/L) before water treatment in 2004 (Dauphine et al. Citation2013).
An average exposure that includes periods of high exposure is unlikely to be equivalent toxicologically to an average exposure involving more constant arsenic water concentrations. In such populations, lagged exposure may be more etiologically relevant to cancer risk than average lifetime exposure; however, USEPA cancer risk assessments for chemicals are based on the assumption of a relatively constant average dose over much of a lifetime.
5.4. Consideration of confounding factors and effect modifiers among studies
Tobacco use (including smoking and betel nut use) and socioeconomic status are established risk factors for bladder and lung cancers, and possibly for non-melanoma skin cancer. Sunlight exposure, particularly in fair-skinned populations, is a major risk factor for skin cancer. Poor diet and nutrition have various effects on arsenic metabolism and biologic effects, and thus may play a role in the development of lung, bladder, and skin cancers and nonmalignant skin lesions (Hsueh et al. Citation1995; Mazumder et al. Citation1998; Gamble et al. Citation2005). All of these risk factors could confound or modify the association between arsenic exposure and risk of these cancers. For example, more rural populations may be more likely to have occupations and lifestyles involving more sunlight exposure, as well as more arsenic exposure from use of private well water. Tobacco use may reduce folate status and affect general methylation capacity, including for arsenic (Tsuji, Perez et al. Citation2014).
Malnutrition may increase internal exposure to the more toxic arsenic intermediate forms for impoverished populations in Bangladesh, West Bengal, and southwestern Taiwan (Hsueh et al. Citation1995; Mazumder et al. Citation1998; Chen et al. Citation2001; Gamble et al. Citation2005, Citation2006; Pilsner et al. Citation2009; Tsuji, Perez et al. Citation2014). Folate deficiency affects one-carbon metabolism, which is required for methylation reactions for proper growth, maintenance, expression and repair of DNA, and many other essential functions throughout the body. Pilsner et al. (Citation2009) noted that increased DNA methylation with greater arsenic exposure may be an adaptive response because decreased methylation of leukocyte DNA was found to be associated with increased risk of skin lesions. Low folate status is a risk factor for cardiovascular disease (summarized by Tsuji, Perez et al. Citation2014) and may also be an independent risk factor for skin cancer (Williams et al. Citation2012), bladder cancer (Schabath et al. Citation2005; Hu et al. Citation2016, He and Shui Citation2014), and possibly lung cancer (Durda et al. Citation2017; Fanidi et al. Citation2018), although evidence for lung cancer, especially in women, is not conclusive (e.g. Zhang, Zhou, et al. Citation2014).
A meta-analysis of randomized controlled trials reported that folic acid supplementation did not affect the risk of cancer, except for reduced risk of melanoma (Qin et al. Citation2013); however, the results may differ in a more folate-deficient population. Low folate status may also impair proper methylation and metabolism of arsenic to less toxic DMAV (Tsuji, Perez et al. Citation2014). Other nutrients of importance for arsenic detoxification may include protein status (methionine), other B vitamins, selenium, and beta-carotene (Hsueh et al. Citation1997; Pierce et al. Citation2011; Tsuji, Perez et al. Citation2014). These potential effect modifiers are of particular importance for consideration when attempting to generalize to the U.S. from studies conducted in foreign countries with poor nutrition and lack of food/nutrient preservation (e.g. refrigeration) or staple food fortification.
At the same drinking water exposure concentrations, populations may differ in the intake of water from direct drinking water and from cooking and in body weight, thereby affecting the arsenic dose per body weight. Populations may also differ in the amount of inorganic arsenic in their food, depending on their diet, and use of contaminated well water to grow crops. Lynch et al. (Citation2017a) adjusted the data from different studies to a standard drinking water intake and body weight. We considered these adjustments and whether differences among populations would affect interpretations of the results of the studies evaluated.
5.5. Study selection results
Given the recent literature review by Lynch et al. (Citation2017a), few additional studies were identified as eligible for dose-response assessment. Of the nine recent studies (six on bladder cancer, one on lung and bladder cancer, one on lung cancer, one on skin cancer) that met the initial study selection criteria (Supplemental Tables 1 and 2), only three (Hsu et al. Citation2017; Koutros, Lenz et al. Citation2018, Koutros, Baris et al. Citation2018) met the eligibility criteria for consideration in the dose-response evaluation. However, all three studies were conducted to evaluate heterogeneity across subgroups of study populations that were analyzed in previous studies included in the Lynch et al. (Citation2017a) dose-response assessment (Chen et al. Citation2010b [lung]; Baris et al. Citation2016 [bladder]). That is, these three studies were focused on evaluating whether the association between arsenic exposure and bladder or lung cancer risk varies by methylation capacity or various polymorphisms related to arsenic methylation, DNA methylation, or DNA repair/tumor suppressor genes. Therefore, the three new studies were considered to provide supplemental information on potential modifiers of the dose-response relationship, but they were not used as primary sources. We also note below a few other studies that were initially considered and why they were excluded, as well as other studies that were worthy of consideration even though they were excluded by Lynch et al. (Citation2017a).
5.5.1. Bladder and lung cancer studies that met eligibility criteria
Koutros, Lenz et al. (Citation2018) is a stratified analysis of a subset of the population-based case-control study of a New England population (Baris et al. Citation2016) that focuses on heterogeneity in the arsenic-bladder cancer association by tumor suppressor immunophenotype, finding an association with cumulative arsenic exposure in cases whose tumors had positive gene expression of either p16 or Rb, but not in those with p16- and Rb-negative tumors. Baris et al. (Citation2016) reported statistically increased bladder cancer risk, with a positive exposure-response trend, with average daily arsenic intake or cumulative intake lagged 40 years, but not when unlagged or for arsenic concentration in well water (either lagged or unlagged). Arsenic intake was calculated by multiplying arsenic water concentration by drinking water intake. The statistically significant positive trend between drinking water intake from all sources and bladder cancer risk (i.e. greater water intake in cases than in controls) may thus be responsible for the apparent dose-response trend with calculated arsenic intake, but not with arsenic water concentration, especially given the small differences in arsenic concentrations among the dose groups.
In another stratified analysis, Koutros, Baris et al. (Citation2018) examined the effect of potential modifying factors on the previously reported association between bladder cancer risk and cumulative lifetime arsenic intake (mg) by Baris et al. (Citation2016). This study focused on factors and individual characteristics that might affect arsenic metabolism, including age, sex, smoking status, body mass index (BMI), alcohol consumption, and folate intake. Although none of these factors were found to be statistically significant effect modifiers of the association between cumulative arsenic exposure and bladder cancer, with or without a 40-year lag, higher odds ratios were observed for current and former smokers compared to never smokers. Unexpectedly, higher cumulative arsenic exposure showed a stronger positive association with bladder cancer risk among those who were folate sufficient than among those with folate insufficiency, and 40-year lagged higher cumulative arsenic exposure was significantly associated with bladder cancer risk among alcohol nondrinkers, but not among drinkers. We used the results in never smokers from Koutros, Baris et al. (Citation2018) in our evaluation of dose-response in never smokers.
Hsu et al. (Citation2017) is an updated analysis of a subset of participants in the Chen et al. (Citation2010b) northeastern Taiwan population-based prospective cohort study of lung cancer. Hsu et al. (Citation2017) restricted their analysis to the 19% of the cohort with speciated urinary arsenic levels, and categorized subjects by methylation capacity based on the ratio of MMA to As or DMA to MMA in urine. Hsu et al. (Citation2017) reported a positive dose-response trend for lung cancer risk with drinking water arsenic concentration in the <100 µg/L range among those with low but not high methylation capacity. However, low folate status in Taiwan (Chen, Pan et al. Citation2011) may reduce methylation capacity and result in higher internal exposure to trivalent arsenic forms. As noted above, low folate may also be an independent risk factor for lung cancer, particularly for men, and lower folate status in this population, combined with effects on methylation, could enhance associations between arsenic exposure and cancer risk. Such effects would not be relevant for populations with folic acid fortification, such as in the U.S.
5.5.2 Recent bladder and lung cancer studies that were excluded after review
Huang et al. (Citation2018) (and the earlier study by Huang et al. Citation2016, both of which were excluded from dose-response assessment because water arsenic concentrations were not measured and could not be derived) conducted a hospital-based study in bladder cancer patients in Taipei City, Taiwan. They reported associations of bladder cancer and upper tract urothelial carcinoma (UTUC) with speciated arsenic in spot urine samples. Participants had no known elevated arsenic exposure (average of 0.7 µg/L in municipal water); therefore, arsenic exposure was dominated by the diet. Conversion of the speciated urinary arsenic concentration to an inorganic arsenic dose is uncertain because of the contribution of DMA from the diet and adjustment for creatinine (see Section 5.3.2). Multivariate models were also adjusted for age, sex, education, smoking, alcohol, tea, coffee, pesticide contact, diabetes, hypertension, and urinary calculus for UTUC only, but not kidney function which could have affected the creatinine-adjusted arsenic concentrations. A related study by Lin et al. (Citation2018) used the same data but did not include the UTUC cases. Another hospital-based case-control study in Taiwan by Chang et al. (Citation2016) reported a higher risk of urothelial carcinoma was associated with higher urinary levels of total arsenic and four other metals. Arsenic in urine was not speciated and no data were presented on environmental exposures. Given the uncertainty in the exposure assessment, none of these studies provides clear evidence on the association between inorganic arsenic exposure and risk of bladder cancer/UTUC.
The study by de la Rosa et al. (Citation2017) on bladder and lung cancer in northern Chile was eliminated from further consideration in our evaluation because exposures were to arsenic drinking water concentrations >200 µg/L.
5.5.3 Other notable bladder and lung cancer studies
Other notable papers many of which were previously reviewed by Lynch et al. (Citation2017a), but not necessarily included in their dose-response assessment, are deserving of comment regarding their evidence on low-level arsenic exposures in association with cancer risk.
Steinmaus et al. (Citation2003) conducted a population-based case-control study of bladder cancer in areas of Nevada and California with elevated arsenic in well water. The dose groups were divided such that those with drinking water exposures to 10 µg/L or less would largely be in the lowest dose group (<10 µg/day) and those exposed to >50 µg/L would be the highest dose group (>80 µg/d), assuming 2L/day drinking water intake. Various odds ratios were calculated (adjusted for age, gender, education, smoking, occupation, and income) for the three dose groups, including a number of measures of average or cumulative exposure and time window analyses. None of the odds ratios for all participants were statistically significantly different from the null. Risks were statistically significantly increased in smokers for the highest dose group for the highest 1-year, 5-year, or 20-year of exposure, but risks were non-statistically significantly decreased in the highest dose group for never smokers. Time window analyses showed statistically significant increases in the highest dose group for smokers for 51 to 60 years and for 61–70 years before diagnosis but not for earlier periods or for 71–80 years. This study was used in the evaluation of the dose-response at low-level exposures in never smokers.
Karagas et al. (Citation2004) investigated an association of bladder cancer with toenail arsenic concentration in a New Hampshire population-based case-control study with water arsenic concentrations of <0.01 to 180 µg/L (Karagas et al. Citation2002). This study could have been included in the dose-response modeling of Lynch et al. (Citation2017a) by conversion of the toenail results to water concentration, based on Karagas et al. (Citation2000) (see Supplemental Information 2). A borderline significant elevated odds ratio was reported for the highest exposure category compared to the lowest (2.17, 95% CI: 0.92 − 5.11) in smokers, whereas no association was apparent in never smokers.
Baastrup et al. (Citation2008) is a prospective population-based cohort study in Denmark that assessed drinking water arsenic levels in association with various cancers including lung, bladder, and skin. The results (reported as incidence rate ratios per 1 µg/L time-weighted average water arsenic concentration or per 5 mg cumulative arsenic exposure) were modeled with arsenic exposure metrics only as continuous variables, thereby assuming a linear, non-threshold dose-response trend. Overall, no association was found between specific cancers examined and time-weighted average arsenic exposure (range: 0.05 to 25.3 µg/L; median: 0.7 µg/L) or cumulative arsenic exposure, with the exception non-melanoma skin cancer, for which statistically significant inverse associations were reported with both exposure metrics.
Steinmaus, Ferreccio, Yuan et al. (Citation2014) reported elevated lung cancer risks in association with greater intake of drinking water arsenic concentrations <100 µg/L, based on 92 lung cancer cases and 288 population-based controls from towns in northern Chile. The study population was likely a subset of a study involving a wider range of exposures (Steinmaus et al. Citation2013). Cumulative, lifetime average, peak, and recent exposures (within 40 years) to arsenic were not significantly associated with lung cancer risk. However, significantly increased risks were reported for the top tertile of the highest single year of arsenic exposure lagged by 40 or more years: odds ratios (adjusted for age, sex, and smoking intensity) of 1.00, 1.43 (90% confidence interval [CI]: 0.82 − 2.52), and 2.01 (90% CI: 1.14 − 3.52) for tertiles with mean arsenic water concentrations of 6.5, 23.0, and 58.6 µg/L, respectively (p for trend =0.02). Odds ratios were higher for subjects younger than 65 years old (1.62 [90% CI: 0.67 − 3.90] and 3.41 [90% CI: 1.51 − 7.70]), indicating possible increased susceptibility to early-life exposure. Use of 90% CIs rather than 95% CIs was justified by an a priori assumption that arsenic increases lung cancer risk, although 95% instead of 90% CIs were reported for odds ratios in relation to smoking by Steinmaus, Ferreccio, Yuan et al. (Citation2014), and for cancer risks at higher arsenic exposure in Steinmaus et al. (Citation2013). The authors also acknowledged that increased risks have been shown at arsenic water concentrations >100 µg/L, whereas they are “less well understood” at lower concentrations. An important limitation of this study that could have affected the results was the high potential for differential exposure misclassification between cases and controls. Arsenic intake (µg/day) was the exposure measure for subjects who could be interviewed regarding their current and past water intake. Such information was obtained for 93% of the controls but only 46% of the cases, many of whom were deceased. For deceased subjects, their arsenic water concentration was used as the exposure measure instead of arsenic intake. Classification of exposure based on arsenic water concentration resulted in the inclusion of more subjects (both cases and controls) in the highest dose group than when exposure was classified based on arsenic intake (supplemental information of Steinmaus, Ferreccio, Yuan et al. Citation2014). Thus, the finding of higher exposure for cases versus controls may have been the result of inclusion of more cases than controls in the highest dose group because exposure was based on arsenic water concentration for 54% of cases but only 7% of controls.
D’lppoliti et al. (2015) investigated lung cancer mortality and arsenic exposure in a large, administrative cohort study using population-based registries in central Italy. This study was clearly an outlier in the modeling of Lynch et al. (Citation2017a), showing a steep dose-response association at relatively low exposures (dose groups of 2, 6, 12, µg/L). The quality of this study was rated as low based on not meeting criteria for adequate adjustment for smoking, exposure measurement, assay accuracy, and adjustment for confounders (Lynch et al. Citation2017a). Lack of individual-level adjustment for smoking in the evaluation of lung cancer resulted in our exclusion of this study for consideration of dose-response assessment.
Lamm et al. (Citation2018), Mendez et al. (Citation2017), Ferdosi et al. (Citation2016), and Lamm et al. (Citation2004) are large ecological studies of lung and/or bladder cancer incidence or mortality in relation to county-level arsenic levels in U.S. drinking water supplies. Although they did not meet our inclusion criteria for assessing individual-level arsenic exposure and adjustment for smoking, these studies are notable for their focus on large U.S. populations. The combined meta-regression analysis of Lamm et al. (Citation2015) also noted little difference in dose-response across studies of drinking water arsenic exposure and lung cancer risk, including ecological studies.
With the exception of Mendez et al. (Citation2017), these studies did not show significantly increased risks of lung or bladder cancer in populations with higher arsenic water concentrations. Unlike the two earlier ecological studies (Lamm et al. Citation2004; Ferdosi et al. Citation2016), Mendez et al. (Citation2017) examined cancer incidence rather than mortality, and had access to data from some states and one USEPA region that at the time were not available to Lamm et al. (Citation2018); they analyzed mean rather than median water concentrations by county (although Lamm et al. [Citation2004] also showed results based on mean arsenic levels); and they included counties with as low as 10% use of groundwater water supplies. Mendez et al. (Citation2017) reported a statistically significant positive log-linear association between mean county-level water arsenic concentration (up to 157.7 µg/L) and lung cancer incidence in women and bladder cancer incidence in men and women, adjusting for county-level confounders.
Lamm et al. (Citation2018) investigated lung cancer incidence in association with median (mostly based on 1 or 2 wells) county groundwater arsenic concentrations <50 µg/L, focusing on counties that had ≥80% population dependency on groundwater supplies. For men, women, and both genders combined, lung cancer incidence was statistically significantly lower in counties with median arsenic water concentrations of 10–50 µg/L compared to those with undetectable arsenic in the water, adjusting for county-level confounders. Negative associations, which were statistically significant among men and both genders combined, also were detected in the full analysis of all counties, regardless of groundwater dependency. Differences in results between this study and that of Mendez et al. (Citation2017) may be related to the restriction by Lamm et al. (Citation2018) to counties with lower drinking water arsenic levels, additional non-publicly-available data in Mendez et al. (Citation2017), differences in adjustment for county-level confounders, several of which were strongly associated with cancer risk, or 80% restriction to groundwater supplies by Lamm et al. (Citation2018) (although their analysis without this restriction had similar results).
Zhang et al. (Citation2016) conducted a large pooled prospective cohort study of rice consumption and cancer risk that included over 200,000 U.S. participants, detailed examination and adjustment for a large number of potential covariates/effect modifiers, and follow up of all reported cancers with medical records. Despite a large number of comparisons (including white or brown rice which has more arsenic), associations were null for total cancers and specific cancer sites including lung, bladder, kidney, prostate, breast, colon and rectum, and melanoma. Null associations were also reported when restricting the analysis to those of European Americans or nonsmokers. Nachman et al. (Citation2018) cite the borderline statistical significance for highest consumption group of total rice intake and bladder cancer (RR of 1.32; 95% CI: 0.99 − 1.76; p = 0.09) as evidence of a causal effect of rice consumption (and arsenic) on bladder cancer. However, a statistically significant association was not reported for brown rice consumption, which has higher levels of inorganic arsenic than white rice (FDA 2013a). Moreover, relative risks for total rice consumption and lung cancer were uniformly less than 1.0 (not statistically significantly), indicating little support for arsenic as a causal agent.
5.5.4. Skin cancer studies
Fewer epidemiological studies have quantified the association between arsenic exposure and skin cancer, compared with lung and bladder cancers. Even considering the non-ecological studies, many of these studies, including the one recent study that met our initial inclusion criteria but not our final eligibility criteria (Kim et al. Citation2017), have major limitations for assessing a causal relationship between skin cancer and arsenic exposure at low doses. Kim et al. (Citation2017) conducted a hospital-based case-control study of non-melanoma skin cancer and speciated urinary arsenic levels in a province of southeastern Korea with low arsenic levels <0.5 µg/L in water supplies. As noted above, speciated urinary arsenic levels are short-term measures of inorganic arsenic exposure that are compromised by the contribution of DMA from consumption of rice, seaweed, and seafood in Asian populations. Other studies considered for dose-response assessment are described briefly below.
Leonardi et al. (Citation2012) is a large, hospital-based case-control study of basal cell carcinoma (BCC) in patients who had lived at least one year in areas of Hungary, Romania, and Slovakia with elevated arsenic in groundwater. No mention was made of squamous cell carcinoma (SCC). Associations with average lifetime water arsenic concentration, peak daily exposure (in mg/day), and cumulative dose (in g) all showed statistically significant trends over the quintile dose groups, with statistically significant or borderline significant associations with the two highest exposure groups (7.10 to 19.43 and 19.54 to 167.29 µg/L). The median lifetime average water concentration was 1.2 µg/L (interquartile range: 0.7 to 13.8 µg/L), 60% of the population had a lifetime average of <7 µg/L. Nevertheless, historical exposures were considerably higher before interventions beginning in the 1980s. Drinking water derived from the alluvial basin on the Hungarian-Rumanian border or the aquifer in central Slovakia may have had concentrations up to 400 and 250 µg/L, respectively (Hough et al. Citation2010). In the examples presented, four locations with arsenic concentrations between 150 and 250 µg/L declined to below 100 µg/L between 1984 and 1995 (Hough et al. Citation2010). Historical arsenic water concentrations thus could have exceeded the low-dose range under consideration for some participants especially in the higher exposure categories, and their average drinking water concentrations are not necessarily representative of low-level exposure.
Lamm et al. (Citation2007) conducted a cross-sectional investigation of clinically determined BCC and squamous cell carcinoma (SCC), as well as nonmalignant skin changes, in association with arsenic concentrations in well water among nearly all residents (3179 out of 3229) of three villages in Inner Mongolia. All participants included in the analysis had information on well-use history and diagnostic data on dermatological diseases (hyperkeratosis, dyspigmentation, and skin cancer) from a clinical survey. Although arsenic concentrations in well water were as high as 2000 µg/L, 69% of the participants had the highest arsenic concentration (for a minimum of 1 year) that was less than 100 µg/L. Only eight skin cancer cases were reported, all of whom had the highest arsenic well water concentration >150 µg/L. The study did not assess sun exposure but noted that skin cancer occurred only in people with skin keratoses or pigmentation changes in non-sun-exposed areas of the body, so the skin cancers were likely related to arsenic exposure. Those with any potential arsenic-related skin changes were 6.3% of the study population.
Karagas et al. (Citation2001a) conducted a population-based case control study of BCC and SCC in relation to arsenic toenail concentration in New Hampshire, using a design similar to that of Karagas et al. (Citation2004). Odds ratios, adjusted for age and gender, were elevated in the highest exposure category, but not statistically significantly so. Based on the correlation between toenail arsenic concentration and drinking water arsenic concentration in this population ( of Karagas et al. Citation2000), the highest exposure category included well water exposures around 100 µg/L and higher. Other factors evaluated as potential confounders but not found to affect the association included education, smoking, skin reaction to sun, radiation exposure, and type of water supply. Amount of sun exposure and occupation were apparently not considered as confounders. Not adjusting for these factors could have biased odds ratios upward, since higher arsenic exposures in New England are associated with the use of private wells, which are more common in rural areas where people may have more sun exposure from outdoor activities or occupations.
Applebaum et al. (Citation2007) subsequently examined the influence of polymorphisms of certain DNA repair genes and found some suggestion of possibly higher risk of BCC and SCC at higher toenail arsenic concentrations (grouped as < or >0.286 µg/g) with certain variants. However, the 95% CIs for nearly all associations included 1.0, and only one statistically significant interaction was found (between a polymorphism in the XPD gene and arsenic in relation to SCC risk).
A separate study of this New Hampshire population reported that glucocorticoid use, and presumably immune modulation, was associated with an increased risk of SCC (adjusted odds ratio =2.31; 95% CI: 1.27 − 4.18) and less so for BCC (adjusted odds ratio =1.49; 95% CI: 0.90 − 2.47) (Karagas et al. Citation2001b). Presumably, no interaction was found with arsenic exposure because glucocorticoid use was not mentioned in the studies of arsenic (or rice) exposure and skin or bladder cancer in this population (Karagas et al. Citation2001a; Citation2004; Gilbert-Diamond et al. Citation2013; Gossai et al. Citation2017). The lack of an effect of glucocorticoid use on arsenic-related cancer risk does not support immune dysregulation as an important factor in the mode of action for arsenic-related carcinogenesis at low exposures.
Gilbert-Diamond et al. (Citation2013) examined associations of SCC with speciated urinary arsenic and individual arsenic species in a population based case control study in New Hampshire (conducted in 2003–2009 rather than 1993–1996 in Karagas et al. Citation2001a). The study noted a greater tendency toward associations of arsenic exposure with SCC than with BCC. Water arsenic concentrations for most of the study population were low: the median was 0.33 (interquartile range: 0.14 − 1.11) µg/L for cases and 0.31 (interquartile range: 0.12 − 0.94) µg/L for controls. Most of the arsenic exposure was thus from the diet, which was likely confounded by organic arsenic forms, especially DMA and organic precursor compounds. People who ate fish within two days of urine collection were excluded, but those who ate rice (22%) were included. Eating rice (which has DMA as well as inorganic As) was associated with higher DMA in urine, although results of a secondary analysis excluding the rice consumers were reported to be generally consistent with the main analysis. The analysis was adjusted for sex, age, BMI, education, smoking status, skin reaction to sun (but not occupation or extent of sun exposure), and urinary creatinine concentration. Associations of SCC with total speciated arsenic or individual arsenic species were statistically significant after natural logarithmic transformation, except for inorganic arsenic. Analyses based on tertiles of urinary arsenic data showed statistically significantly elevated risks for the third tertile for mainly MMA in urine. Duration of consuming current water source (< or >17 years) did not seem to modify these associations; and paradoxically, odds ratios tended to be stronger in those with shorter duration of exposure. As noted above, the short-term nature of urinary arsenic, which was measured after SCC diagnosis and corrected for urinary creatinine, adds to the uncertainty and low reliability of these measurements for characterizing inorganic arsenic exposure as the causal agent in development of SCC.
In a cross-sectional study, Knobeloch et al. (Citation2006) evaluated 19 townships in Wisconsin that had elevated arsenic in well water. The mean water arsenic concentration was 12.0 µg/L (standard deviation: 91.2 µg/L; maximum: 3,100 µg/L; 20% of samples ≥10 µg/L). Skin cancer was self-reported by questionnaire, with no information on the type of skin cancer (i.e. without exclusion of melanoma) and no diagnostic confirmation; additionally, no information was considered on sun exposure, skin response to sunlight, pigmentation, or occupation. Thus, the results of this study are of questionable validity. Increased skin cancer risk with greater arsenic exposure was statistically significant only among smokers aged 65 years or older.
Baastrup et al. (Citation2008), as noted above, is a Danish prospective cohort study that found a statistically significant inverse association between time-weighted average arsenic exposure (0.05 to 25.3 µg/L) and risk of non-melanoma skin cancer. The data presented in this study, which assume a linear trend based on the modeling of arsenic exposure only as a continuous variable, could not be used in quantitative dose-response modeling.
Hsueh et al. (Citation1997) is a prospective cohort study of a population in three villages with high arsenic-related disease rates in the arseniasis-endemic area of southwestern Taiwan. Unlike the larger ecological studies of this area (e.g. Morales et al. Citation2000), Hsueh et al. (Citation1997) tracked villages where individuals lived and tied individual exposures to individual outcomes. Use of village average well-water arsenic concentrations could have resulted in some exposure misclassification for individuals who lived in villages with multiple wells with a large difference in arsenic water concentrations (NRC Citation1999). This study is not informative of risks at low-level exposures because of the high doses included in the arsenic exposure groups (e.g. water concentrations of 0, 0 − 700, >710 µg/L). The significant inverse associations of skin cancer risk with higher serum beta-carotene levels and greater percentages of MMA and lower percentages of DMA in urine of cases, likewise involve these high exposures and the impoverished conditions of this population.
Gossai et al. (Citation2017) conducted a population-based case-control study of the New Hampshire Skin Cancer Study Population to investigate the association of rice intake (based on a questionnaire of food frequency over the past year) with non-melanoma skin cancer. An odds ratio of 1.5 (95% CI: 1.1 − 2.0) was reported for SCC for any rice consumption compared to those reporting no rice consumption and appeared to be based largely on those with arsenic drinking water concentrations <1 µg/L. However, a clear dose response for SCC risk was not observed with greater amounts of rice intake.
5.5.5. Nonmalignant skin lesion studies
Because of the few studies that have examined associations with skin cancer, we also reviewed evidence from studies of arsenic exposure and nonmalignant skin lesions. Skin lesions are an obvious and prevalent sign in populations exposed to elevated arsenic levels in drinking water that also experience other health effects including skin, bladder, and lung cancer (Karagas et al. Citation2015). Skin lesions including hyper- or hypopigmentation and hyperkeratosis occur after a shorter latency than skin cancer, and some skin lesions are thought to involve pre-malignant changes that can lead to skin cancer (NRC Citation1999; Tsuji et al. Citation2004; Seow et al. Citation2012; Cohen et al. Citation2013). Case-control, cohort, and cross-sectional studies of skin lesions with individual assessments of arsenic exposure (including some with exposures <150 µg/L) reviewed by Karagas et al. (Citation2015), as well as more recent publications, were predominantly conducted in Inner Mongolia, West Bengal, Bangladesh, and Pakistan. Overall, statistically significant increases in skin lesions generally occur at arsenic water exposure concentrations >50 µg/L or >100 µg/L, although some studies show a positive trend in risk beginning at drinking water arsenic concentrations as low as >5 µg/L. Such associations may be affected by exposure measurement error, as well as variation in the diagnosis of arsenic-related skin lesions. The influence of these uncertainties—in particular, the importance of assessing lifetime drinking water arsenic exposure and reevaluating the presence of skin lesions over time in relation to changing arsenic exposure levels—is illustrated by the series of detailed studies in the West Bengal population.
A cross-sectional study of 7683 participants in West Bengal in 1995–1996 noted that hyperkeratosis and hyperpigmentation of the skin were rare at arsenic drinking water levels <50 µg/L (measured in the primary current drinking water source), infrequent at 50 to 100 µg/L, and more common with water concentrations above this level (up to 3400 µg/L; Mazumder et al. Citation1998). A higher prevalence of skin lesions (especially keratoses) was observed in those with low body weight, suggesting a role of malnutrition. A follow-up case-control study conducted in 1998–2000 in West Bengal selected cases (those with skin lesions diagnosed previously; N = 192) and controls (those without skin lesions diagnosed previously; N = 213) from those in the earlier cross-sectional survey whose primary drinking water source contained <500 µg/L arsenic (Haque et al. Citation2003). Cases and controls had similar sociodemographics and body mass index. This study investigated participants’ drinking water exposure history for at least the prior 20 years. Skin lesions were examined by a physician, as before, and were also photographed for subsequent consensus review by four physicians to confirm whether skin lesions were likely to be related to arsenic exposure.
Observations from Haque et al. (Citation2003) and Lamm et al. (Citation2007) (see above) indicate the importance of obtaining a complete drinking water history and diagnosing skin lesions related to arsenic exposure. The occurrence of skin lesions was associated with past peak arsenic exposures, indicating that current or average exposures may be misleading when used to characterize risk of skin lesions. In Haque et al. (Citation2003), 25 of the controls lacked skin lesions in 1995–1996 but had skin lesions in 1998–2000. The drinking water history of these individuals revealed “high” past arsenic exposures (average peak of 253 µg/L; prior 5-year average of 140 µg/L). Conversely, of the 192 cases diagnosed in 1995–1996, 72 no longer had arsenic-related skin lesions in 1998–2000, including 57 who had consumed water with an arsenic concentration <50 µg/L in the intervening period. Thus, skin lesions appear to have resolved with low recent exposure, although misdiagnoses may also have occurred. The average latency for the observation of skin lesions in this study was 23 years (range 10–42) from first exposure to an arsenic water concentration >100 µg/L, and 19 years (range 3–42) from each case’s first exposure to their peak concentration, which ranged from 115 to 1,113 µg/L). The average peak drinking water arsenic concentration was 325 µg/L in cases and 180 µg/L in controls. All confirmed skin lesion cases in 1998–2000 had consumed water with an arsenic concentration >100 µg/L at some point in their life.
A recent cross-sectional study examined 398 children and adults who had lived for at least the past 5 years in 6 previously unstudied villages in Pakistan with a wide range in well water arsenic levels (<1 to 3090 µg/L) (Rasheed et al. Citation2018). The authors reported that the prevalence of arsenic-related skin lesions was 0.68%, 13.82%, and 60% in associations with current water arsenic concentrations of 10–50, 50–100, and >100 µg/L, respectively. Skin lesions were more prevalent for older participants (age >16 years), men versus women, intensive versus non-intensive laborers, and those with higher body mass (likely related to age and sex), higher water intake, or less efficient methylation capacity (indicated by higher percentage of MMA or inorganic arsenic and lower percentage of DMA in urine). Because a complete exposure history was not reported, the one case of skin lesions in the current 10–50 µg/L group could not reliably be attributed to this exposure level. Similarly, past exposures more than 5 years previously for cases in the 50–100 and >100 µg/L groups were unknown.
A nested case-control study of 876 incident skin lesion cases and individually matched controls from a prospective cohort study in Bangladesh also reported statistically significant increases in skin lesions with higher arsenic well water concentrations, beginning with the 10 − 50 µg/L exposure group (compared to <10 µg/L) and extending to the highest exposure group (>200 µg/L) with a monotonic positive trend (Niedzwiecki et al. Citation2018). Past drinking water concentrations were not mentioned. This study also reported an increased risk of skin lesions in association with increased serum homocysteine (an indicator of impaired one-carbon metabolism from deficiency of folate and other nutrients), whereas risks were decreased for greater percentage of DMA in urine.
5.6. Assessment of dose-response
5.6.1. Bladder and lung cancer
Our updated analysis found very few informative low-dose epidemiological studies that would add studies to the dose-response modeling of bladder and lung cancer conducted by Lynch et al. (Citation2017a). Lynch et al. (Citation2017a) reported that sequential elimination of highest doses showed a trend toward an increasing slope and widening of the confidence intervals of the meta-regression analysis (see Supplemental information from Lynch et al. Citation2017a). We reexamined the data for the studies considered by Lynch et al. (Citation2017a) by focusing on data at lower doses (i.e. <200 µg/L). Compared to studies with low-level arsenic exposure evaluated by Lynch et al. (Citation2017a), the earlier meta-regression analysis for lung cancer (non-ecological studies of Lamm et al. Citation2015), and meta-analysis of bladder cancer (Tsuji, Alexander et al. Citation2014), studies included in our low-level dose-response evaluation of bladder and lung cancer were most similar to those selected by Lynch et al. (Citation2017a) given similar study selection criteria, the recent publication, and focus on both bladder and lung cancer ().
Three studies in Lynch et al. (Citation2017a) were excluded because they involved only high-dose risk comparisons for bladder cancer (Huang et al. Citation2008; Wang, Yeh et al. Citation2009) and bladder and lung cancer (Chiou et al. Citation1995). These studies from the southwestern Taiwan arseniasis-endemic area involved a malnourished population with increased susceptibility for arsenic-related health risks (Chen et al. Citation2001), and also used median village well water arsenic concentrations, which would underestimate potential exposure in some villages (e.g. median of 30 µg/L for a range in concentrations of 10 to 770 µg/L within a village; NRC Citation1999).
For lung cancer, we also eliminated D’Ippoliti et al. (Citation2015) and the results exclusively among smokers from Mostafa et al. (Citation2008) because of concerns regarding residual confounding or interactions with smoking. Lynch et al. (Citation2017a) also excluded D’Ippoliti from their meta-regression analysis because this study did not adjust for smoking in the statistical analysis. We included the results of only male nonsmokers from Mostafa et al. (Citation2008) because this study stratified by smoking for men, but did not present stratified data for women or smoking-adjusted data for the overall study population. For bladder cancer, we added the study of Karagas et al. (Citation2004) by converting the arsenic toenail concentrations to water concentrations based on data presented in Karagas et al. (Citation2000) (see Supplemental Information 2 for conversion). For the other studies, with the exception of Chen et al. (Citation2010a), we used the arsenic dose and cancer risk data for bladder and lung cancer presented by Lynch et al. (Citation2017a) (truncated to the low dose range <200 µg/L). For Chen et al. (Citation2010a) we selected the results based on all urinary cancers instead of only urothelial cancers. This selection is consistent with the association between arsenic exposure and all urinary tract cancers, even though the other studies reviewed focused on urothelial cancer or “bladder” cancer. However, including the other urinary cancers likely has little effect on the results because urothelial carcinomas are the predominant cancer type (Chen et al. Citation2010a).
Lynch et al. (Citation2017a) estimated midpoint arsenic water exposure concentrations of the dose groups, adjusted to account for differences in water consumption rates and body weight in some foreign populations as compared to the U.S. For consistency with calculations of USEPA cancer risk assessments, they also selected results for lifetime average risk estimates from studies or the most similar measure available. To estimate midpoint exposures for open-ended lowest or highest dose groups presented as less than or greater than a value, Lynch et al. (Citation2017a) assumed the midpoint between 0 and the lowest value or the midpoint between the highest value and two times the highest value, respectively.
Adjustment for differences in water consumption rates was based on study/population-specific values that were available for most populations, except for northeastern Taiwan (Chen et al. Citation2010a,b) and Bangladesh (Mostafa et al. Citation2008; Mostafa and Cherry Citation2015), for which Lynch et al. (Citation2017a) assumed default water consumption rates of 2.75 L/day and 3 L/day, respectively. Although some data, particularly for Bangladesh, indicate that combined direct drinking and cooking water intakes maybe 1.5 to 2 times higher than the assumed intake rates (Tsuji, Perez et al. Citation2014), we used the assumptions of Lynch et al. (Citation2017a) for Taiwan and Bangladesh to facilitate comparisons with their earlier work. Underestimation of drinking water intake rates is conservative and will tend to overestimate risk per dose or cancer potency. Adjustment for differences in body weight assumed a population average of 70 kg for non-Asian populations, 50 kg for Taiwan, and 60 kg for Bangladesh (Lynch et al. Citation2017a). As noted by Lynch et al. (Citation2017a) a sensitivity analysis omitting these adjustments tended to overestimate the slope of the dose-response relationship because of underestimation of dose per body weight in the Asian populations who have higher water intake rates and lower body weights on average.
We reviewed the water intake rates selected for the studies included by Lynch et al. (Citation2017a) and made a few changes to improve overall consistency with the results reported in certain studies (Bates et al. Citation2004; Meliker et al. Citation2010; Baris et al. Citation2016). For example, Lynch et al. (Citation2017a) apparently assumed a default water consumption rate of 1.2 L/day for some U.S. studies (Meliker et al. Citation2010; Baris et al. Citation2016). In general, when reported, we selected mean or median water intake rates for the control group and averaged rates reported at different time periods.
The study reported dose groups, drinking water intake assumptions, adjusted midpoint arsenic water concentrations, and relative risks and case numbers for each included study for the current dose-response evaluation are summarized in for bladder cancer and for lung cancer.
Table 4. Studies included in the current dose-response evaluation of arsenic water concentration and bladder cancer risk.
Table 5. Studies included in the current dose-response evaluation of arsenic water concentration and lung cancer risk.
The dose-response for the bladder cancer studies in the low-dose range shows considerable variability with an overall lack of dose-response (). One outlier is the statistically significant increased risk (RR = 1.52; 95% CI: 1.08 − 2.14) at 53 µg/L, compared with the reference group of 8.75 µg/L, in the Bangladesh population studied by Mostafa and Cherry (Citation2015). However, the next highest exposure group in this study, at 131 µg/L, had a nearly null RR of 1.07 (95% CI: 0.73 − 1.57) as did the next highest dose group (262.5 µg/L) above the low-level range with RR of 0.99 (95% CI: 0.69 − 1.41). At the higher end of the low exposure range (), a statistically significant increased risk of bladder cancer was reported in the northern Chile population for the 80 to 197 µg/L (125 µg/L midpoint) average lifetime exposure group (odds ratio of 2.62, 95% CI: 1.53 − 4.50; Steinmaus et al. Citation2013). However, participants with average lifetime exposures in this range may have experienced higher exposures considerably above 100 µg/L in the past because of greatly elevated arsenic water concentrations in some Chilean towns before installation of water treatment (i.e. prior to 1971 or 1971–1977). Risks were lower and not statistically significant (odds ratio of 1.36; 95% CI: 0.78 − 2.37) for lifetime average arsenic concentration of 11 to 90 µg/L before 1971 (Steinmaus et al. Citation2013).
Figure 4. Relative risks (95% confidence intervals) of bladder cancer at low-level average arsenic water concentrations (a. <12 µg/L; b. <180 µg/L).
The lung cancer data likewise indicate a lack of dose-response below 60 to 100 µg/L arsenic in drinking water (), with statistically non-significant relative risks near 1.0. Although each of the available studies has only one dose group below 100 µg/L to compare with the reference group (all below 12 µg/L), the relative risks are both above and below 1.0, indicating that attenuation from exposure misclassification cannot explain the lack of a positive dose response. Two studies show a significantly increased risk in their top exposure group with a midpoint concentration of about 120 µg/L and upper-end limits around 200 µg/L (). Both studies are of the northern Chile population (Smith et al. Citation2009; Steinmaus et al. Citation2013). As for bladder cancer, lifetime average exposure in the larger study (Steinmaus et al. Citation2013) involving longer follow-up (diagnosis in 2007–2010 versus 1994–1996 in Smith et al. Citation2009) likely underestimates the impact of higher past exposures. For lifetime average exposure of 11 to 90 µg/L before 1971, lung cancer risks were not statistically significantly elevated (odds ratio of 1.27; 95% CI: 0.81 − 1.98). The earlier results presented by the smaller study by Smith et al. (Citation2009) were based on average exposure concentration from 1958 to 1970. However, even for average exposures during this period of higher arsenic water concentrations for some towns, migration among towns could have resulted in higher (or lower) past exposures than reflected by the average concentration during this period, with those in the higher exposure category (60 to 199 µg/L) being more likely to have consumed some water in the towns with exposures in excess of the low-level range.
Figure 5. Relative risks (95% confidence intervals) of lung cancer at low-level average arsenic water concentrations.
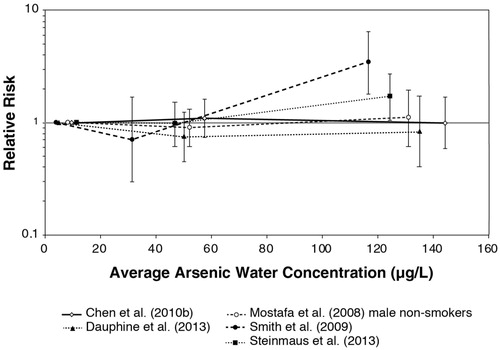
Limitations of our presentation of dose-response data in and are the inability to include evidence from studies that used arsenic exposure metrics other than drinking water concentration, and the potential for residual confounding from incomplete adjustment for tobacco use. Additional insight can thus be obtained by examination of the dose-response associations in never smokers. For this analysis, we considered other dose metrics in addition to arsenic drinking water concentration from the available studies that reported data in the low-level range of exposure for never smokers. Studies included for this analysis were all that met the inclusion criteria, with the exception of the restriction to studies reporting exposure as arsenic concentration in water. For completeness, presents the never smoker results for the different types of dose metrics reported in the studies. The forest plot of findings for never smokers in illustrates representative results for each study (see footnotes of for details on selection of results from studies with multiple exposure metrics). The results for Dauphine et al. (Citation2013) could not be included because of the lack of quantitative precise odds ratio estimates (see ).
Compared to the results of models that included smokers and nonsmokers with adjustment for smoking (e.g. nearly all of the studies in and , and ), even less of a dose-response pattern is apparent at low doses for never smokers. One exception is reported by Ferreccio et al. (Citation2013) for bladder cancer, but not for lung cancer (). In the 11 to 91 µg/L (lifetime average before 1971) dose group, the bladder cancer risk was nearly statistically significantly increased with a higher odds ratio among never smokers (2.66; 95% CI: 0.91 − 7.83) than among smokers and never smokers combined as presented by Steinmaus et al. (Citation2013) (1.36; 95% CI: 0.78 − 2.37). Overall, however, for studies that reported associations between arsenic exposure and bladder and lung cancer in never smokers (, ), few statistically significant associations and inconsistent dose-response patterns are apparent in the low dose range of exposure (i.e. approximately <100–150 µg/L) or even when comparing this low dose range to the next higher dose group. In fact, a number of relative risk estimates (mostly odds ratios from case-control studies) are less than 1. As noted above, the occurrence of such associations in the negative direction, suggests that regression toward the null is not the sole explanation for the lack of a statistically significant positive association between low-level arsenic exposure and risk of lung or bladder cancer among never smokers. Quite the opposite, exposure misclassification among groups will, in fact, produce more of a monotonically increasing apparent dose-response relationship, even when the underlying data are threshold in nature (Crump Citation2006; Rhomberg et al. Citation2011). The evidence from results in never smokers thus supports a lack of dose-response in the low dose region.
Figure 6. Odds ratios (except as noted) for lung and bladder cancer at low-level arsenic exposures reported for never smokers. *Converted from toenail concentration ranges based on Karagas et al. (Citation2000) (see ).
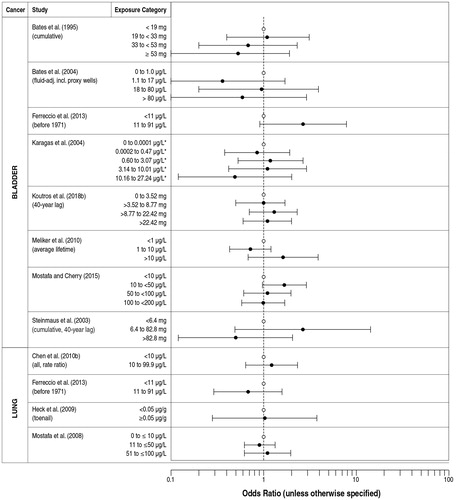
Table 6. Results of epidemiological studies of low-level arsenic exposure and risk of bladder or lung cancer among never smokers. Results in italics are not directly relevant to the dose-response assessment of low-level drinking water arsenic and bladder or lung cancer risk but are included for completeness.
5.6.2. Skin cancer
Of the few studies meeting most of the study eligibility criteria, there are considerable limitations for assessing the association between low-dose inorganic arsenic exposure and skin cancer risk because of potential confounding influences of other risk factors, such as sun exposure, and inclusion of historical exposures at high doses in the study of BCC by Leonardi et al. (Citation2012). Studies with some of the lowest arsenic exposures that were based on short-term biomarkers or a consumption survey of arsenic from food exposures that include DMA (Gilbert-Diamond et al. Citation2013; Gossai et al. Citation2017), likewise diminish confidence that such findings are actually related to inorganic arsenic exposure at these exposure levels per se.
A notable limitation of the skin cancer studies is that with the exception of Lamm et al. (Citation2007), these studies do not note whether the skin cancers observed occurred on parts of the body that were not sun-exposed or whether arsenic-related skin lesions were specifically diagnosed in the population. The most informative study of skin cancer by Lamm et al. (Citation2007) included diagnosis of arsenic-related skin cancer and skin lesions and detailed investigation of historical arsenic exposures in a population in Inner Mongolia, exposed to arsenic in drinking water above and below 100 µg/L. Based on threshold dose-response modeling, Lamm et al. (Citation2007) reported a threshold for skin cancer at 122 µg/L and a threshold for nonmalignant skin changes at 40 to 50 µg/L. The detailed assessment of skin lesions and current and historical arsenic exposures in West Bengal indicated an exposure concentration around 100 µg/L and above at some point in life as a threshold for increased occurrence of skin lesions related to arsenic exposure (Haque et al. Citation2003). Thus, the overall evidence, based on skin cancer and skin lesions and the likely mechanistic relationship between these effects, suggests a threshold for skin cancer risk probably around 100 µg/L. However, it may be as low as 50 µg/L based on increased risk of skin lesions in populations with lower nutritional status and likely higher drinking water intake than for U.S. populations.
5.6.3. Individual susceptibility
Many of the relevant epidemiological studies have been conducted in populations with increased risk of arsenic-related health effects from greater dose of inorganic arsenic and its reactive trivalent form and metabolites due to external factors (i.e. increased drinking water intake) and internal factors (i.e. reduced methylation capacity from nutritional deficiencies) (Cohen et al. Citation2013; Tsuji, Perez et al. Citation2014; Lynch et al. Citation2017a; Rasheed et al. Citation2018; Sharma and Flora, Citation2018). In some of these populations (e.g. in Bangladesh), impoverished living conditions, which can be correlated with use of wells with higher arsenic concentrations, are also associated with less resistance to arsenic toxicity resulting from impaired anti-oxidant defense and DNA repair. Such lifestyle factors are also independent risk factors for several of the common diseases that are associated with arsenic exposure.
Thus, extrapolation of results from these study populations is expected to be protective of the relatively well-nourished U.S. population with lower water intakes (and who generally do not use local water to grow all of their rice and vegetables). As noted above, much of the most recent epidemiological research on arsenic has focused on assessing genetic or behavioral heterogeneity in individual susceptibility within previously studied populations. Several studies suggest that positive associations between inorganic arsenic exposure and risk of bladder, lung, or skin cancer or lesions are more likely to be observed in those with reduced methylation capacity or various polymorphisms in genes related to arsenic methylation, DNA methylation, or DNA repair/tumor suppression (NRC Citation2013; Bhattacharjee et al. Citation2018; Bjørklund et al. Citation2018), but results for specific genotypes or phenotypes are sparse and allow no conclusions.
Associations of cancer risk with arsenic methylation capacity (i.e. proportions of urinary metabolites; e.g. Gilbert-Diamond et al. Citation2013; Melak et al. Citation2014; Hsu et al. Citation2017; Gamboa-Loira et al. Citation2017 and studies reviewed therein), as well as variations in the arsenic-cancer association by methylation capacity, are particularly uncertain for interpreting causal relationships. Arsenic methylation, as well as general health and resistance to cancer and other diseases, requires one-carbon metabolism, which involves nutritional co-factors and multiple genes coding for enzymes and other biochemical factors. Individual health conditions or exposure to other substances may also influence arsenic methylation capacity. Thus, assessment of arsenic methylation capacity as a susceptibility factor for arsenic-related disease in retrospective case-control or cross-sectional studies is less robust than in prospective cohort studies. Extrapolation of findings on methylation-mediated susceptibility from nutritionally deficient populations to U.S. populations is also uncertain.
Studies investigating various polymorphisms for arsenic-related disease susceptibility may indicate areas for additional research, but currently do not comprise a consistent body of evidence for specifically determining individual susceptibility, particularly for associations at low-level exposures in nutritionally sufficient populations. For example, a hospital-based case-control study in Taiwan with no known source of excess arsenic water exposure reported associations of certain polymorphisms in the As3mt gene with bladder cancer risk or with arsenic methylation efficiency (Huang et al. Citation2018). This study also found a few associations with polymorphisms in the glutathione S-transferase omega gene, but not the purine nucleoside phosphorylase gene. By contrast, in an Inner Mongolia population exposed to an average drinking water arsenic concentration of 124 µg/L, Luo et al. (Citation2018) found that some of the same polymorphisms in the genes coding for glutathione S-transferase omega and purine nucleoside phosphorylase, but not As3mt, were associated with risk of skin lesions.
The nested case-control study of skin lesion cases in Bangladesh examined associations with 26 single nucleotide polymorphisms (SNPs) in 13 one-carbon metabolism genes (Niedzwiecki et al. Citation2018). Although cases exhibited greater impairment of one-carbon metabolism and methylation capacity (independent of one-carbon metabolism), little association was found with SNPs in one-carbon metabolism genes. The most commonly studied SNP in a key enzyme in one-carbon metabolism, methylenetetrahydrofolate reductase (677 C->T polymorphism), was associated with higher homocysteine levels, higher %MMA, and lower %DMA, but not with risk of skin lesions. Two other SNPs (in the methionine synthase reductase and folate receptor 1 genes) were inversely associated with risk of skin lesions at water arsenic concentrations <50 µg/L but positively associated at concentrations ≥50 µg/L, while a third SNP (in the thymidylate synthase gene) was positively associated with skin lesion risk only at concentrations ≥50 µg/L. Another case-control study of 540 skin lesion cases and 400 controls in Bangladesh reported increased risk of skin lesions in association with current water exposure concentrations above 50 µg/L in carriers without a minor G allele in SNP rs1133400 of the squamous cell carcinoma gene INPP5A, whereas an increased risk of skin lesions was not observed until water arsenic levels exceeded 200 µg/L in those with this allele (Seow et al. Citation2015).
In general, the lack of consistent results for interactions between genetic polymorphisms and arsenic exposure may reflect differences in populations, individual-level susceptibility factors, target tissue, dose, research focus, or random findings. Lastly, it is expected that genetic polymorphisms or other individual differences in arsenic methylation would be less important for lower arsenic exposures and in never smokers (e.g. Beebe-Dimmer et al. Citation2012; Karagas et al. Citation2012), and less likely to lead to lack of comparability among populations than differences in water consumption, inorganic arsenic sources and intake (e.g. use of contaminated water to grow crops), and malnutrition.
Some epidemiological evidence comparing individuals exposed at different life stages suggests increased susceptibility for early life exposures (e.g. ≤15 years old; Steinmaus, Ferreccio, Acevedo et al. Citation2014) compared to exposure later in life. In particular, in northern Chile, where high arsenic water concentrations in some towns occurred from 1958 to 1970, increased risk of cancer incidence and mortality was later observed in those with high early life exposures during this period compared to those who were older at the time of high exposure (Smith et al. Citation2012; Steinmaus, Ferreccio, Acevedo et al. Citation2014). Interpretation of findings from short-term exposures in terms of the implications for lifetime exposures is problematic, as discussed in the Section on early life exposure studies in experimental animals. Fortunately, most epidemiological studies of arsenic in drinking water include populations with long-term, multi-generational arsenic exposure from drinking water including in utero and early childhood periods.
6. Estimation of threshold level in humans
Arsenic is one of the few substances for which comprehensive in vitro, experimental animal, and human data are available for assessment of the mode of action and dose response for cancer, as envisioned by NRC (Citation2007) in its integrated approach to toxicity testing. The collective evidence strongly supports a mode of action that involves a threshold. A linear, non-threshold approach to risk assessment is virtually excluded by the fact that arsenicals do not react with DNA and are therefore not directly mutagenic (Nesnow et al. Citation2002). The in vitro evidence indicates that a concentration >0.2 µM is required to produce a toxic effect, and a threshold has also been demonstrated in animal studies with a NOAEL >2 ppm of the diet or drinking water for inorganic arsenic. This in vivo NOAEL correlates well with the in vitro findings as urine and tissue levels in the studies associated with adverse effects exceed 0.1 µM. Based on the in vitro and in vivo findings, drinking water levels >60 µg/L are required in humans to achieve these effects. Epidemiology studies corroborate this threshold level expectation in humans for urinary bladder, lung, and skin cancer and skin lesions, indicating a similar threshold at around 100 µg/L, although this could range from 50 to 150 µg/L.
The mode of action for arsenic carcinogenesis does not fit the traditional linear dose-response model of direct interaction with DNA. Although a low-dose linear dose-response has been postulated for arsenic based on hypothetical simultaneous occurrence of multiple modes of action, the evidence for other modes of action is that they occur at considerably higher doses than for cytotoxicity and regenerative proliferation, have little substantiation in vivo, or are of unclear importance for toxicity, particularly at low doses.
Overall, the in vitro evidence for inorganic arsenic and its trivalent metabolites, in established immortalized cell lines and in primary human cells, demonstrates a concentration of 0.1 µM below which any changes are adaptive, and above which various gene/protein expression changes related to toxicity and proliferation occur (Gentry et al. Citation2010, Citation2014b; Clewell et al. Citation2011; Cohen et al. Citation2013; Yager et al. Citation2013). Other effects such as oxidative stress have also been demonstrated, although sufficient toxicity to result in DNA damage requires much higher concentrations (e.g. >10 µM) and thus are more likely a consequence rather than a cause of cytotoxicity. In vivo evidence of genomic changes in the mouse urinary bladder with arsenic dosing over time likewise supports the in vitro findings (Clewell et al. Citation2011). In general, a 0.1 µM concentration is likely conservative because in vitro conditions lack the ameliorating conditions that occur in vivo.
Animal models have limitations as cancer bioassays of inorganic arsenic in humans because of species differences in protein binding, enzyme reactivity, and pharmacokinetics. Nevertheless, although the administered doses must be higher (>2 ppm in diet or water) to achieve similar tissue levels of reactive trivalent arsenic metabolites, the cellular concentration that causes cytotoxicity and proliferation in animal studies is similar (i.e. >0.1 µM). With prolonged dosing, such concentrations can also cause a mild tumorigenic response. The DMAV rat urothelial regenerative hyperplasia model demonstrates the potential for trivalent arsenicals to cause cancer in epithelial tissues. This does not occur in mice, and human cells appear to react to DMAIII at similar or higher concentrations than rat urothelial cells. These effects lead to proliferative changes in the bladder epithelium, thereby increasing the chance of malignancy. Cytotoxicity and regenerative hyperplasia leading to bladder cancer in humans can also result from chronic inflammatory damage to the bladder epithelium, such as by bacterial cystitis and schistosomiasis (Cohen et al. Citation2000), consistent with a non-DNA-reactive and, thus, a threshold mode of action.
Lung epithelial tissue is highly perfused with blood and covered with lung surfactant proteins that are rich in sulfhydryl-containing cysteine for binding trivalent arsenicals. The increased occurrence of bronchitis and bronchiectasis in populations exposed to high levels of arsenic in drinking water suggests that these diseases involving hyperplasia may be precursors to lung carcinoma in such settings. Similarly, like hair and nails, which bind arsenic, skin is rich in sulfhydryl-containing keratin. Hyperkeratoses in skin representing proliferative effects of arsenic are thought to be pre-malignant changes. Arsenic-related cancer thus appears to result from prolonged non-genotoxic effects on lung and bladder epithelial tissues and the skin.
The overall epidemiological evidence for these arsenic-related cancers indicates a likely threshold of 50–100 µg/L for inorganic arsenic in drinking water, based largely on studies in foreign populations. As discussed earlier, several of these populations may be more susceptible to arsenical toxicity because of higher water intake, malnutrition, and/or the lack of dietary fortification with essential nutrients involved in arsenic methylation, such as folate. Overall, smoking-adjusted epidemiological studies of bladder and lung cancers indicate less consistency in dose response and often do not demonstrate a significant increase in cancer risk at exposures equivalent to drinking water levels below 100 µg/L. Analyses of bladder and lung cancers in never smokers provide additional evidence that is less subject to residual confounding from incomplete adjustment for smoking. These findings show no clear increase in dose-response below 100 µg/L. At higher concentrations (e.g. 300 µg/L; Dauphine et al. Citation2013; Ferreccio et al. Citation2013), stratified analyses in never smokers also indicate that positive associations between arsenic exposure and cancer risk can be demonstrated at a sufficient dose.
The most informative studies for assessing the dose-response for skin effects were those that evaluated the lifetime arsenic exposure history of participants and considered whether diagnoses of skin lesions or skin cancer were probably related to arsenic exposure. These studies indicate a likely threshold of approximately 100 µg/L, with the possible occurrence of nonmalignant skin lesions at 50–100 µg/L in populations with less sufficient nutrition than in the U.S. (e.g. West Bengal, Bangladesh, and Inner Mongolia).
An arsenic drinking water concentration of 100–150 µg/L in humans results in urinary concentrations similar to those in rats and mice exposed to >2 ppm in the diet or drinking water, the no-effect level for cytotoxicity and regenerative proliferation (Cohen et al. Citation2013). These doses also result in similar urinary concentrations >0.1 µM, which is an approximate no effect level in vitro. Thus, the collective evidence on effect levels from in vitro, animal, and human studies, along with the mode of action information, support a similar threshold of 50–100 µg/L as a point of departure for deriving health protective criteria.
7. Comparison with other risk assessments of inorganic arsenic
The original Maximum Contaminant Level (MCL) for inorganic arsenic in drinking water in the U.S. was set at 0.05 mg/L, based on a potable water standard developed by the Public Health Service in the 1940s to protect against the acute toxicity of inorganic arsenic, and was selected to limit the intake of inorganic arsenic in drinking water (at a water consumption rate of 2L/day) to less than 10% of the intake of arsenic in food, which was assumed at the time to be on the order of 1 mg/day (USPHS Citation1943). Subsequently, the USEPA (Citation2001) reduced the MCL to the current value of 0.01 mg/L, on the basis of dose-response modeling of bladder and lung cancer in a population in Taiwan chronically exposed to concentrations of arsenic in drinking water ranging as high as 1.75 mg/L (NRC Citation1999, Morales et al. Citation2000). These dose-response calculations were performed under the USEPA (Citation1986) default assumption of linearity and no threshold, despite the existing evidence at that time that the mode of action for the carcinogenicity of arsenic was more supportive of nonlinearity. Indeed, the NRC (Citation1999) review had concluded that the mechanisms associated with arsenic-induced cancer most likely have a sub-linear character, implying that linear models would overestimate risk.
The oral cancer slope factor for inorganic arsenic currently in USEPA’s Integrated Risk Information System (IRIS) database (USEPA Citation1995) is 1.5 (mg/kg/day)−1, based upon the prevalence of skin cancer reported in the earlier southwestern Taiwanese drinking water studies (Tseng et al. Citation1968; Tseng Citation1977). The associated lifetime risk predicted for ingestion of an arsenic concentration in drinking water of 0.01 mg/L (the current MCL) would be 0.4 per thousand, assuming the USEPA Office of Water drinking water intake rate of 2 L/day and 70 kg lifetime average body weight. At the time the USEPA (Citation1995) risk assessment was performed, the agency felt that there was insufficient dose-response data to develop a risk estimate based upon the incidence of internal tumors.
In 2001, the USEPA lowered its drinking water standard from 50 µg/L to 10 µg/L based on a revised risk assessment by the USEPA Office of Water (66 CFR 6976 – 7066; USEPA Citation2001) that assessed risks of lung and bladder cancer mortality in the southwestern Taiwanese population using the no-threshold extrapolation of Morales et al. (Citation2000) and assessment of NRC (Citation1999). Although the USEPA (Citation2006) considered DMA in pesticides to be a threshold carcinogen, they assessed the environmental degradation product, inorganic arsenic, using the USEPA Office of Water risk assessment. The combined cancer slope from this assessment was 3.67 (mg/kg/day)−1. USEPA (Citation2008) also used this slope factor in their risk assessment of arsenic exposure from treated wood play structures.
NRC (Citation2001) reassessed the Morales et al. (Citation2000) data and estimated cancer risk of 3 per thousand at 10 µg/L (assumed 1 L/day and 70 kg body weight). This estimate assumed 52% of the risk from bladder cancer and 48% of the risk from lung cancer. The predicted excess risk of bladder cancer by the NRC (Citation2001), however, was shown to be statistically inconsistent with low-level bladder cancer risks for the U.S. population based on a meta-analysis of epidemiological studies, especially for never smokers (Tsuji, Alexander et al. Citation2014).
In a subsequent draft reevaluation based on internal tumors, USEPA (Citation2005b) again relied upon the linear default approach for low dose extrapolation and the same data, stating that it lacked a full understanding of the arsenic modes of carcinogenic action. In a review of these analyses (USEPA Citation2007), the Science Advisory Board (SAB) recommended reconsideration of the evidence from inorganic arsenic animal toxicology, pharmacokinetics, and pharmacodynamics research that suggested other than a linear bladder cancer dose response. Another draft IRIS assessment was then completed (USEPA 2010, 2011) which provided an extensive literature search on the mode of action data spanning a three-year period (2005–2007). However, there was no structured mode of action analysis with pre-defined criteria to evaluate causality in an explicit weight-of-evidence process and no attempt to integrate the relevant understanding into the dose-response assessment, which was still based principally on a linear dose-response from Morales et al. (Citation2000). The resulting combined lung and bladder cancer slope factor in this revised assessment was considerably higher (the higher slope factor of 25.7 per mg/kg/day for females was recommended) than previously based on revised model selection, exposure assumptions, and use of more recent U.S. mortality and incidence data (USEPA Citation2010, Citation2011). Following a request from Congress for an independent review, the draft risk assessment was withdrawn by the USEPA in early 2012.
The FDA (Citation2013b) developed a draft risk assessment of arsenic for setting action levels for juice, which was later revised in the risk assessment of arsenic in rice (FDA Citation2016). Both risk assessments used the traditional approach of focusing on health risks in one population, in this case, the lung and bladder cancer risks in northeastern Taiwan (Chen et al. Citation2010a,b). Although these studies have individual-specific exposure data and were adjusted for smoking, unlike those used in the USEPA’s assessments of southwestern Taiwan (e.g. Morales et al. Citation2000), the FDA did not integrate mechanistic data and evidence from animal studies in the assessment, which relied on linear extrapolation to assess health risks at low doses. Most notably, the low dose lung cancer data for this population, which had a much larger number of cases at low doses than for bladder cancer, showed evidence of a threshold for increased risk below 100 µg/L in drinking water (Chen et al. Citation2010b). The slope factor from this assessment was similar to that derived by the USEPA Office of Water (3.67 {mg/kg/day}−1; USEPA Citation2006, Citation2008).
Other global authoritative bodies have provided recommendations for acceptable drinking water exposures to arsenic or considered the dose-response relationships for carcinogenicity. The World Health Organization (WHO) has a current recommended guideline value of 10 µg/L arsenic in drinking water (WHO Citation2018). This guideline is considered “provisional” as it is based on water treatment performance and analytical achievability, recognizing the practical difficulties in removing arsenic from drinking water. The Joint Food and Agriculture Organization of the United Nations (FAO)/World Health Organization (WHO) Expert Committee on Food Additives (JECFA), developed a lower benchmark dose associated with a 0.5% risk of cancer at 3 µg/kg/day based on the northeastern Taiwanese lung cancer study of Chen et al. (Citation2010b) (JECFA Citation2011). Linear extrapolations from this benchmark dose to lower acceptable risk levels for arsenic in food is equivalent to a slope factor of 1.67 (mg/kg/day)−1. Health Canada (Citation2008) has estimated 0.3 µg/L as the target acceptable concentration of arsenic in drinking water that would present an “essentially negligible” level of risk. In the context of drinking water guidelines, Health Canada has defined the term "essentially negligible" to characterize risk on the order of 1 in 105 to 106.
Based on the mode of action of inorganic arsenic, the current approach indicates that doses that are protective of non-cancer effects related to cytotoxicity and regenerative hyperplasia would also be protective of cancer. USEPA’s reference dose for noncancer risk assessment of inorganic arsenic (0.3 µg/kg/day) is based on skin lesions in southwestern Taiwan as the sensitive endpoint. Assuming lifetime consumption of 2 L/day of water and 70 kg average body, exposure to arsenic at the drinking water standard of 10 µg/L would not exceed USEPA’s arsenic reference dose.
8. Strengths and weaknesses of this assessment
The main strength of this assessment lies in its reliance on an abundance of complementary and consistent evidence supporting the presence of a threshold for the cancer effects of inorganic arsenic based on mode of action and supported by extensive in vitro and animal in vivo investigations. The evidence is strong from mechanistic, toxicologic and epidemiologic investigations. For any chemical, all three evidence streams have limitations for justifying a threshold approach for cancer, and arsenic is no exception. In vitro studies lack the complexity of the whole organism, and experimental animals may not be appropriate models for humans. Epidemiological studies are generally not well designed to identify doses at which no excess risk of cancer would occur, and they often have uncertainties in exposure assessment, control for confounding, and evaluation of effect modifiers. Thus, the weaknesses of this assessment stem from the limitations of the underlying scientific evidence, especially the potential lack of human relevance of toxicological studies and problems with methodological error, bias, and heterogeneity in epidemiological studies. The integration of the overall evidence, however, supports conclusions regarding the likely doses at which a threshold for arsenic-related cancer risk might occur. The presence of a threshold in the cancer effects of numerous non-DNA reactive chemicals have been demonstrated, whether involving mitogenic effects secondary to interactions with specific receptors, or due to cytotoxicity with regenerative proliferation (Andersen et al. Citation2000; Cohen and Arnold Citation2011; Corton et al. Citation2014, Citation2018; Haney Citation2015).
The inorganic arsenic epidemiological evidence includes a large number of studies in various populations, primarily in Inner Mongolia, China, Taiwan, Bangladesh, West Bengal, Chile, Argentina, Mexico, and the U.S. Key uncertainties in these studies include the accuracy, reliability, and etiological relevance (e.g. with respect to timing) of exposure assessment, control for confounding and modifying factors (e.g. smoking, nutrition, sunlight exposure), and methodological and population-specific differences that impede comparison of results across populations. Among the various arsenic exposure metrics used in epidemiological studies, such as water concentration, biomarkers in urine or nails, and arsenic daily or cumulative dose based on reported water intake, most studies have estimated exposure based on water concentration. Use of this metric is clouded by the fact that drinking water intake rates were likely higher historically in non-U.S. populations than in the U.S. The contribution of inorganic arsenic in food to exposures at low doses may also undermine assessments of exposure based solely on water concentrations; however, dietary inorganic arsenic exposure is considerably lower than exposure to arsenic in drinking water at 10 µg/L (Tsuji et al. Citation2007; Xue et al. Citation2010), particularly in non-U.S. populations with higher water intake rates. Therefore, dietary sources are not expected to affect exposure meaningfully at the water concentration range identified as a likely threshold for cancer risk. Use of contaminated well water to grow crops, including rice such as in Bangladesh and Taiwan, however, would increase arsenic exposure at a given arsenic water concentration compared to the situation in U.S. populations.
The interpretation of epidemiological results is also complicated by potential individual variation in genetic susceptibility to the health effects of arsenic exposure. Potential genetic susceptibility is an area of current research interest, particularly for genes related to arsenic metabolism or DNA repair. Some studies have identified heterogeneity in arsenic-cancer associations across polymorphisms in such genes (e.g. Applebaum et al. Citation2007; Huang et al. Citation2016, Citation2018; de La Rosa et al. Citation2017; Lin et al. Citation2018). However, findings thus far are inconsistent and, like many other epidemiological studies of gene-environment interactions, suffer from multiple hypothesis testing and limited sample sizes. Available evidence on the variation in toxicokinetics and toxicodynamics associated with drinking water exposure to inorganic arsenic is consistent with default expectations for human inter-individual variability (see Section 4). Moreover, in many foreign populations, genetic variation in arsenic metabolism is likely to be less influential at lower arsenic doses than nutritional deficiencies that cause impairment of one-carbon metabolism, which reduces arsenic methylation and can independently increase disease susceptibility. Because much of the available epidemiological data to assess cancer risk associated with low-dose arsenic is in non-U.S. populations with greater inorganic arsenic exposure at a given water concentration and likely increased susceptibility to arsenic toxicity, risks are likely overestimated and threshold doses are likely to be underestimated for the general U.S. population, which provides some additional protection for more susceptible individuals.
9. Conclusions
The overall evidence on the mode of action supports the hypothesis that the critical biologic effects of inorganic arsenic are secondary to the interaction of trivalent arsenicals with sulfhydryl groups in critical proteins in the target tissues. Since these proteins are constantly regenerating, this will be a threshold effect requiring the level of interaction to be greater than the active regenerative process of the proteins. A threshold concentration for the biologic effects in tissues is supported by mechanistic, in vitro and animal investigations (). In animal studies, pre-neoplastic and neoplastic changes require administration of high levels in either the drinking water or diet, with no adverse effect levels greater than 2 ppm (2 mg/kg of diet or 2 mg/L in drinking water), which corresponds to a level produced in the urine of >0.1 µM. Extensive investigations in vitro have demonstrated a lack of adverse biologic effects of trivalent arsenicals below 0.2 µM, which as a level in urine for exposure to the bladder epithelium is estimated to be equivalent to a drinking water threshold in humans of 65 µg/L. Human exposure to inorganic arsenic in the drinking water at high levels is associated with increased incidences of urinary bladder, lung and skin cancer along with other non-neoplastic toxicities. Analysis of populations exposed to low levels of inorganic arsenic in the drinking water indicates a threshold effect between 50 and 100 µg/L (ppb) for all three of these cancers and also for the pre-neoplastic skin changes associated with arseniasis, although most of the evidence suggests 100 µg/L as the threshold. This value based on epidemiology investigations is consistent with the estimated calculated drinking water threshold based on mechanistic, in vitro and animal studies.
Table 7. Convergence of evidence: inorganic arsenic.
Therefore, we conclude that there is a convincing biologic basis for a threshold cancer risk assessment for a non-DNA reactive chemical such as inorganic arsenic that can be the same as for non-cancer endpoints, utilizing a threshold. The non-DNA reactive trivalent arsenicals do not induce cancer directly, rather they produce non-cancer toxicities, some of which lead to epithelial regenerative proliferation and ultimately carcinomas. This evaluation provides an alternative mode of action-based approach for assessing health-protective levels for oral arsenic exposure based on the collective in vitro, in vivo, and human evidence rather than the use of a linear low dose extrapolation based on assumptions and theories. Based on the relevant epidemiological studies with individual-level data, a threshold level for inorganic arsenic in the drinking water for these cancers is estimated to be around 100 µg/L, with strong evidence that the threshold is between 50 and 150 µg/L, consistent with the value calculated based on mechanistic, in vitro and in vivo investigations.
Declaration of interest
The employment affiliations of the authors are shown on the cover page. This review paper was prepared at the request of the Texas Commission on Environmental Quality (TCEQ), a regulatory agency of the State of Texas. The team of consultants was organized by S.M. Cohen with funding provided by TCEQ to the University of Nebraska Medical Center (UNMC). The UNMC, in turn, provided funds to Exponent, Inc for J.S. Tsuji and E.T. Chang and to Ramboll US. Corp. for P.R. Gentry and H.J. Clewell, with funds provided to P. Boffetta through a subcontract with Ramboll US Corp. Each of the six authors participated in the preparation of this review paper as independent professionals. The views expressed and opinions offered are those of the authors and not necessarily those of their employers.
Although the work involved with this specific project was supported by TCEQ, the investigators involved with this project have received support from numerous other sources for work on arsenic toxicity and carcinogenicity. No funding from these other sources was used in the current review, evaluation, or preparation of this manuscript.
J.S. Tsuji has provided scientific consultation to the Arsenic Science Task Force (ASTF), the Wood Preservatives Science Council (WPSC), and the Electric Power Research Institute (EPRI) regarding arsenic dose-response issues, including providing public comments on USEPA Integrated Risk and Information System (IRIS) assessments for arsenic. The ASTF represents trade associations of industries, manufacturers, and agricultural producers with interests in the scientific and regulatory developments on arsenic. WPSC is a trade organization funded by manufacturers of wood preservative chemicals including those containing arsenic. EPRI is a nonprofit organization that conducts research, development, and demonstration projects on scientific topics of interest to electric utilities. Its members are mostly electric utilities, and also include businesses, government agencies, regulators, and public and private entities. Some of J.S. Tsuji and E.T. Chang’s published papers on arsenic were partially funded by the ASTF and EPRI. Some of J.S. Tsuji’s publications were also partially funded by Rio Tinto (a mining company), the American Chemistry Council (specifically, companies within the anti-microbial sector of this chemical trade organization that funds research on health, safety, and the environment), or ILSI North America Technical Committee on Food and Chemical Safety. ILSI is a nonprofit foundation (funded by member food companies) focused on scientific issues related to nutritional quality and safety of the food supply. The arsenic biomonitoring study in New York published by J.S. Tsuji was funded by FMC Corporation (a former manufacturer of arsenic-based pesticides); FMC had no role in providing comments on the reporting of the biomonitoring study results or in funding its publication. J.S. Tsuji has also provided public comments on arsenic exposure and health risks from treated wood on behalf of the above parties, and scientific consultation to a number of mining companies, the Tennessee Valley Authority, and the USEPA for conducting health risk assessments of arsenic at contaminated sites, and in guiding cleanup of sites. J.S. Tsuji has also provided expert testimony and E.T. Chang has provided scientific consultation on behalf of both defendants and plaintiffs on arsenic exposure and health risk in legal cases.
H.J. Clewell and P.R. Gentry have previously conducted arsenic research, including research discussed in this manuscript, and have provided scientific consultation in the development of public comments related to USEPA IRIS assessments for arsenic with funding from the Electric Power Research Institute. However, no EPRI funding was received for the preparation of this manuscript.
The research of P. Boffetta has been supported by the US National Cancer Institute and the US Centers for Disease Control and Prevention. He has received financial support from the ASTF for analysis of arsenic exposure and cancer risk. He received a subcontract from Ramboll US Corp. for his role in the preparation of this manuscript.
The research of S.M. Cohen has been supported by the US National Cancer Institute, ILSI North America Technical Committee on Food and Chemical Safety, EPRI, ASTF, the Organic Arsenical Products Task Force (OAPTF) (trade organization of companies producing methylated arsenicals for pesticide use), USEPA, Alberta Health (from the Canadian Province of Alberta), Alberta Innovates (from the Canadian Province of Alberta), Canada Research Chair's Program, Canadian Institute of Health Research, and Natural Sciences and Engineering Research Council of Canada. S.M. Cohen has also provided public comments on USEPA IRIS assessment for arsenic.
Supplemental material
Supplemental material for this article is available online here.
Supplemental Material
Download MS Word (31.6 KB)Supplemental_Tables_-_clean_-_1-18-19.docx
Download MS Word (52.1 KB)Supplemental_Information_2.docx
Download MS Word (14.4 KB)Acknowledgments
The work involved with this evaluation and the development of this manuscript was supported by a contract from the Texas Commission on Environmental Quality (TCEQ). No other sources of support were used for this project. TCEQ reviewed the manuscript and suggested minor edits, but the content is that of the authors. We gratefully acknowledge Lora Arnold, Jeanne Bradford, and Betty Dowd for their assistance with the preparation of this manuscript. We are very appreciative of the many helpful comments and suggestions provided by the peer reviewers that improved the manuscript.
Notes
1 DMA in the urine is usually measured as total DMA without separately identifying DMAV and DMAIII; likewise for MMA. In this report, DMA and MMA without the oxidation state refers to the total of +3 and +5 oxidation states.
References
- Abernathy CO, Chappell WR, Meek ME, Gibb H, Guo H-R. 1996. Is ingested inorganic arsenic a “threshold carcinogen”? Fundam Appl Toxicol. 29:168–175.
- Agency for Toxic Substances and Disease Registry [ATSDR]. 2007. Toxicological profile for arsenic. Atlanta (GA): U.S. Department of Health and Human Services. [accessed 2018 Oct 6]. https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=22&tid=3.
- Andersen ME, Meek ME, Boorman GA, Brusick DJ, Cohen SM, Dragan YP, Frederick CB, Goodman JI, Hard GC, O'Flaherty EJ, et al. 2000. Lessons learned in applying the U.S. EPA proposed cancer guidelines to specific compounds. Toxicol Sci. 53:159–172.
- Andrew AS, Mason RA, Kelsey KT, Schned AR, Marsit CJ, Nelson HH, Karagas MR. 2009. DNA repair genotype interacts with arsenic exposure to increase bladder cancer risk. Toxicol Lett. 187:10–14.
- Aposhian HV. 1997. Enzymatic methylation of arsenic species and other new approaches to arsenic toxicity. Annu Rev Pharmacol Toxicol. 37:397–419.
- Applebaum KM, Karagas MR, Hunter DJ, Catalano PJ, Byler SH, Morris S, Nelson HH. 2007. Polymorphisms in nucleotide excision repair genes, arsenic exposure, and non-melanoma skin cancer in New Hampshire. Environ Health Perspect. 115:1231–1236.
- Arnold LL, Eldan M, Nyska A, van Gemert MM, Capen CC, Cohen SM. 2003. Chronic studies evaluating the carcinogenicity of monomethylarsonic acid in rats and mice. Toxicology. 190:197–219.
- Arnold LL, Eldan M, Nyska A, van Gemert M, Cohen SM. 2006. Dimethylarsinic acid: results of chronic toxicity/oncogenicity studies in F344 rats and in B6C3F1 mice. Toxicology. 223:82–100.
- Arnold LL, Suzuki S, Yokohira M, Kakiuchi-Kiyota S, Pennington KL, Cohen SM. 2014. Time course of urothelial changes in rats and mice orally administered arsenite. Toxicol Pathol. 42:855–862.
- Aylward LL, Ramasamy S, Hays SM, Schoeny R, Kirman CR. 2014. Evaluation of urinary speciated arsenic in NHANES: issues in interpretation in the context of potential inorganic arsenic exposure. Regul Toxicol Pharmacol. 69:49–54.
- Baastrup R, Sørensen M, Balstrøm T, Frederiksen K, Larsen CL, Tjønneland A, Overvad K, Raaschou-Nielsen O. 2008. Arsenic in drinking waterand risk for cancer in Denmark. Environ Health Perspect. 116:231–237.
- Bailey KA, Hester SD, Knapp GW, Owen RD, Thai SF. 2010. Gene expression of normal human epidermal keratinocytes modulated by trivalent arsenicals. Mol Carcinog. 49:981–998.
- Bailey KA, Wallace K, Smeester L, Thai SF, Wolf DC, Edwards SW, Fry RC. 2012. Transcriptional modulation of the ERK1/2 MAPK and NF-κB pathways in human urothelial cells after trivalent arsenical exposure: implications for urinary bladder cancer. J Can Res Updates. 1:57–68.
- Barchowsky A, Roussel R, Klei L, James P, Ganju N, Smith K, Dudek E. 1999. Low levels of arsenic trioxide stimulate proliferative signals in primary vascular cells without activating stress effector pathways. Toxicol Appl Pharmacol. 159:65–75.
- Baris D, Waddell R, Beane Freeman LE, Schwenn M, Colt JS, Ayotte JD, Ward MH, Nuckols J, Schned A, Jackson B. 2016. Elevated bladder cancer in northern New England: the role of drinking water and arsenic. J Natl Cancer Inst. 108:5.
- Barr DB, Wilder LC, Caudill SP, Gonzalez AJ, Needham LL, Pirkle JL. 2005. Urinary creatinine concentrations in the U.S. population: implications for urinary biologic monitoring measurements. Environ Health Perspect. 113:192–200.
- Basu A, Ghosh P, Das JK, Banerjee A, Ray K, Giri AK. 2004. Micronuclei as biomarkers of carcinogen exposure in populations exposed to arsenic through drinking water in West Bengal, India: A comparative study in three cell types. Cancer Epidemiol Biomark Prev. 13:820–827.
- Bates MN, Rey OA, Biggs ML, Hopenhayn C, Moore LE, Kalman D, Steinmaus C, Smith AH. 2004. Case-control study of bladder cancer and exposure to arsenic in Argentina. Am J Epidemiol. 159:381–389.
- Bates MN, Smith AH, Cantor KP. 1995. Case-control study of bladder cancer and arsenic in drinking water. Am J Epidemiol. 141:523–530.
- Beebe-Dimmer JL, Iyer PT, Nriagu JO, Keele GR, Mehta S, Meliker JR, Lange EM, Schwartz AG, Zuhlke KA, Schottenfeld D, et al. 2012. Genetic variation in glutathione S-transferase omega-1, arsenic methyltransferase and methylene-tetrahydrofolate reductase, arsenic exposure and bladder cancer: a case-control study. Environ Health. 11:43.
- Bhattacharjee P, Paul S, Bhattacharjee S, Giri AK, Bhattacharjee P. 2018. Association of H3K79 monomethylation (an epigenetic signature) with arsenic-induced skin lesions. Mutat Res. 807:1–9.
- Bjørklund G, Aaseth J, Chirumbolo S, Urbina MA, Uddin R. 2018. Effects of arsenic toxicity beyond epigenetic modifications. Environ Geochem Health. 40:955–965.
- Bojkowska K, Santoni de Sio F, Barde I, Offner S, Verp S, Heinis C, Johnsson K, Trono D. 2011. Measuring in vivo protein half-life. Chem Biol. 18:805–815.
- Bomhard EM, Gelbke HP, Schenk H, Williams GM, Cohen SM. 2013. Evaluation of the carcinogenicity of gallium arsenide. Crit Rev Toxicol. 43:436–466.
- Boobis AR, Cohen SM, Dellarco V, McGregor D, Meek ME, Vickers C, Willcocks D, Farland W. 2006. IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol. 36:781–792.
- Boobis AR, Doe JE, Heinrich-Hirsch B, Meek ME, Munn S, Ruchirawat M, Schlatter J, Seed J, Vickers C. 2008. IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit Rev Toxicol. 38:87–96.
- Burgoon L, Druwe IL. 2016. Revisiting Cohen et al 2015, Cohen et al 2014 and Waalkes et al 2014: A Bayesian re-analysis of tumor incidences. Arch Toxicol. 90:2047–2048.
- Button M, Jenkin GR, Harrington CF, Watts MJ. 2009. Human toenails as a biomarker of exposure to elevated environmental arsenic. J Environ Monit. 11:610–617.
- Cascio C, Raab A, Jenkins RO, Feldmann J, Meharg AA, Haris PI. 2011. The impact of a rice based diet on urinary arsenic. J Environ Monit. 13:257–265.
- Chang CH, Liu CS, Liu HJ, Huang C-P, Huang C-Y, Hsu H-T, Liou S-H, Chung C-J. 2016. Association between levels of urinary heavy metals and increased risk of urothelial carcinoma. Int J Urol. 23:233–239.
- Chen B, Arnold LL, Cohen SM, Thomas DJ, Le XC. 2011. Mouse arsenic (+3 oxidation state) methyltransferase genotype affects metabolism and tissue dosimetry of arsenicals after arsenite administration in drinking water. Toxicol Sci. 124:320–326.
- Chen CL, Chiou HY, Hsu LI, Hsueh YM, Wu MM, Wang YH, Chen CJ. 2010b. Ingested arsenic, characteristics of well water consumption and risk of different histological types of lung cancer in northeastern Taiwan. Environ Res. 110:455–462.
- Chen CJ, Chuang YC, You SL, Lin TM, Wu HY. 1986. A retrospective study on malignant neoplasms of bladder, lung and liver in blackfoot disease endemic area in Taiwan. Br J Cancer. 53:399–405.
- Chen CJ, Chuang YC, Lin TM, Wu HY. 1985. Malignant neoplasms among residents of a blackfoot disease-endemic area in taiwan: high-arsenic artesian well water and cancers. Cancer Res. 45:5895–5899.
- Chen KJ, Pan WH, Lin YC, Lin BF. 2011. Trends in folate status in the Taiwanese population aged 19 years and older from the Nutrition and Health Survey in Taiwan 1993-1996 to 2005-2008. Asia Pac J Clin Nutr. 20:275–282.
- Chen LX, Schumacher HR. 2008. Gout: an evidence-based review. J Clin Rheumatol. 14:S55–S62.
- Chen CL, Chiou HY, Hsu LI, Hsueh YM, Wu MM, Wang YH, Chen CJ. 2010a. Arsenic in drinking water and risk of urinary tract cancer: a follow-up study from northeastern Taiwan. Cancer Epidemiol Biomarkers Prev. 19:101–110.
- Cheng P-S, Weng S-F, Chiang C-H, Lai F-J. 2016. Relationship between arsenic-containing drinking water and skin cancers in the arseniasis endemic areas in Taiwan. J Dermatol. 43:181–186.
- Chen CJ, Hsueh YM, Tseng MP, Lin YC, Hsu LI, Chou WL, Chiou HY, Wang IH, Chou YL, Tseng CH, et al. 2001. Individual susceptibility to arseniasis. In: Chappell WR, Abernathy CO, Calderon RL, editors. Proceedings of the Fourth Annual International Conference on Arsenic Exposure and HealthEffects, 18–22 June 2000. London: Elsevier Science Ltd.; p. 135–143.
- Chen CJ, Kuo TL, Wu MM. 1988. Arsenic and cancers. Lancet. 1:414–415.
- Chiou HY, Hsueh YM, Liaw KF, Horng SF, Chiang MH, Pu YS, Lin JS, Huang CH, Chen CJ. 1995. Incidence of internal cancers and ingested inorganic arsenic: a seven-year follow-up study in Taiwan. Cancer Res. 55:1296–1300.
- Choi B-S, Choi S-J, Kim D-W, Huang M, Kim N-Y, Park K-S, Kim C-Y, Lee H-M, Yum Y-N, Han E-S, et al. 2010. Effects of repeated seafood consumption on urinary excretion of arsenic species by volunteers. Arch Environ Contam Toxicol. 58:222–229.
- Clewell HJ, Thomas RS, Gentry PR, Crump KS, Kenyon EM, El-Masri HA, Yager JW. 2007. Research toward the development of a biologically based dose response assessment for inorganic arsenic carcinogenicity: a progress report. Toxicol Appl Pharmacol. 222:388–398.
- Clewell HJ, Thomas RS, Kenyon EM, Hughes MF, Adair BM, Gentry PR, Yager JW. 2011. Concentration- and time-dependent genomic changes in the mouse urinary bladder following exposure to arsenate in drinking water for up to 12 weeks. Toxicol Sci. 123:421–432.
- Cohen PR. 2017. Do infants fed rice and rice products have an increased risk for skin cancer? Cutis. 100:250.
- Cohen SM. 2018a. Letter to the editor, re: Lynch et al., 2017. Environ Int. 111:316.
- Cohen SM. 2018b. Screening for human urinary bladder carcinogens: two-year bioassay is unnecessary. Toxicol Res (Camb).7:565–575.
- Cohen SM, Arnold LL. 2011. Chemical carcinogenesis. Toxicol Sci. 120:S76–S92.
- Cohen SM, Arnold LL, Beck BD, Lewis AS, Eldan M. 2013. Evaluation of the carcinogenicity of inorganic arsenic. Crit Rev Toxicol. 43:711–752.
- Cohen SM, Arnold LL, Eldan M, Lewis AS, Beck BD. 2006. Methylated arsenicals: the implications of metabolism and carcinogenicity studies in rodents to human risk assessment. Crit Rev Toxicol. 36:99–133.
- Cohen SM, Arnold LL, Klaunig JE, Goodman JI. 2014. Re: Waalkes et al.: Lung tumors in mice induced by “whole-life” inorganic arsenic exposure at human-relevant doses, Arch Toxicol, 2014 . Arch Toxicol. 88:2061–2062.
- Cohen SM, Arnold LL, Klaunig JE, Goodman JI. 2015. Response to the Waalkes et al., Letter to the editor concerning our “letter to the editor, Re: Lung tumors in mice induced by “whole-life” inorganic arsenic exposure at human relevant doses, Waalkes et al., Arch Toxicol, 2014”. Arch Toxicol. 89:2167–2168.
- Cohen SM, Arnold LL, Klaunig JE, Goodman JI. 2016. Response to Druwe and Burgoon: Re: Druwe, I.L. and Burgoon, L.: revisiting Cohen et al. 2015, Cohen et al. 2014 and Waalkes et al. 2014: a bayesian re-analysis of tumor incidences. Arch Toxicol. 90:3129–3130.
- Cohen SM, Arnold LL, Uzvolgyi E, Cano M, St John M, Yamamoto S, Lu X, Le XC. 2002. Possible role of dimethylarsinous acid in dimethylarsinic acid-induced urothelial toxicity and regeneration in the rat. Chem Res Toxicol. 15:1150–1157.
- Cohen SM, Chowdhury A, Arnold LL. 2016. Inorganic arsenic: A non-genotoxic carcinogen. J Environ Sci (China).49:28–37.
- Cohen SM, Shirai T, Steineck G. 2000. Epidemiology and etiology of premalignant and malignant urothelial changes. Scand J Urol Nephrol. 34:105–115.
- Corton J, Cunningham ML, Hummer BT, Lau C, Meek B, Peters JM, Popp JA, Rhomberg L, Seed J, Klaunig JE. 2014. Mode of action framework analysis for receptor-mediated toxicity: the peroxisome proliferator-activated receptor alpha (PPARα) as a case study. Crit Rev Toxicol. 44:1–49.
- Corton JC, Peters JM, Klaunig JE. 2018. The PPARα-dependent rodent liver tumor response is not relevant to humans: addressing misconceptions. Arch Toxicol. 92:83–119.
- Coryell M, McAlpine M, Pinkham NV, McDermott TR, Walk ST. 2018. The gut microbiome is required for full protection against acute arsenic toxicity in mouse models. Nature Comm. 9:5424.
- Crump KS. 2006. The effect of random error in exposure measurement upon the shape of the exposure response. Dose Response. 3:456–464.
- Cui X, Wakai T, Shirai Y, Hatakeyama K, Hirano S. 2006. Chronic oral exposure to inorganic arsenate interferes with methylation status of p16INK4a and RASSF1A and induces lung cancer in A/J mice. Toxicol Sci. 91:372–381.
- Cullen WR. 2008. Is arsenic an aphrodisiac? the sociochemistry of an element. Cambridge: Royal Society of Chemistry.
- Cullen WR, Reimer KJ. 2017. Arsenic is Everywhere: Cause for Concern? Cambridge (UK): Royal Society of Chemistry.
- Currier JM, Svoboda M, de Moraes DP, Matousek T, Dĕdina J, Stýblo M. 2011. Direct analysis of methylated trivalent arsenicals in mouse liver by hydride generation-cryotrapping-atomic absorption spectrometry. Chem Res Toxicol. 24:478–480.
- D’Ippoliti D, Santelli E, De Sario M, Scortichini M, Davoli M, Michelozzi P. 2015. Arsenic in drinking water and mortality for cancer and chronic diseases in central Italy, 1990-2010. PLoS One. 10:e0138182.
- Dai YC, Wang SC, Haque MM, Lin WH, Lin LC, Chen CH, Liu YW. 2017. The interaction of arsenic and N-butyl-N-(4-hydroxybutyl)nitrosamine on urothelial carcinogenesis in mice. PLoS One. 12:e0186214.
- Dauphine DC, Smith AH, Yuan Y, Balmes JR, Bates MN, Steinmaus C. 2013. Case-control study of arsenic in drinking water and lung cancer in california and nevada. Ijerph. 10:3310–3324.
- de la Rosa R, Steinmaus C, Akers NK, Conde L, Ferreccio C, Kalman D, Zhang KR, Skibola CF, Smith AH, Zhang L, Smith MT. 2017. Associations between arsenic (+3 oxidation state) methyltransferase (AS3MT) and N-6 adenine-specific DNA methyltransferase 1 (N6AMT1) polymorphisms, arsenic metabolism, cancer risk in a Chilean population. Environ Mol Mutagen. 58:411–422.
- Del Razo LM, Garcia-Vargas GG, Valenzuela OL, Castellanos EH, Sanchez-Pena LC, Currier JM, Drobna Z, Loomis D, Styblo M. 2011. Exposure to arsenic in drinking water is associated with increased prevalence of diabetes: a cross-sectional study in the Zimapán and Lagunera regions in Mexico. Environ Health. 10:73–71.
- Del Razo LM, Garcia-Vargas GG, Vargas H, Albores A, Gonsebatt ME, Montero R, Ostrosky-Wegman P, Kelsh M, Cebrian ME. 1997. Altered profile of urinary arsenic metabolites in adults with chronic arsenicism. A pilot study. Arch Toxicol. 71:211–217.
- Diamond GL, Bradham KD, Brattin WJ, Burgess M, Griffin S, Hawkins CA, Juhasz AL, Klotzbach JM, Nelson C, Lowney YW, et al. 2016. Predicting oral relative bioavailability of arsenic in soil from in vitro bioaccessibility. J Toxicol Environ Health Part A. 79:165–173.
- Dodmane PR, Arnold LL, Kakiuchi-Kiyota S, Qiu F, Liu X, Rennard SI, Cohen SM. 2013. Cytotoxicity and gene expression changes induced by inorganic and organic trivalent arsenicals in human cells. Toxicology. 312:18–29.
- Dodmane PR, Arnold LL, Muirhead DE, Suzuki S, Yokohira M, Pennington KL, Dave BJ, Lu X, Le XC, Cohen SM. 2014. Characterization of intracellular inclusions in the urothelium of mice exposed to inorganic arsenic. Toxicol Sci. 137:36–46.
- Dodmane PR, Arnold LL, Pennington KL, Cohen SM. 2014. Orally administered nicotine induces urothelial hyperplasia in rats and mice. Toxicology. 315:49–54.
- Doherty MK, Hammond DE, Clague MJ, Gaskell SJ, Beynon RJ. 2009. Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. J Proteome Res. 8:104–112.
- Donehower L, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr Butel JS, Bradley A. 1992. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 356:215.
- Driscoll KE, Costa DL, Hatch G, Henderson R, Oberdorster G, Salem H, Schlesinger RB. 2000. Intratracheal instillation as an exposure technique for the evaluation of respiratory tract toxicity: uses and limitations. Toxicol Sci. 55:24–35.
- Drobna Z, Naranmandura H, Kubachka KM, Edwards BC, Herbin-Davis K, Styblo M, Le XC, Creed JT, Maeda N, Hughes MF, et al. 2009. Disruption of the arsenic (+3 oxidation state) methyltransferase gene in the mouse alters the phenotype for methylation of arsenic and affects distribution and retention of orally administered arsenate. Chem Res Toxicol. 22:1713–1720.
- Drobna Z, Walton FS, Paul DS, Xing W, Thomas DJ, Styblo M. 2010. Metabolism of arsenic in human liver: the role of membrane transporters. Arch Toxicol. 84:3–16.
- Drobna Z, Waters SB, Walton FS, LeCluyse EL, Thomas DJ, Styblo M. 2004. Interindividual variation in the metabolism of arsenic in cultured primary human hepatocytes. Toxicol Appl Pharmacol. 201:166–177.
- Drobna Z, Walton FS, Harmon AW, Thomas DJ, Styblo M. 2010. Interspecies differences in metabolism of arsenic by cultured primary hepatocytes. Toxicol Appl Pharmacol. 245:47–56.
- Durda K, Kąklewski K, Gupta S, Szydłowski M, Baszuk P, Jaworska-Bieniek K, Sukiennicki G, Kaczmarek K, Waloszczyk P, Narod S, et al. 2017. Serum folate concentration and the incidence of lung cancer. PLoS One. 12:e0177441.
- Ebert F, Weiss A, Bultemeyer M, Hamann I, Hartwig A, Schwerdtle T. 2011. Arsenicals affect base excision repair by several mechanisms. Mutat Res. 715:32–41.
- Efremenko AY, Seagrave J, Clewell HJ, Van Landingham C, Gentry PR, Yager JW. 2015. Evaluation of gene expression changes in human primary lung epithelial cells following 24-hr exposures to inorganic arsenic and its methylated metabolites and to arsenic trioxide. Environ Mol Mutagen. 56:477–490.
- EFSA Scientific Committee. 2017. Clarification of some aspects related to genotoxicity assessment. EFSA J. 15:5113.
- El-Masri HA, Hong T, Henning C, Mendez W, Jr, Hudgens EE, Thomas DJ, Lee JS. 2018. Evaluation of a physiologically based pharmacokinetic (PBPK) model for inorganic arsenic exposure using data from two diverse human populations. Environ Health Perspect. 126:077004.
- El-Masri HA, Kenyon EM. 2008. Development of a human physiologically based pharmacokinetic (PBPK) model for inorganic arsenic and its mono- and di-methylated metabolites. J Pharmacokinet Pharmacodyn. 35:31–68.
- Erraguntla NK, Sielken RL, Jr, Valdez-Flores C, Grant RL. 2012. An updated inhalation unit risk factor for arsenic and inorganic arsenic compounds based on a combined analysis of epidemiological studies. Reg Toxicol Pharmacol. 64:329–341.
- Faita F, Cori L, Bianchi F, Andreassi MG. 2013. Arsenic-induced genotoxicity and genetic susceptibility to arsenic-related pathologies. IJERPH. 10:1527–1546.
- Fanidi A, Muller DC, Yuan JM, Stevens VL, Weinstein SJ, Albanes D, Prentice R, Thomsen CA, Pettinger M, Cai Q. 2018. Circulating folate, Vitamin B6, and methionine in relation to lung cancer risk in the lung cancer cohort consortium (LC3). J Natl Cancer Inst. 110:57–67.
- Ferdosi H, Dissen EK, Afari-Dwamena NA, Li J, Chen R, Feinleib M, Lamm SH. 2016. Arsenic in drinking water and lung cancer mortality in the United States: an analysis based on US Counties and 30 years of observation (1950-1979). J Environ Public Health. 2016:1.
- Ferreccio C, Smith AH, Duran V, Barlaro T, Benitez H, Valdes R, Aguirre JJ, Moore LE, Acevedo J, Vasquez MI, et al. 2013. Case-control study of arsenic in drinking water and kidney cancer in uniquely exposed Northern Chile. Am J Epidemiol. 178:813–818.
- Forman HJ, Zhang H, Rinna A. 2009. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 30:1–12.
- Fraser JL, F, Sede CP. Vendetti 2016. Disorders of cobalamin and folate metabolism. In: Hollak CEM, Lachmann R, editors. Inherited metabolic disease in adults: a clinical guide. New York (NY): Oxford University Press; p. 171–180.
- Frost F, Harter L, Milham S, Royce R, Smith AH, Hartley J, Enterline P. 1987. Lung cancer among women residing close to an arsenic emitting copper smelter. Arch Environ Health. 42:148–152.
- Fu S, Wu J, Li Y, Liu Y, Gao Y, Yao F, Qiu C, Song L, Wu Y, Liao Y, Sun D. 2014. Urinary arsenic metabolism in a Western Chinese population exposed to high-dose inorganic arsenic in drinking water: influence of ethnicity and genetic polymorphisms. Toxicol Appl Pharmacol. 274:117–123.
- Gamble MV, Liu X, Ahsan H, Pilsner JR, Ilievski V, Slavkovich V, Parvez F, Chen Y, Levy D, Factor-Litvak P, et al. 2006. Folate and arsenic metabolism: a double-blind, placebo-controlled folic acid-supplementation trial in Bangladesh. Am J Clin Nutr. 84:1093–1101.
- Gamble MV, Liu X, Ahsan H, Pilsner R, Ilievski V, Slavkovich V, Parvez F, Levy D, Factor-Litvak P, Graziano JH. 2005. Folate, homocysteine, and arsenic metabolism in arsenic-exposed individuals in Bangladesh. Environ Health Perspect. 113:1683–1688.
- Gamboa-Loira B, Cebrian ME, Franco-Marina F, Lopez-Carrillo L. 2017. Arsenic metabolism and cancer risk: a meta-analysis. Environ Res. 156:551–558.
- Garland DL. 1978. Kinetics and mechanism of colchicine binding to tubulin: evidence for ligand-induced conformational changes. Biochemistry. 17:4266–4272.
- Garrett S, Belcastro M, Sens M, Somji S, Sens D. 2001. Acute exposure to arsenite induces metallothionein isoform-specific gene expression in human proximal tubule cells. J Toxicol Environ Health A. 64:343–355.
- Garry MR, Santamaria AB, Williams AL, DeSesso JM. 2015. In utero arsenic exposure in mice and early life susceptibility to cancer. Regul Toxicol Pharmacol. 73:378–390.
- Gentry PR, McDonald TB, Sullivan DE, Shipp AM, Yager JW, Clewell HJ. 2010. Analysis of genomic dose-response information on arsenic to inform key events in a mode of action for carcinogenicity. Environ Mol Mutagen. 51:1–14.
- Gentry PR, Yager JW, Clewell RA, Clewell HJ. 2014. Use of mode of action data to inform a dose–response assessment for bladder cancer following exposure to inorganic arsenic. Toxicol in Vitro. 28:1196–1205.
- Gentry PR, Clewell HJ, Greene TB, Franzen AC, Yager JW. 2014. The impact of recent advances in research on arsenic cancer risk assessment. Regul Toxicol Pharmacol. 69:91–104.
- Germolec DR, Spalding J, Yu HS, Chen GS, Simeonova PP, Humble MC, Bruccoleri A, Boorman GA, Foley JF, Yoshida T, et al. 1998. Arsenic enhancement of skin neoplasia by chronic stimulation of growth factors. Am J Pathol. 153:1775–1785.
- Gift JS, Caldwell JC, Jinot J, Evans MV, Cote I, Vandenberg JJ. 2013. Scientific considerations for evaluating cancer bioassays conducted by the Ramazzini Institute. Environ Health Perspect. 121:1253–1263.
- Gilbert-Diamond D, Li Z, Perry AE, Spencer SK, Gandolfi AJ, Karagas MR. 2013. A population-based case-control study of urinary arsenic species and squamous cell carcinoma in New Hampshire, USA. Environ Health Perspect. 121:1154–1160.
- Goodman JI, Augustine KA, Cunnningham ML, Dixon D, Dragan YP, Falls JG, Rasoulpour RJ, Sills RC, Storer RD, Wolf DC, Pettit SD. 2010. What do we need to know prior to thinking about incorporating an epigenetic evaluation info safety assessments? Toxicol Sci. 116:375–381.
- Gossai A, Zens MS, Punshon T, Jackson BP, Perry AE, Karagas MR. 2017. Rice consumption and squamous cell carcinoma of the skin in a United States population. Environ Health Perspect. 125:097005–092018.
- Hamadeh H, Trouba K, Amin R, Afshari C, Germolec D. 2002. Coordination of altered DNA repair and damage pathways in arsenite-exposed keratinocytes. Toxicol Sci. 69:306–316.
- Haney J. 2015. Consideration of non-linear, non-threshold and threshold approaches for assessing the carcinogenicity of oral exposure to hexavalent chromium. Regul Toxicol Pharmacol. 73:834–852.
- Haque R, Mazumder DN, Samanta S, Ghosh N, Kalman D, Smith MM, Mitra S, Santra A, Lahiri S, Das S, et al. 2003. Arsenic in drinking water and skin lesions: dose-response data from West Bengal, India. Epidemiology. 14:174–182.
- Haseman JK. 1983. A reexamination of false-positive rates for carcinogenesis studies. Fundam Appl Toxicol. 3:334–339.
- Hayashi H, Kanisawa M, Yamanaka K, Ito T, Udaka N, Ohji H, Okudela K, Okada S, Kitamura H. 1998. Dimethylarsinic acid, a main metabolite of inorganic arsenics, has tumorigenicity and progression effects in the pulmonary tumors of A/J mice. Cancer Lett. 125:83–88.
- He H, Shui B. 2014. Folate intake and risk of bladder cancer: a meta-analysis of epidemiological studies. Int J Food Sci Nutr. 65:286–292.
- Health Canada. 2008. Guidelines for Canadian drinking water quality: guideline technical document – arsenic. https://www.canada.ca/en/health-canada/services/publications/healthy-living/guidelines-canadian-drinking-water-quality-guideline-technical-document-arsenic/page-11-guidelines-canadian-drinking-water-quality-guideline-technical-document-arsenic.html#a10.
- Heck JE, Andrew AS, Onega T, Rigas JR, Jackson BP, Karagas MR, Duell EJ. 2009. Lung cancer in a U.S. population with low to moderate arsenic exposure. Environ Health Perspect. 117:1718–1723.
- Hindmarsh JT, McCurdy RF. 1986. Clinical and environmental aspects of arsenic toxicity. Crit Rev Clin Lab Sci. 23:315–347.
- Hinkson IV, Elias JE. 2011. The dynamic state of protein turnover: It's about time. Trends Cell Biol. 21:293–303.
- Hirano S, Cui X, Li S, Kanno S, Kobayashi Y, Hayakawa T, Shraim A. 2003. Difference in uptake and toxicity of trivalent and pentavalent inorganic arsenic in rat heart microvessel endothelial cells. Arch Toxicol. 77:305–312.
- Hong Y-S, Ye B-J, Kim Y-M, Kim B-G, Kang G-H, Kim J-J, Song K-H, Kim Y-H, Seo J-W. 2017. Investigation of health effects according to the exposure of low concentration arsenic contaminated ground water. Int J Environ Res Pub Health. 14:1461.
- Hough RL, Fletcher T, Leonardi GS, Goessler W, Gnagnarella P, Clemens F, Gurzau E, Koppova K, Rudnai P, Kumar R, et al. 2010. Lifetime exposure to arsenic in residential drinking water in Central Europe. Int Arch Occup Environ Health. 83:471–481.
- Hsu KH, Tsui KH, Hsu LI, Chiou HY, Chen CJ. 2017. Dose-response relationship between inorganic arsenic exposure and lung cancer among arseniasis residents with low methylation capacity. Cancer Epidemiol Biomarkers Prev. 26:756–761.
- Hsueh Y-M, Cheng G-S, Wu M-M, Yu H-S, Kuo T-L, Chen C-J. 1995. Multiple risk factors associated with arsenic-induced skin cancer: effects of chronic liver disease and malnutrition status. Br J Cancer. 71:109–114.
- Hsueh Y-M, Chiou H-Y, Huang Y-L, Wu W-L, Huang C-C, Yang M-H, Lue L-C, Chen G-S, Chen C-J. 1997. Serum beta-carotene level, arsenic methylation capability, and incidence of skin cancer. Cancer Epidemiol Biomarkers Prev. 6:589–596.
- Hu J, Juan W, Sahyoun NR. 2016. Intake and biomarkers of folate and risk of cancer morbidity in older adults, NHANES 1999-2002 with Medicare linkage. PLoS One. 11:e0148697.
- Huang YK, Huang YL, Hsueh YM, Yang MH, Wu MM, Chen SY, Hsu LI, Chen CJ. 2008. Arsenic exposure, urinary arsenic speciation, and the incidence of urothelial carcinoma: a twelve-year follow-up study. Cancer Causes Control. 19:829–839.
- Huang C-Y, Lin Y-C, Shiue H-S, Chen W-J, Su C-T, Pu Y-S, Ao P-L, Hsueh Y-M. 2018. Comparison of arsenic methylation capacity and polymorphisms of arsenic methylation genes between bladder cancer and upper tract urothelial carcinoma. Toxicol Lett. 295:64–73.
- Huang C-Y, Pu Y-S, Shiue H-S, Chen W-J, Lin Y-C, Hsueh Y-M. 2016. Polymorphisms of human 8-oxoguanine DNA glycosylase 1 ad 8-hydroxydeoxyguanosine increase susceptibility to arsenic methylation capacity-related urothelial carcinoma. Arch Toxicol. 90:1917–1927.
- Hughes MF, Edwards BC, Herbin-Davis KM, Saunders J, Styblo M, Thomas DJ. 2010. Arsenic (+3 oxidation state) methyltransferase genotype affects steady-state distribution and clearance of arsenic in arsenate-treated mice. Toxicol Appl Pharmacol. 249:217–223.
- Hwang YH, Bornschein RL, Grote J, Menrath W, Roda S. 1997a. Environmental arsenic exposure of children around a former copper smelter site. Environ Res. 72:72–81.
- Hwang YH, Bornschein RL, Grote J, Menrath W, Roda S. 1997b. Urinary arsenic excretion as a biomarker of arsenic exposure in children. Arch Environ Health. 52:139–147.
- International Agency for Research on Cancer[IARC]. 1973. IARC monographs on the evaluation of carcinogenic risk of chemicals to man: some inorganic and organometallic compounds. Vol. 2. Lyon: IARC Press.
- International Agency for Research on Cancer[IARC]. 1980. IARC monographs on the evaluation of carcinogenic risk of chemicals to humans: some metals and metallic compounds. Vol. 23. Lyon: IARC Press.
- International Agency for Research on Cancer[IARC]. 2009. IARC Special Report: Policy. Vol. 10. Lyon: IARC Press.
- International Programme on Chemical Safety[IPCS]. 2005. Chemical-specific adjustment factors for interspecies differences and human variability: guidance document for use of data in dose/concentration-response assessment. harmonization project document No. 2. Geneva: IPCS, World Health Organization. [accessed 2018 Oct 6]. http://www.who.int/ipcs/methods/harmonization/areas/uncertainty/en/index.html.
- Jaspers I, Samet J, Reed W. 1999. Arsenite exposure of cultured airway epithelial cells activates kappaB-dependent interleukin-8 gene expression in the absence of nuclear factor-kappaB nuclear translocation. J Biol Chem. 274:31025–31033.
- Joint FAO/WHO Expert Committee on Food Additives[JECFA]. 2011. Safety evaluation of certain contaminants in food. WHO Food Additives Series: 63. FAO JECFA Monographs 8. Geneva: World Health Organization. Rome: Food and Agriculture Organization of the United Nations.
- Kao Y, Yu C, Chang L, Yu H. 2003. Low concentrations of arsenic induce vascular endothelial growth factor and nitric oxide release and stimulate angiogenesis in vitro. Chem Res Toxicol. 16:460–468.
- Karagas MR, Andrew AS, Nelson HH, Li Z, Punshon T, Schned A, Marsit CJ, Morris JS, Moore JH, Tyler AL, et al. 2012. SLC39A2 and FSIP1 polymorphisms as potential modifiers of arsenic-related bladder cancer. Hum Genet. 131:453–461.
- Karagas MR, Stukel TA, Morris JS, Tosteson TD, Weiss JE, Spencer SK, Greenberg ER. 2001. Skin cancer risk in relation to toenail arsenic concentrations in a US population-based case-control study. Am J Epidemiol. 153:559–565.
- Karagas MR, Stukel TA, Tosteson TD. 2002. Assessment of cancer risk and environmental levels of arsenic in New Hampshire. Int J Hyg Environ Health. 205:85–94.
- Karagas MR, Tosteson TD, Blum J, Klaue B, Weiss JE, Stannard V, Spate V, Morris JS. 2000. Measurement of low levels of arsenic exposure: a comparison of water and toenail concentrations. Am J Epidemiol. 152:84–90.
- Karagas MR, Tosteson TD, Morris JS, Demidenko E, Mott LA, Heaney J, Schned A. 2004. Incidence of transitional cell carcinoma of the bladder and arsenic exposure in New Hampshire. Cancer Causes Control. 15:465–472.
- Karagas MR, Cushing GL, Greenberg ER, Mott LA, Spencer SK, Nierenberg DW. 2001. Non-melanoma skin cancers and glucocorticoid therapy. Br J Cancer. 85:683–686.
- Karagas MR, Gossai A, Pierce B, Ahsan H. 2015. Drinking water arsenic contamination, skin lesions, and malignancies: a systematic review of the global evidence. Curr Envir Health Rpt. 2:52–68.
- Kenyon EM, Klimecki WT, El-Masri H, Conolly RB, Clewell HJ, Beck BD. 2008. How can biologically-based modeling of arsenic kinetics and dynamics inform the risk assessment process? – a workshop review. Toxicol Appl Pharmacol. 232:359–368.
- Kim T-H, Seo J-W, Hong Y-S, Song K-H. 2017. Case-control study of chronic low-level exposure of inorganic arsenic species and non-melanoma skin cancer. J Dermatol. 44:1374–1379.
- Kinoshita A, Wanibuchi H, Morimura K, Wei M, Nakae D, Arai T, Minowa O, Noda T, Nishimura S, Fukushima S. 2007. Carcinogenicity of dimethylarsinic acid in Ogg1-deficient mice. Cancer Sci. 98:803–814.
- Kirkland DJ, Müller L. 2000. Interpretation of the biological relevance of genotoxicity test results: the importance of thresholds. Mutat Res. 464:137–147.
- Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, Bernal AL, Reid KB, Madan T, Chakraborty T. 2006. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol. 43:1293–1315.
- Kitchin KT, Conolly R. 2010. Arsenic-induced carcinogenesis-oxidative stress as a possible mode of action and future research needs for more biologically based risk assessment. Chem Res Toxicol. 23:327–335.
- Kitchin KT, Wallace K. 2005. Arsenite binding to synthetic peptides based on the Zn finger region and the estrogen binding region of the human estrogen receptor-alpha. Toxicol Appl Pharmacol. 206:66–72.
- Kitchin KT, Wallace K. 2006. Dissociation of arsenite-peptide complexes: triphasic nature, rate constants, half-lives, and biological importance. J Biochem Mol Toxicol. 20:48–56.
- Kitchin KT, Wallace K. 2008a. Evidence against the nuclear in situ binding of arsenicals—oxidative stress theory of arsenic carcinogenesis. Toxicol Appl Pharmacol. 232:252–257.
- Kitchin KT, Wallace K. 2008b. The role of protein binding of trivalent arsenicals in arsenic carcinogenesis and toxicity. J Inorg Biochem. 102:532–539.
- Kligerman AD, Doerr CL, Tennant AH, Harrington-Brock K, Allen JW, Winkfield E, Poorman-Allen P, Kundu B, Funasaka K, Roop BC, et al. 2003. Methylated trivalent arsenicals as candidate ultimate genotoxic forms of arsenic: induction of chromosomal mutations but not gene mutations. Environ Mol Mutagen. 42:192–205.
- Kligerman AD, Tennant AH. 2007. Insights into the carcinogenic mode of action of arsenic. Toxicol Appl Pharmacol. 222:281–288.
- Knobeloch LM, Zierold KM, Anderson HA. 2006. Association of arsenic-contaminated drinking-water with prevalence of skin cancer in Wisconsin’s Fox River Valley. J Health Popul Nutr. 24:206–213.
- Koutros S, Baris D, Waddell R, Beane Freeman LE, Colt JS, Schwenn M, Johnson A, Ward MH, Hosain GM, Moore LE, et al. 2018. Potential effect modifiers of the arsenic-bladder cancer risk relationship. Int J Cancer. 143:2640–2646.
- Koutros S, Lenz P, Hewitt SM, Kida M, Jones M, Schned AR, Baris D, Pfeiffer R, Schwenn M, Johnson A, et al. 2018. Elevated bladder cancer in northern New England: the role of drinking water and arsenic. J Natl Cancer Inst. 110:1273–1274.
- Kuo YC, Lo YS, Guo HR. 2017. Lung cancer associated with arsenic Ingestion: cell-type specificity and dose response . Epidemiology. 28:S106–S112.
- Kuo CC, Moon KA, Wang SL, Silbergeld E, Navas-Acien A. 2017. The association of arsenic metabolism with cancer, cardiovascular disease, and diabetes: a systematic review of the epidemiological evidence. Environ Health Perspect. 125:087001.
- Kurttio P, Pukkala E, Kahelin H, Auvinen A, Pekkanen J. 1999. Arsenic concentrations in well water and risk of bladder and kidney cancer in Finland. Environ Health Perspect. 107:705–710.
- Lamm SH, Boroje IJ, Ferdosi H, Ahn J. 2018. Lung cancer risk and low (≤50 μg/L) drinking water arsenic levels for US Counties (2009-2013)-a negative Association. Ijerph. 15:1200.
- Lamm SH, Engel A, Kruse MB, Feinleib M, Byrd DM, Lai S, Wilson R. 2004. Arsenic in drinking water and bladder cancer mortality in the United States: an analysis based on 133 U.S. counties and 30 years of observation. J Occup Environ Med. 46:298–306.
- Lamm SH, Ferdosi H, Dissen EK, Li J, Ahn J. 2015. A systematic review and meta-regression analysis of lung cancer risk and inorganic arsenic in drinking water. Int J Environ Res Public Health. 12:15498–15515.
- Lamm SH, Luo Z-D, Bo F-B, Zhang G-Y, Zhang Y-M, Wilson R, Byrd DM, Lai S, Li F-X, Polkanov M. 2007. An epidemiological study of asenic-related skin disorders and skin cancer and the consumption of arsenic-related skin disorders and skin cancer and the consumption of arsenic-contaminated well waters in Huhhot, Inner Mongolia, China. Human Ecol Risk Asesss. 13:713–746.
- Lantz RC, Hays AM. 2006. Role of oxidative stress in arsenic-induced toxicity. Drug Metab Rev. 38:791–804.
- Lau A, Li M, Xie R, He Q, Chiu J. 2004. Opposed arsenite-induced signaling pathways promote cell proliferation or apoptosis in cultured lung cells. Carcinogenesis. 25:21–28.
- Le XC, Lu X, Ma M, Cullen WR, Aposhian HV, Zheng B. 2000. Speciation of key arsenic metabolic intermediates in human urine. Anal Chem. 72:5172–5177.
- Leonardi G, Vahter M, Clemens F, Goessler W, Gurzau E, Hemminki K, Hough R, Koppova K, Kumar R, Rudnai P, et al. 2012. Inorganic arsenic and basal cell carcinoma in areas of Hungary, Romania, and Slovakia: a case-control study. Environ Health Perspect. 120:721–726.
- Lewis AS, Beyer LA, Zu K. 2015. Considerations in deriving quantitative cancer criteria for inorganic arsenic exposure via inhalation. Environ Int. 74:258–273.
- Lewis DR, Southwick JW, Ouellet-Hellstrom R, Rench J, Calderon RL. 1999. Drinking water arsenic in Utah: a cohort mortality study. Environ Health Perspect. 107:359–365.
- Li J, Packianathan C, Rossman TG, Rosen BP. 2017. Nonsynonymous polymorphisms in the human AS3MT arsenic methylation gene: Implications for arsenic toxicity. Chem Res Toxicol. 30:1481–1491.
- Liao W, Chang K, Yu C, Chen G, Chang L, Yu H. 2004. Arsenic induces human keratinocyte apoptosis by the FAS/FAS ligand pathway, which correlates with alterations in nuclear factor-j B, activator protein-1 activity. J Invest Dermatol. 122:125–129.
- Lin Y-C, Chen W-J, Huang C-Y, Shiue H-S, Su C-T, Ao P-L, Pu Y-S, Hsueh Y-M. 2018. Polymorphisms of arsenic (+3 oxidation state) methyltransferase and arsenic methylation capacity affect the risk of bladder cancer. Toxicol Sci. 164:328–338.
- Lock EA, Hard GC. 2004. Chemically induced renal tubule tumors in the laboratory rat and mouse: Review of the NCI/NTP database and categorization of renal carcinogens based on mechanistic information. Crit Rev Toxicol. 34:211–299.
- Lu X, Arnold LL, Cohen SM, Cullen WR, Le XC. 2003. Speciation of dimethylarsinous acid and trimethylarsine oxide in urine from rats fed with dimethylarsinic acid and dimercaptopropane sulfonate. Anal Chem. 75:6463–6468.
- Lu K, Cable PH, Abo RP, Ru H, Graffam ME, Schlieper KA, Parry NM, Levine S, Bodnar WM, Wishnok JS, et al. 2013. Gut microbiome perturbations induced by bacterial infection affect arsenic biotransformation. Chem Res Toxicol. 26:1893–1903.
- Lu K, Mahbub R, Cable PH, Ru H, Parry NM, Bodnar WM, Wishnok JS, Styblo M, Swenberg JA, Fox JG, et al. 2014. Gut microbiome phenotypes driven by host genetics affect arsenic metabolism. Chem Res Toxicol. 27:172–174.
- Lu M, Wang H, Li XF, Arnold LL, Cohen SM, Le XC. 2007. Binding of dimethylarsinous acid to cys-13alpha of rat hemoglobin is responsible for the retention of arsenic in rat blood. Chem Res Toxicol. 20:27–37.
- Lu M, Wang H, Li XF, Lu X, Cullen WR, Arnold LL, Cohen SM, Le XC. 2004. Evidence of hemoglobin binding to arsenic as a basis for the accumulation of arsenic in rat blood. Chem Res Toxicol. 7:1733–1742.
- Luo L, Li Y, Gao Y, Zhao L, Feng H, Wei W, Qiu C, He Q, Zhang Y, Fu S, Sun D. 2018. Association between arsenic metabolism gene polymorphisms and arsenic-induced skin lesions in individuals exposed to high-dose inorganic arsenic in northwest China. Sci Rep. 118:413.
- Lynch HN, Zu K, Kennedy EM, Lam T, Liu X, Pizzurro DM, Loftus CT, Rhomberg LR. 2017a. Quantitative assessment of lung and bladder cancer risk and oral exposure to inorganic arsenic: Meta-regression analyses of epidemiological data. Environ Int. 106:178–206.
- Lynch HN, Zu K, Kennedy EM, Lam T, Liu X, Pizzurro DM, Loftus CT, Rhomberg LR. 2017b. Corrigendum to quantitative assessment of lung and bladder cancer risk and oral exposure to inorganic arsenic: meta-regression analyses of epidemiological data Environmental International 106:178-206. Environ Int. 109:195–196.
- MacGregor JT, Frötschl R, White PA, Crump KS, Eastmond DA, Fukushima S, Guérard M, Hayashi M, Soeteman-Hernández LG, Kasamatsu T, et al. 2015. IWGT report on quantitative approaches to genotoxicity risk assessment. II. Use of point-of-departure (POD) metrics in defining acceptable exposure limits and assessing human risk. Mutat Res. 783:55–65.
- Mandal BK, Ogra Y, Suzuki KT. 2001. Identification of dimethylarsinous and monomethylarsonous acids in human urine of the arsenic-affected areas in West Bengal, India. Chem Res Toxicol. 14:371–378.
- Marafante E, Vahter M, Dencker L. 1984. Metabolism of arsenocholine in mice, rats and rabbits. Sci Total Environ. 34:223–240.
- Mayer JE, Goldman RH. 2016. Arsenic and skin cancer in the USA: the current evidence regarding arsenic-contaminated drinking water. Int J Dermatol. 55:e585–e591.
- Mazumder DN. 2007. Arsenic and non-malignant lung disease. J Environ Sci Health A Tox Hazard Subst Environ Eng. 42:1859–1867.
- Mazumder DNG, Haque R, Ghosh N, De BK, Santra A, Chakraborty D, Smith AH. 1998. Arsenic levels in drinking water and the prevalence of skin lesions in West Bengal, India. Int J Epidemiol. 27:871–877.
- Mazumder DN, Steinmaus C, Bhattacharya P, von Ehrenstein OS, Ghosh N, Gotway M, Sil A, Balmes JR, Haque R, Hira-Smith MM, et al. 2005. Bronchiectasis in persons with skin lesions resulting from arsenic in drinking water. Epidemiology. 16:760–765.
- Meek ME, Bucher JR, Cohen SM, Dellarco V, Hill RN, Lehman-McKeeman LD, Longfellow DG, Pastoor T, Seed J, Patton DE. 2003. A framework for human relevance analysis of information on carcinogenic modes of action. Crit Rev Toxicol. 33:591–653.
- Melak D, Ferreccio C, Kalman D, Parra R, Acevedo J, Pérez L, Cortés S, Smith AH, Yuan Y, Liaw J, et al. 2014. Arsenic methylation and lung and bladder cancer in a case-control study in northern Chile. Toxicol Appl Pharmacol. 274:225–231.
- Meliker JR, Slotnick MJ, Avruskin GA, Schottenfeld D, Jacquez GM, Wilson ML, Goovaerts P, Franzblau A, Nriagu JO. 2010. Lifetime exposure to arsenic in drinking water and bladder cancer: a population-based case-control study in Michigan, USA. Cancer Causes Control. 21:745–757.
- Mendez WM, Jr, Eftim S, Cohen J, Warren I, Cowden J, Lee JS, Sams R. 2017. Relationships between arsenic concentrations in drinking water and lung and bladder cancer incidence in U.S. counties. J Expo Sci Environ Epidemiol. 27:235–243.
- Mengesdorf T, Althausen S, Paschen W. 2002. Genes associated with pro-apoptotic and protective mechanisms are affected differently on exposure of neuronal cell cultures to arsenite. No indication for endoplasmic reticulum stress despite activation of grp78 and gadd153 expression. Brain Res Mol Brain Res. 104:227–239.
- Michaud DS, Wright ME, Cantor KP, Taylor PR, Virtamo J, Albanes D. 2004. Arsenic concentrations in prediagnostic toenails and the risk of bladder cancer in a cohort study of male smokers. Am J Epidemiol. 160:853–859.
- Mink PJ, Alexander DD, Barraj LM, Kelsh MA, Tsuji JS. 2008. Low-level arsenic exposure in drinking water and bladder cancer: a review and meta-analysis. Regul Toxicol Pharmacol. 52:299–310.
- Morales KH, Ryan L, Kuo TL, Wu MM, Chen CJ. 2000. Risk of internal cancers from arsenic in drinking water. Environ Health Perspect. 108:655–661.
- Morikawa T, Wanibuchi H, Morimura K, Ogawa M, Fukushima S. 2000. Promotion of skin carcinogenesis by dimethylarsinic acid in Keratin (K6)/ODC transgenic mice. Jpn J Cancer Res. 91:579–581.
- Mostafa MG, Cherry N. 2015. Arsenic in drinking water, transitional cell cancer and chronic cystitis in rural Bangladesh. Int J Enviorn Res Public Health. 12:13739–13749.
- Mostafa MG, McDonald JC, Cherry NM. 2008. Lung cancer and exposure to arsenic in rural Bangladesh. Occup Environ Med. 65:765–768.
- Nachman KE, Punshon T, Rardin L, Signes-Pastor AJ, Murray CJ, Jackson BP, Guerinot ML, Burke TA, Chen CY, Ahsan H, et al. 2018. Opportunities and challenges for dietary arsenic intervention. Environ Health Perspect. 126:84503.
- Namgung U, Xia Z. 2001. Arsenic induces apoptosis in rat cerebellar neurons via activation of JNK3 and p38 MAP kinases. Toxicol Appl Pharmacol. 174:130–138.
- National Research Council [NRC]. 1999. Arsenic in drinking water. Washington (DC): National Academies Press.
- National Research Council [NRC]. 2001. Arsenic in drinking water: an update. Washington (DC): National Academies Press.
- National Research Council [NRC]. 2007. Toxicity testing in the 21st century: a vision and a strategy. Washington (DC): National Academies Press.
- National Research Council [NRC]. 2013. Critical aspects of the EPA’s IRIS assessment of inorganic arsenic: interim report. Committee on inorganic arsenic, board on environmental studies and toxicology, division on earth and life studies. Washington (DC): National Academies Press.
- National Toxicology Program [NTP]. 2000. Toxicology and Carcinogenesis Studies of Gallium Arsenide (CAS NO. 1303-00-0) in F344/N Rats and B6C3F1 Mice (Inhalation Studies).
- National Toxicology Program [NTP]. 2011. Summary report of the National Toxicology Program and Environmental Protection Agency‐sponsored review of pathology materials from selected Ramazzini Institute rodent cancer bioassays.
- Nearing MM, Koch I, Reimer KJ. 2014. Arsenic speciation in edible mushrooms. Environ Sci Technol. 48:14203–14210.
- Nesnow S, Roop BC, Lambert G, Kadiiska M, Mason RP, Cullen WR, Mass MJ. 2002. DNA damage induced by methylated trivalent arsenicals is mediated by reactive oxygen species. Chem Res Toxicol. 15:1627–1634.
- Ng JC, Seawright AA, Qi L, Garnett CM, Chiswell B, Moore MR. 1999. Tumors in mice induced by exposure to sodium arsenate in drinking water. In: Chappell WR, Abernathy CO, Calderon RL, editors. Arsenic Exposure and Health Effects. Amsterdam: Elsevier Science, p. 217–223.
- Niedzwiecki MM, Liu X, Zhu H, Hall MN, Slavkovich V, Ilievski V, Levy D, Siddique AB, Kibriya MG, Parvez F, et al. 2018. Serum homocysteine, arsenic methylation, and arsenic-induced skin lesion incidence in Bangladesh: a one-carbon metabolism candidate gene study. Environ Int. 113:133–142.
- Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, Fraire AE, Gabrielson EW, Gunning WT, Haines DC, et al. 2004. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 64:2307–2316.
- Nohara K, Tateishi Y, Suzuki T, Okamura K, Murai H, Takumi S, Maekawa F, Nishimura N, Kobori M, Ito T. 2012. Late-onset increases in oxidative stress and other tumorigenic activities and tumors with a Ha-ras mutation in the liver of adult male C3H mice gestationally exposed to arsenic. Toxicol Sci. 129:293–304.
- Nunez O, Fernandez-Navarro P, Martin-Mendez I, Bel-Lan A, Locutura JF, Lopez-Abente G. 2016. Arsenic and chromium topsoil levels and cancer mortality in Spain. Environ Sci Pollut Res Int. 23:17664–17675.
- Organisation for Economic Co-operation and Development[OECD]. 2010. OECD guidance document n° 116 on the design and conduct of chronic toxicity and carcinogenicity studies, supporting tg 451, 452, and 453, section 4: statistical and dose response analysis, including benchmark dose and linear extrapolation, NOAELS and NOELS, LOAELS and LOELS. Paris: OECD.
- Organisation for Economic Co-operation and Development[OECD]. 2015. Guidance document on revisions to OECD genetic toxicology test guidelines. August 31, 2015. Paris: OECD.
- Ott MG, Holder BB, Gordon HL. 1974. Respiratory cancer and occupational exposure to arsenicals. Arch Environ Health. 29:250–255.
- Packianathan C, Kandavelu P, Rosen BP. 2018. The Structure of an As(III) S-adenosylmethionine methyltransferase with 3-coordinately bound As(III) depicts the first step in catalysis. Biochemistry. 57:4083–4092.
- Palmieri MA, Molinari BL. 2015. Effect of sodium arsenite on mouse skin carcinogenesis. Toxicol Pathol. 43:704–714.
- Parrish A, Zheng X, Turney K, Younis H, Gandolfi A. 1999. Enhanced transcription factor DNA binding and gene expression induced by arsenite or arsenate in renal slices. Toxicol Sci. 50:98–105.
- Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Aposhian HV. 2000. Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicol Appl Pharmacol. 163:203–207.
- Pierce BL, Argos M, Chen Y, Melkonia S, Parvez F, Islam T, Ahmed A, Hasan R, Rathouz PJ, Ahsan H. 2011. Arsenic exposure, dietary patterns, and skin lesion risk in Bangladesh: a prospective study. Am J Epidemiol. 173:345–354.
- Pilsner JR, Liu X, Ahsa H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. 2009. Folate deficiency, hyperhomocysteinemia, low urinary creatinine, and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ Health Perspect. 117:254–260.
- Pinyayev TS, Kohan MJ, Herbin-Davis K, Creed JT, Thomas DJ. 2011. Preabsorptive metabolism of sodium arsenate by anaerobic microbiota of mouse cecum forms a variety of methylated and thiolated arsenicals. Chem Res Toxicol. 24:475–477.
- Proctor DM, Suh M, Chappell G, Borghoff SJ, Thompson CM, Wiench K, Finch L, Ellis-Hutchings R. 2018. An Adverse Outcome Pathway (AOP) for forestomach tumors induced by non-genotoxic initiating events. Regul Toxicol Pharmacol. 96:30–40.
- Qin X, Cui Y, Shen L, Sun N, Zhang Y, Li J, Xu X, Wang B, Xu X, Huo Y, Wang X. 2013. Folic acid supplementation and cancer risk: A meta-analysis of randomized controlled trials. Int J Cancer. 133:1033–1041.
- Rager JE, Auerbach SS, Chappell GA, Martin E, Thompson CM, Fry RC. 2017. Benchmark dose modeling estimates of the concentrations of inorganic arsenic that induce changes to the neonatal transcriptome, proteome, and epigenome in a pregnancy cohort. Chem Res Toxicol. 30:1911–1920.
- Rasheed H, Kay P, Slack R, Gong YY. 2018. The effect of association between inefficient arsenic methylation capacity and demographic characteristics on the risk of skin lesions. Toxicol Appl Pharm. 339:42–51.
- Rea M, Gregg J, Qin Q, Phillips M, Rice R. 2003. Global alteration of gene expression in human keratinocytes by inorganic arsenic. Carcinogenesis. 24:747–756.
- Rehman K, Naranmandura H. 2012. Arsenic metabolism and thioarsenicals. Metallomics. 4:881–892.
- Rhomberg LR, Goodman JE, Haber LT, Dourson M, Andersen ME, Klaunig JE, Meek B, Price PS, McClellan RO, Cohen SM. 2011. Linear low-dose extrapolation for noncancer heath effects is the exception, not the rule. Crit Rev Toxicol. 41:1–21.
- Rossman TG, Uddin AN, Burns FJ, Bosland MC. 2001. Arsenite is a cocarcinogen with solar ultraviolet radiation for mouse skin: an animal model for arsenic carcinogenesis. Toxicol Appl Pharmacol. 176:64–71.
- Saint-Jacques N, Brown P, Nauta L, Boxall J, Parker L, Dummer TJB. 2018. Estimating the risk of bladder and kidney cancer from exposure to low-levels of arsenic in drinking water, Nova Scotia, Canada. Environ Int. 110:95–104.
- Salim EI, Wanibuchi H, Morimura K, Wei M, Mitsuhashi M, Yoshida K, Endo G, Fukushima S. 2003. Carcinogenicity of dimethylarsinic acid in p53 heterozygous knockout and wild-type C57BL/6J mice. Carcinogenesis. 24:335–342.
- Schabath MB, Spitz MR, Lerner SP, Pillow PC, Hernandez LM, Delclos GL, Grossman HB, Wu X. 2005. Case-control analysis of dietary folate and risk of bladder cancer. Nutr Cancer. 53:144–151.
- Schoof RA, Yost LJ, Eickhoff J, Crecelius EA, Cragin DW, Meacher DM, Menzel DB. 1999. A market basket survey of inorganic arsenic in food. Food Chem Toxicol. 37:839–846.
- Seed J, Carney EW, Corley RA, Crofton KM, DeSesso JM, Foster PM, Kavlock R, Kimmel G, Klaunig J, Meek ME, et al. 2005. Overview: using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit Rev Toxicol. 35:664–672.
- Seow WJ, Pan WC, Kile ML, Baccarelli AA, Quamruzzaman Q, Rahman M, Mahiuddin G, Mostofa G, Lin X, Christiani DC. 2012. Arsenic reduction in drinking water and improvement in skin lesions: a follow-up study in Bangladesh. Environ Health Perspect. 120:1733–1738.
- Seow WJ, Pan WC, Kile ML, Lin T, Baccarelli A, Quamruzzaman Q, Rahman M, Mostofa G, Rakibuz-Zaman M, Kibriya M, et al. 2015. A distinct and replicable squamous cell carcinoma gene INPPA5 variant modifies susceptibility of arsenic-associated skin lesions in Bangladesh. Cancer. 121:2222–2229.
- Shain AH, Bastian BC. 2016. From melanocytes to melanomas. Nat Rev Cancer. 16:345.
- Sharma A, Flora SJS. 2018. Nutritional management can assist a significant role in alleviation of arsenicosis. J Trace Elem Med Biol. 45:11–20.
- Shen J, Wanibuchi H, Salim EI, Wei M, Kinoshita A, Yoshida K, Endo G, Fukushima S. 2003. Liver tumorigenicity of trimethylarsine oxide in male Fischer 344 rats-association with oxidative DNA damage and enhanced cell proliferation. Carcinogenesis. 24:1827–1835.
- Shimizu M, Hochadel J, Fulmer B, Waalkes M. 1998. Effect of glutathione depletion and metallothionein gene expression on arsenic-induced cytotoxicity and c-myc expression in vitro. Toxicol Sci. 45:204–211.
- Shirachi DY, Johansen MG, McGowan JP, Tu SH. 1983. Tumorigenic effect of sodium arsenite in rat kidney. Proc West Pharmacol Soc. 26:413–415.
- Simeonova PP, Wang S, Toriuma W, Kommineni V, Matheson J, Unimye N, Kayama F, Harki D, Dng M, Vallyathan V, Luster MI. 2000. Arsenic mediates cell proliferation and gene expression in the bladder epithelium: association with activating protein-1 transactivation. Cancer Res. 60:3445–3453.
- Smeester L, Rager JE, Bailey KA, Guan X, Smith N, García-Vargas G, Del Razo L-M, Drobná Z, Kelkar H, Stýblo M, Fry RC. 2011. Epigenetic changes in individuals with arsenicosis. Chem Res Toxicol. 24:165–167.
- Smith AH, Ercumen A, Yuan Y, Steinmaus CM. 2009. Increased lung cancer risks are similar whether arsenic is ingested or inhaled. J Expo Sci Environ Epidemiol. 19:343–348.
- Smith AH, Marshall G, Liaw J, Yuan Y, Ferreccio C, Steinmaus C. 2012. Mortality in young adults following in utero and childhood exposure to arsenic in drinking water. Environ Health Perspect. 120:1527–1531.
- Smith AH, Marshall G, Roh T, Ferreccio C, Liaw J, Steinmaus C. 2018. Lung, bladder, and kidney cancer mortality 40 years after arsenic exposure reduction. J Natl Cancer Inst. 110:241–249.
- Snow ET, Sykora P, Durham TR, Klein CB. 2005. Arsenic, mode of action at biologically plausible low doses: what are the implications for low dose cancer risk? Toxicol Appl Pharmacol. 207:557–564.
- Soffritti M, Belpoggi F, Degli Esposti D, Lambertini L. 2006. Results of a long-term carcinogenicity bioassay on Sprague-Dawley rats exposed to sodium arsenite administered in drinking water. Ann N Y Acad Sci. 1076:578–591.
- Sonich-Mullin C, Fielder R, Wiltse J, Baetcke K, Dempsey J, Fenner-Crisp P, Grant D, Hartley M, Knaap A, Kroese D, et al. 2001. IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis. Regul Toxicol Pharmacol. 34:146–152.
- Stanek EJIII, Calabrese EJ, Zorn M. 2001. Soil ingestion distributions for Monte Carlo risk assessment in children. Human Ecol Risk Assess. 7:357–368.
- Steinmaus C, Bates MN, Yuan Y, Kalman D, Atallah R, Rey OA, Biggs ML, Hopenhayn C, Moore LE, Hoang BK, Smith AH. 2006. Arsenic methylation and bladder cancer risk in case-control studies in Argentina and the United States. J Occup Environ Med. 48:278–488.
- Steinmaus C, Ferreccio C, Yuan Y, Acevedo J, González F, Perez L, Cortés S, Balmes JR, Liaw J, Smith AH. 2014. Elevated lung cancer in younger adults and low concentrations of arsenic in water. Am J Epidemiol. 180:1082–1087.
- Steinmaus C, Yuan Y, Bates MN, Smith AH. 2003. Case-control study of bladder cancer and drinking water arsenic in the western United States. Am J Epidemiol.Epidemiol. 158:1193–1201.
- Steinmaus CM, Yuan Y, Smith AH. 2005. The temporal stability of arsenic concentrations in well water in western Nevada. Environ Res. 99:164–168.
- Steinmaus C, Ferreccio C, Acevedo J, Yuan Y, Liaw J, Duran V, Cuevas S, Garcia J, Meza R, Valdes R, et al. 2014. Increased lung and bladder cancer incidence in adults after in utero and early-life arsenic exposure. Cancer Epidemiol Biomarkers Prev. 23:1529–1538.
- Steinmaus CM, Ferreccio C, Romo JA, Yuan Y, Cortes S, Marshall G, Moore LE, Balmes JR, Liaw J, Golden T, Smith AH. 2013. Drinking water arsenic in northern chile: high cancer risks 40 years after exposure cessation. Cancer Epidemiol Biomarkers Prev. 22:623–630.
- Stengel B. 2010. Chronic kidney disease and cancer: a troubling connection. J Nephrol. 23:253–262.
- Storer RD, French JE, Haseman J, Hajian G, LeGrand EK, Long GG, Mixson LA, Ochoa R, Sagartz JE, Soper KA. 2001. P53+/− hemizygous knockout mouse: overview of available data. Toxicologic Path. 29:30–50.
- Strupp C, Bomann W, Cohen SM, Weber K. 2016. Relationship of metabolism and cell proliferation to the mode of action of fluensulfone-induced mouse lung tumors. II: additional mechanistic studies. Toxicol Sci. 154:296–308.
- Sturlan S, Baumgartner M, Roth E, Bachleitner-Hofmann T. 2003. Docosahexaenoic acid enhances arsenic trioxide-mediated apoptosis in arsenic trioxide-resistant HL-60 cells. Blood. 101:4990–4997.
- Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ. 2000. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol. 74:289–299.
- Suzuki S, Arnold LL, Ohnishi T, Cohen SM. 2008. Effects of inorganic arsenic on the rat and mouse urinary bladder. Toxicol Sci. 106:350–363.
- Suzuki S, Arnold LL, Pennington KL, Chen B, Naranmandura H, Le XC, Cohen SM. 2010. Dietary administration of sodium arsenite to rats: relations between dose and urinary concentrations of methylated and thio-metabolites and effects on the rat urinary bladder epithelium. Toxicol Appl Pharmacol. 244:99–105.
- Suzuki S, Arnold LL, Pennington KL, Kakiuchi-Kiyota S, Cohen SM. 2009. Effects of co-administration of dietary sodium arsenite and an NADPH oxidase inhibitor on the rat bladder epithelium. Toxicology. 261:41–46.
- Suzuki S, Cohen SM, Arnold LL, Kato H, Fuji S, Pennington KL, Nagayasu Y, Naiki-Ito A, Yamashita Y, Takahashi S. 2018. Orally administered nicotine effects on rat urinary bladder proliferation and carcinogenesis. Toxicology. 398–399:31–40.
- Takumi S, Aoki Y, Sano T, Suzuki T, Nohmi T, Nohara K. 2014. In vivo mutagenicity of arsenite in the livers of gpt delta transgenic mice. Mutat Res Genet Toxicol Environ Mutagen. 760:42–47.
- Takumi S, Okamura K, Suzuki T, Hano H, Nohara K, Yanagisawa H. 2015. (Abstract). Gestational arsenic exposure affects gene expression in the kidney and lung in the F1 and F2 mice. The Toxicologist. 422.
- Taylor V, Li Z, Sayarath V, Palys TJ, Morse KR, Scholz-Bright RA, Karagas MR. 2017. Distinct arsenic metabolites following seaweed consumption in humans. Sci Rep. 7:3920.
- Thomas DJ. 2010. Arsenolysis and thiol-dependent arsenate reduction. Toxicol Sci. 117:249–252.
- Thomas DJ. 2015. The chemistry and metabolism of arsenic. In: J. C. States, editor. Arsenic: Exposure Sources, Health Risks and Mechanisms of Toxicity. New York, NY: Wiley; p. 149–201.
- Thomas DJ, Bradham K. 2016. Role of complex organic arsenicals in food in aggregate exposure to arsenic. J Environ Sci (China). 49:86–96.
- Thomas RS, Philbert MA, Auerbach SS, Wetmore BA, Devito MJ, Cote I, Rowlands JC, Whelan MP, Hays SM, Andersen ME, et al. 2013. Incorporating new technologies into toxicity testing and risk assessment: moving from 21st century vision to a data-driven framework. Toxicol Sci. 136:4–18.
- Thomas DJ, Styblo M, Lin S. 2001. The cellular metabolism and systemic toxicity of arsenic. Toxicol Appl Pharmacol. 176:127–144.
- Thomas RS, Wesselkamper SC, Wang NCY, Zhao QJ, Petersen DD, Lambert JC, Cote I, Yang L, Healy E, Black MB, et al. 2013. Temporal concordance between apical and transcriptional points of departure for chemical risk assessment. Toxicol Sci. 134:180–194.
- Thompson CM, Wolf JC, McCoy A, Suh M, Proctor DM, Kirman CR, Haws LC, Harris MA. 2017. Comparison of toxicity and recovery in the duodenum of B6C3F1 mice following treatment with intestinal carcinogens captan, folpet, and hexavalent chromium. Toxicol Pathol. 45:1091–1101.
- Tokar EJ, Benbrahim-Tallaa L, Ward JM, Lunn R, Sams RL, II, Waalkes MP. 2010a. Cancer in experimental animals exposed to arsenic and arsenic compounds. Crit Rev Toxicol. 40:912–927.
- Tokar EJ, Diwan BA, Thomas DJ, Waalkes MP. 2012. Tumors and proliferative lesions in adult offspring after maternal exposure to methylarsonous acid during gestation in CD1 mice. Arch Toxicol. 86:975–982.
- Tokar EJ, Diwan BA, Ward JM, Delker DA, Waalkes MP. 2011. Carcinogenic effects of whole-life exposure to inorganic arsenic in CD1 mice. Toxicol Sci. 119:73–83.
- Tollestrup K, Frost FJ, Harter LC, McMillan GP. 2003. Mortality among children residing near the American Smelting and Refining Company (ASARCO) copper smelter in Ruston, Washington. Arch Environ Health. 58:683–691.
- Tseng WP. 1977. Effects and dose response relationships of skin cancer and blackfoot disease with arsenic. Environ Health Perspect. 19:109–119.
- Tseng WP, Chu HM, How SW, Fong JM, Lin CS, Yeh S. 1968. Prevalence of skin cancer in an endemic area of chronic arsenicism in Taiwan. J Natl Cancer Inst. 40:453–463.
- Tsuji JS, Alexander DD, Perez V, Mink PJ. 2014. Arsenic exposure and bladder cancer: quantitative assessment of studies in human populations to detect risks at low doses. Toxicology. 317:17–30.
- Tsuji JS, Benson R, Schoof RA, Hook GC. 2004. Health effect levels for risk assessment of childhood exposure to arsenic. Regul Toxicol Pharmacol. 39:99–110.
- Tsuji JS, Garry MR, Perez V, Chang ET. 2015. Low-level arsenic exposure and developmental neurotoxicity in children: a systematic review and risk assessment. Toxicology. 337:91–107.
- Tsuji JS, Perez V, Garry MR, Alexander DD. 2014. Association of low-level arsenic exposure in drinking water with cardiovascular disease: a systematic review and risk assessment. Toxicology. 323:78–94.
- Tsuji JS, Yost LJ, Barraj LM, Scrafford CG, Mink PJ. 2007. Use of background inorganic arsenic exposures to provide perspective on risk assessment results. Regul Toxicol Pharmacol. 48:59–68.
- Tsuji JS, Van Kerkhove MD, Kaetzel RS, Scrafford CG, Mink PJ, Barraj LM, Crecelius EA, Goodman M. 2005. Evaluation of exposure to arsenic in residential soil. Environ Health Perspect 113:1736–1740.
- Tynkevich E, Flamant M, Haymann JP, Metzger M, Thervet E, Boffa JJ, Vrtovsnik F, Houillier P, Froissart M, Stengel B. 2014. Decrease in urinary creatinine excretion in early stage chronic kidney disease. PLoS One. 9:e111949.
- U.S. Environmental Protection Agency [USEPA], Science Advisory Committee [SAB]. 2011. Review Comments on EPA’s Responsiveness to SAB 2007 Recommendations for the Revision of Cancer Assessment of Inorganic Arsenic. Washington (DC): USEPA Science Advisory Committee.
- U.S. Environmental Protection Agency [USEPA]. 1984. Health assessment document for arsenic EPA/600/8-83/021F. Washington (DC): Office of Health and Environmental Assessment, USEPA.
- U.S. Environmental Protection Agency [USEPA]. 1986. Guidelines for carcinogen risk assessment. Federal Register. 51:33992–34003.
- U.S. Environmental Protection Agency [USEPA]. 1988. Special report on ingested inorganic arsenic. Skin cancer; nutritional essentiality. EPA/625/3-87/013. Washington (DC): Risk Assessment Forum, USEPA.
- U.S. Environmental Protection Agency [USEPA]. 1994. Methods for derivation of inhalation reference concentrations and application of inhalation dosimetry EPA/600/8-90/066F. Washington (DC): Office of Health and Environmental Assessment, USEPA.
- U.S. Environmental Protection Agency [USEPA]. 1995. Arsenic, inorganic CASRN 7440-38-2. Integrated Risk Information System Chemical Summary. Washington (DC): National Center for Environmental Assessment. [accessed 2018 Oct 7]. https://cfpub.epa.gov/ncea/iris2/chemicalLanding.cfm?substance_nmbr=278.
- U.S. Environmental Protection Agency [USEPA]. 2001. National primary drinking water regulations: arsenic and clarifications to compliance and new source contaminants monitoring. Final Rule. 40 CFR Parts 141 and 142. Fed Reg. 66:6976–7066.
- U.S. Environmental Protection Agency [USEPA]. 2005a. Guidelines for carcinogen risk assessment. EPA/630/P-03/001B. Washington (DC): Risk Assessment Forum, USEPA.
- U.S. Environmental Protection Agency [USEPA]. 2005b. Toxicological review of ingested inorganic arsenic (CAS No. 7440-38-2). In support of summary information on the Integrated Risk Information System (IRIS). July. IRIS review draft for the Science Advisory Board. Washington (DC): USEPA.
- U.S. Environmental Protection Agency [USEPA]. 2006. Revised reregistration eligibility decision for MSMA, DSMA, CAMA, and Cacodylic Acid.
- U.S. Environmental Protection Agency [USEPA]. 2007. Advisory on EPA’s Assessments of Carcinogenic Effects of Organic and Inorganic Arsenic: a report of the US EPA Science Advisory Board. (EPA-SAB-07-008) http://yosemite.epa.gov/sab/sabproduct.nsf/EADABBF40DED2A0885257308006741EF/$File/sab-07-008.pdf.
- U.S. Environmental Protection Agency [USEPA]. 2008. Probabilistic risk assessment for children who contact CCA-treated playsets and decks. Prepared by J. Chen, N. Mottl, T. Lindheimer, N. Cook. Office of Pesticide Programs, Antimicrobials Division. Washington (DC): USEPA.
- U.S. Environmental Protection Agency [USEPA]. 2010. Toxicological review of inorganic arsenic (CAS No. 7440-38-2). In support of summary information on the Integrated Risk Information System (IRIS). IRIS review draft for the Science Advisory Board. Washington (DC): USEPA. http://nepis.epa.govatend.
- U.S. Environmental Protection Agency [USEPA]. 2018. Regional Screening levels (RSLs)—Users Guide. U.S. Environmental Protection Agency. https://www.epa.gov/risk/regional-screening-levels-rsls-users-guide.
- U.S. Food and Drug Administration [FDA]. 2001. Guidance for industry statistical aspects of the design, analysis, and interpretation of chronic rodent carcinogenicity studies of pharmaceuticals. Bethesda, MD.
- U.S. Food and Drug Administration [FDA]. 2013a Analytical results for arsenic in rice. Bethesda, MD.
- U.S. Food and Drug Administration [FDA]. 2013b. Arsenic in juice. Bethesda, MD.
- U.S. Food and Drug Administration [FDA]. 2016. Risk assessment of arsenic in rice. Bethesda, MD.
- U.S. Public Health Service [USPHS]. 1943. Public Health Service drinking water standards. Public Health Rep. 58:69–111.
- Valenzuela OL, Borja-Aburto VH, Garcia-Vargas GG, Cruz-Gonzalez MB, Garcia-Montalvo EA, Calderon-Aranda ES, Del Razo LM. 2005. Urinary trivalent methylated arsenic species in a population chronically exposed to inorganic arsenic. Environ Health Perspect. 113:250–254.
- Vega L, Styblo M, Patterson R, Cullen W, Wang C, Germolec D. 2001. Differential effects of trivalent and pentavalent arsenicals on cell proliferation and cytokine secretion in normal human epidermal keratinocytes. Toxicol Appl Pharmacol. 172:225–232.
- Vogt B, Rossman T. 2001. Effects of arsenite on p53, p21 and cyclin D expression in normal human fibroblasts - a possible mechanism for arsenite's comutagenicity. Mutat Res. 478:159–168.
- Waalkes MP, Keefer LK, Diwan BA. 2000. Induction of proliferative lesions of the uterus, testes, and liver in swiss mice given repeated injections of sodium arsenate: Possible estrogenic mode of action. Toxicol Appl Pharmacol. 166:24–35.
- Waalkes MP, Liu J, Germolec DR, Trempus CS, Cannon RE, Tokar EJ, Tennant RW, Ward JM, Diwan BA. 2008. Arsenic exposure in utero exacerbates skin cancer response in adulthood with contemporaneous distortion of tumor stem cell dynamics. Cancer Res. 68:8278–8285.
- Waalkes MP, Liu J, Ward JM, Diwan BA. 2006. Enhanced urinary bladder and liver carcinogenesis in male CD1 mice exposed to transplacental inorganic arsenic and postnatal diethylstilbestrol or tamoxifen. Toxicol Appl Pharmacol. 215:295–305.
- Waalkes MP, Liu J, Ward JM, Powell DA, Diwan BA. 2006. Urogenital carcinogenesis in female CD1 mice induced by in utero arsenic exposure is exacerbated by postnatal diethylstilbestrol treatment. Cancer Res. 66:1337–1345.
- Waalkes MP, Qu W, Tokar EJ, Kissling GE, Dixon D. 2014. Lung tumors in mice induced by whole-life inorganic arsenic exposure at human-relevant doses. Arch Toxicol. 88:1–11.
- Waalkes MP, Ward JM, Diwan BA. 2004. Induction of tumors of the liver, lung, ovary and adrenal in adult mice after brief maternal gestational exposure to inorganic arsenic: promotional effects of postnatal phorbol ester exposure on hepatic and pulmonary, but not dermal cancers. Carcinogenesis. 25:133–141.
- Waalkes MP, Ward JM, Liu J, Diwan BA. 2003. Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol Appl Pharmacol. 186:7–17.
- Wang YX, Feng W, Zeng Q, Sun Y, Wang P, You L, Yang P, Huang Z, Yu SL, Lu WQ. 2016. Variability of metal levels in spot, first morning, and 24-hour urine samples over a 3-month period in healthy adult Chinese men. Environ Health Perspect. 124:468–476.
- Wang A, Kligerman AD, Holladay SD, Wolf DC, Robertson JL. 2009. Arsenate and dimethylarsinic acid in drinking water did not affect DNA damage repair in urinary bladder transitional cells or micronuclei in bone marrow. Environ Mol Mutagen. 50:760–770.
- Wang X, Mandal AK, Saito H, Pulliam JF, Lee EY, Ke ZJ, Lu J, Ding S, Li L, Shelton BJ, et al. 2012. Arsenic and chromium in drinking water promote tumorigenesis in a mouse colitis-associated colorectal cancer model and the potential mechanism is ROS-mediated Wnt/beta-catenin signaling pathway. Toxicol Appl Pharmacol. 262:11–21.
- Wang X, Proud C. 1997. p70 S6 kinase is activated by sodium arsenite in adult rat cardiomyocytes: Roles for phosphatidylinositol 3-kinase and p38 MAP kinase. Biochem Biophys Res Commun. 238:207–212.
- Wang YH, Yeh SD, Shen KH, Shen CH, Juang GD, Hsu LI, Chiou HY, Chen CJ. 2009. A significantly joint effect between arsenic and occupational exposures and risk genotypes/diplotypes of CYP2E1, GSTO1 and GSTO2 on risk of urothelial carcinoma. Toxicol Appl Pharmacol. 241:111–118.
- Wanibuchi H, Yamamoto S, Chen H, Yoshida K, Endo G, Hori T, Fukushima S. 1996. Promoting effects of dimethylarsinic acid on N-butyl-N-(4-hydroxybutyl)nitrosamine-induced urinary bladder carcinogenesis in rats. Carcinogenesis. 17:2435–2439.
- Wedel WR, Muirhead DE, Arnold LL, Dodmane PR, Lele SM, Maness-Harris L, Hoyt R, Cohen SM. 2013. Urothelial cell intracytoplasmic inclusions after treatment of promyelocytic leukemia with arsenic trioxide. Toxicol Sci. 134:271–275.
- Wei M, Arnold L, Cano M, Cohen SM. 2005. Effects of co-administration of antioxidants and arsenicals on the rat urinary bladder epithelium. Toxicol Sci. 83:237–245.
- Wei M, Yamada T, Yamano S, Kato M, Kakehashi A, Fujioka M, Tago Y, Kitano M, Wanibuchi H. 2013. Diphenylarsinic acid, a chemical warfare-related neurotoxicant, promotes liver carcinogenesis via activation of aryl hydrocarbon receptor signaling and consequent induction of oxidative DNA damage in rats. Toxicol Appl Pharmacol. 273:1–9.
- Wei M, Wanibuchi H, Yamamoto S, Li W, Fukushima S. 1999. Urinary bladder carcinogenicity of dimethylarsinic acid in male F344 rats. Carcinogenesis. 20:1873–1876.
- Wijeweera J, Gandolfi A, Parrish A, Lantz R. 2001. Sodium arsenite enhances AP-1 and NFjB DNA binding and induces stress protein expression in precision-cut rat lung slices. Toxicol Sci. 61:283–294.
- Williams JD, Jacobson EL, Kim H, Kim M, Jacobson MK. 2012. Folate in skin cancer prevention. Subcell Biochem. 56:181–197.
- Wong G, Staplin N, Emberson J, Baigent C, Turner R, Chalmers J, Zoungas S, Pollock C, Cooper B, Harris D. 2016. Chronic kidney disease and the risk of cancer: an individual patient data meta-analysis of 32,057 participants from six prospective studies. BMC Cancer 16:488.
- World Health Organization[WHO]. 2018. Arsenic fact sheet. Geneva: World Health Organization. [accessed Oct 6]. http://www.who.int/news-room/fact-sheets/detail/arsenic.
- Xu Y, Wang Y, Zheng Q, Li B, Li X, Jin Y, Lv X, Qu G, Sun G. 2008. Clinical manifestations and arsenic methylation after a rare subacute arsenic poisoning accident. Toxicol Sci. 103:278–284.
- Xue J, Zartarian V, Wang SW, Liu SV, Georgopoulos P. 2010. Probabilistic modeling of dietary arsenic exposure and dose and evaluation with 2003-2004 NHANES data. Environ Health Perspect. 118:345–350.
- Yager JW, Erdei E, Myers O, Siegel M, Berwick M. 2016. Arsenic and ultraviolet radiation exposure: melanoma in a New Mexico non-Hispanic white population. Environ Geochem Health. 38:897–910.
- Yager JW, Gentry PR, Thomas RS, Pluta L, Efremenko A, Black M, Arnold LL, McKim JM, Wilga P, Gill G, et al. 2013. Evaluation of gene expression changes in human primary uroepithelial cells following 24-hr exposures to inorganic arsenic and its methylated metabolites. Environ Mol Mutagen. 54:82–98.
- Yamada T, Kondo M, Miyata K, Ogata K, Kushida M, Sumida K, Kawamura S, Osimitz TG, Lake BG, Cohen SM. 2017. An evaluation of the human relevance of the lung tumors observed in female mice treated with permethrin based on mode of action. Toxicol Sci. 157:465–486.
- Yamamoto A, Hisanaga A, Ishinishi N. 1987. Tumorigenicity of inorganic arsenic compounds following intratracheal instillations to the lungs of hamsters. Int J Cancer. 40:220–223.
- Yamamoto S, Konishi Y, Matsuda T, Murai T, Shibata MA, Matsui-Yuasa I, Otani S, Kuroda S, Endo G, Fukushima S. 1995. Cancer induction by an organic arsenic compound, dimethylarsinic acid (cacodylic acid), in F344/DuCrj rats after pretreatment with five carcinogens. Cancer Res. 55:1271–1276.
- Yamanaka K, Ohtsubo K, Hasegawa A, Hayashi H, Ohji H, Kanisawa M, Okada S. 1996. Exposure to dimethylarsinic acid, a main metabolite of inorganic arsenics, strongly promotes tumorigenesis initiated by 4-nitroquinoline 1-oxide in the lungs of mice. Carcinogenesis. 17:767–770.
- Yamanaka K, Mizol M, Kato K, Hasegawa A, Nakano M, Okada S. 2001. Oral administration of dimethylarsinic acid, a main metabolite of inorganic arsenic, in mice promotes skin tumorigenesis initiated by dimethylbenz(a)anthracene with or without ultraviolet B as a promoter. Biol Pharm Bull. 24:510–514.
- Yih L, Lee T. 2000. Arsenite induces p53 accumulation through an ATM-dependent pathway in human fibroblasts. Cancer Res. 60:6346–6352.
- Yokohira M, Arnold LL, Pennington KL, Suzuki S, Kakiuchi-Kiyota S, Herbin-Davis K, Thomas DJ, Cohen SM. 2011. Effect of sodium arsenite dose administered in the drinking water on the urinary bladder epithelium of female arsenic (+3 oxidation state) methyltransferase knockout mice. Toxicol Sci. 121:257–266.
- Zhang R, Zhang X, Wu K, Wu H, Sun Q, Hu FB, Han J, Willett WC, Giovannucci EL. 2016. Rice consumption and cancer incidence in US men and women. Int J Cancer. 138:555–564.
- Zhang Y-F, Zhou L, Zhang H-W, Hou A-J, Gao H-F, Zhou Y-H. 2014. Association between folate intake and the risk of lung cancer: a dose-response meta-analysis of prospective studies. PLoS ONE. 9:e93465.
- Zhang Q, Bhattacharya S, Conolly RB, Clewell HJ, Kaminski NE, Andersen ME. 2014. Molecular signaling network motifs provide a mechanistic basis for cellular threshold responses. Environ Health Perspect. 122:1261–1270.
- Zhou X, Sun X, Cooper KL, Wang F, Liu KJ, Hudson LG. 2011. Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J Biol Chem. 286:22855–22863.
- Zhou Q, Xi S. 2018. A review on arsenic carcinogenesis: epidemiology, metabolism, genotoxicity and epigenetic changes. Regul Toxicol Pharmacol. 99:78–88.