
Abstract
TP53 encodes a transcription factor that is centrally-involved in several pathways, including the control of metabolism, the stress response, DNA repair, cell cycle arrest, senescence, programmed cell death, and others. Since the discovery of TP53 as the most frequently-mutated tumor suppressor gene in cancer over four decades ago, the field has focused on uncovering target genes of this transcription factor that are essential for tumor suppression. This search has been fraught with red herrings, however. Dozens of p53 target genes were discovered that had logical roles in tumor suppression, but subsequent data showed that most were not tumor suppressive, and were dispensable for p53-mediated tumor suppression. In this review, we focus on p53 transcriptional targets in two categories: (1) canonical targets like CDKN1A (p21) and BBC3 (PUMA), which clearly play critical roles in p53-mediated cell cycle arrest/senescence and cell death, but which are not mutated in cancer, and for which knockout mice fail to develop spontaneous tumors; and (2) a smaller category of recently-described p53 target genes that are mutated in human cancer, and which appear to be critical for tumor suppression by p53. Interestingly, many of these genes encode proteins that control broad cellular pathways, like splicing and protein degradation, and several of them encode proteins that feed back to regulate p53. These include ZMAT3, GLS2, PADI4, ZBXW7, RFX7, and BTG2. The findings from these studies provide a more complex, but exciting, potential framework for understanding the role of p53 in tumor suppression.
Introduction
The tumor suppressor gene TP53 encodes a transcription factor that plays a pivotal role in preventing cancer formation in multicellular organisms. p53 is well understood to serve as a master transcriptional regulator of genotoxic stress, DNA damage, senescence, cell cycle arrest, and cell death. In addition, this protein is now associated with the control of several more cellular pathways, including metabolism, immune function, and splicing (Levine and Oren Citation2009). Upwards of 3000 transcriptional targets have been identified for p53, and yet there is limited understanding as to the roles of these target genes in tumor suppression. While some targets are robustly and ubiquitously expressed upon p53 activation and play essential roles in senescence and programmed cell death, many of these targets are not bona fide tumor suppressors; that is, they do not suppress tumor development when overexpressed, they are not mutated in sporadic tumors, and/or studies in knockout mice for these genes indicates that they are dispensable for tumor suppression by p53. Other transcriptional targets of p53 are transactivated by this protein in more tissue-specific or context-specific manner, and some of these possess intrinsic tumor suppressor activity. For the purposes of this review, we focus on p53 transcriptional targets that are known tumor suppressor genes (that is, are mutated in human tumors) and whose protein products suppress tumor progression in mouse models, and which thus qualify as bona fide tumor suppressor genes.
Functional domains of p53
p53 is a zinc-coordinated transcription factor that binds to DNA in a sequence-specific manner and controls the transcriptional regulation of up to 3000 target genes. The structure of p53 is composed of an intrinsically disordered N terminal region that includes two transactivation domains, followed by a proline-rich domain, a central DNA binding domain, an oligomerization domain, and an intrinsically disordered C terminal domain (). Within the C terminus of p53 lies a nuclear localization signal and a nuclear export signal. Notably, each domain of p53 has been shown to be critical for transcriptional activity and tumor suppressor function. The N terminal domain of p53 facilitates the interaction of p53 with various transcriptional co-activators and regulatory proteins, thus modulating its transcriptional activity (Vousden and Lu Citation2002). Within the N terminus are two transactivation domains of p53. The first transactivation domain (TAD1), spanning approximately amino acids 20–40, is crucial for the transcriptional activity of p53. TAD1 interacts with several transcriptional regulators, including the general transcription machinery and co-activators like p300/CBP (Laptenko and Prives Citation2006). The second transactivation domain (TAD2) covers approximately amino acids 40–60 and supports the function of TAD1. While TAD2 plays a more subsidiary role compared to TAD1, it contributes to the overall stability and efficacy of transcriptional activation by p53 (Vousden and Lu Citation2002; Laptenko and Prives Citation2006; Levine and Oren Citation2009).
The Proline-Rich Domain (PRD) of p53, located approximately between amino acids 60–100, contains an abundance of proline residues, likely forming a rigid helix upon which the transactivation domains rest. The PRD is thought to interact with SH3 domain-containing proteins, although the exact mechanism and significance of these interactions are not fully understood (Walker and Levine Citation1996). The central core of p53 comprises the DNA-binding domain (DBD), which spans approximately amino acids 100–300. This domain is responsible for sequence-specific DNA binding. p53 recognizes a response element that consists of two half site palindromes with the consensus PuPuPuC(A/T)(T/A)GPyPyPy (Sullivan et al. Citation2018). The DBD is the most conserved region across species and is the site of the majority of mutations in human cancers, thus highlighting the importance of transactivation to p53-mediated tumor suppression (Ho et al. Citation2006).
The tetramerization domain, located approximately between amino acids 325–355, is essential for the oligomerization of p53. This domain facilitates the formation of a homo-tetramer, the functional form of p53 necessary for its DNA-binding and transcriptional activities. The tetramerization domain is crucial for the cooperative binding of p53 to DNA and significantly enhances p53’s sequence specificity and affinity for DNA (Jeffrey et al. Citation1995; Natan et al. Citation2011). The remainder of the C terminus of p53 spans amino acid 356 to 393. This C terminal region possesses strong nonspecific DNA binding activity, allowing p53 to scan the genome for sites of DNA damage, such as double strand breaks (Laptenko et al. Citation2015). Finally, the activity of p53 is finely tuned through various post-translational modifications (for example, phosphorylation, acetylation, methylation, and others) and interactions with other cellular proteins, which determine the stability, localization, and ability of p53 to activate transcription. These post-translational modifications are the topic of excellent reviews (Meek and Anderson Citation2009; Nguyen et al. Citation2014).
The importance of transactivation for tumor suppression by p53
There is considerable evidence that the transactivation function of p53 is critical for tumor suppression, including data that tumor-derived mutants lack this activity (for review, Pfister and Prives Citation2017). Perhaps the most compelling data come from the Attardi group, who generated a series of knock-in mice with mutations in the p53 transcriptional activation domains (TADs). The mutations were either in TAD1 (p5325,26), TAD2 (p5353,54), or both TADs (p5325,26,53,54). The TAD2 mutant was minimally compromised for p53 target gene expression, suggesting that TAD1 is the critical mediator of transcriptional activity. They found that the TAD1 mutant is severely compromised for p53 target gene transactivation, and this mutant is unable to induce growth arrest or apoptosis in response to DNA damaging agents. However, the p5325,26 mutant retained the ability to upregulate some p53 target genes, including those involved in senescence, and this mutant possesses tumor suppressor function in some mouse models. But when both transactivation domains were mutated, p53 was transcriptionally dead and mice with the p5325,26,53,54 mutant develop tumors to similar levels as p53 null mice (Brady et al. Citation2011). Surprisingly however, many of the “canonical” p53 targets involved in growth arrest and apoptosis do not appear to be required for tumor suppression.
CDKN1A (p21), growth arrest, and senescence
One of the first p53 target genes identified was CDKN1A, encoding the cyclin-dependent kinase inhibitor p21 (el-Deiry et al. Citation1993). This protein is essential for the ability to induce growth arrest and senescence in cells (Harper et al. Citation1993). Although p53 has been shown to transcriptionally activate other genes involved in G1 arrest such as 14-3-3σ and Killin, p21 remains the dominant p53 target gene that regulates the cell cycle pathway (Luo et al. Citation1995; Chen Citation2016). Mechanistically, p21 serves as a master regulator of the G1 checkpoint by binding to cyclin E/CDK2 and cyclin D/CDK4 to inhibit the cell cycle (Luo et al. Citation1995; Chen Citation2016). The inhibition of CDKs cooperates with the tumor suppressor protein Rb, and shifts Rb binding to E2F1 to induce transcriptional repression of cell cycle genes (). This p21-CDK-Rb axis is clearly a critical tumor suppressive checkpoint in cancer, as exemplified by its near universal mutational abrogation in tumors, and by the success of CDK inhibitors in human cancer (Rubin et al. Citation2020). p21 is also essential for the ability of p53 to repress gene transcription, mediated by the DREAM complex. The DREAM complex has been shown to directly bind to multiple cell cycle genes that are downregulated in a p53-dependent manner (Fischer Citation2017; Engeland Citation2018). At present, the contribution of the DREAM complex, and of p53-mediated transcriptional repression (either direct or indirect) to tumor suppression by p53 is not known. Knockout of CDKN1A in mice causes loss of most, but not all, mechanisms of G1 arrest, as well as the loss of the majority of p53-dependent gene repression (Brugarolas et al. Citation1995; Fischer Citation2016). Although p21 plays a critical role in G1 arrest, senescence, and transcriptional repression by the DREAM complex, CDKN1A is infrequently mutated in cancer, and p21 knockout mice develop normally and are not cancer-prone (Deng et al. Citation1995; Martín-Caballero et al. Citation2001; El-Deiry Citation2016).
Figure 1. Gene and domain organization of p53. TP53 gene and corresponding full-length protein (corresponds to human p53 major isoform α) is comprised of several functional domains. TAD1, transactivation domain 1; TAD2, transactivation domain 2; PRD, proline rich domain; DBD, DNA binding domain; NLS, nuclear localization signal; TETD, tetramerization domain; NES, nuclear export signal.
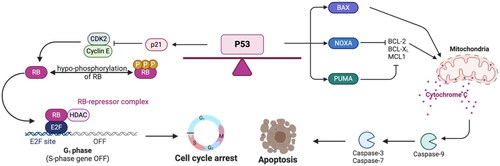
Figure 2. Canonical p53 stress response: cell cycle arrest and apoptosis. Growth arrest and apoptosis are well-understood p53 pathways in response to stress. Cell cycle arrest is mediated by transcriptional upregulation of p21, a cyclin-dependent kinase inhibitor that binds and inhibits CDK2. This causes decreased phosphorylation of the tumor suppressor RB, and RB is able to form the RB-E2F repressor complex to inhibit the upregulation of cell cycle genes. In order to induce apoptosis, p53 transcriptionally activates genes that include BAX, NOXA, and PUMA. BAX oligomerizes at the mitochondria to induce cytochrome c release to trigger the activation of caspase-9, which cleaves and activates caspase-3 and -7 to induce apoptosis. NOXA and PUMA inhibit the anti-apoptotic proteins BCL-2, BCL-XL, and MCL1, and this inhibition promotes BAK and BAX oligomerization at the mitochondria.
PMAIP1 (NOXA), BBC3 (PUMA) and BAX: p53 target genes involved in programmed cell death
Programmed cell death, or apoptosis, is a highly regulated process essential for maintaining cellular homeostasis and eliminating damaged or potentially cancerous cells. Apoptosis is critical for the protection against DNA damage to avoid the accumulation of mutations, many of which can be oncogenic. Apoptosis can be separated into two primary pathways: the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway. Both pathways converge on the activation of caspases, the family of proteases that are the central executioners of apoptosis, and both pathways are regulated by p53.
In the intrinsic pathway, the pro-apoptotic Bcl2 family proteins BAK and BAX oligomerize at the mitochondrial membrane to generate mitochondrial outer membrane permeabilization. This results in the release of cytochrome c into the cytosol. Cytochrome c interacts with apoptotic protease activating factor-1 (Apaf-1) and pro-caspase-9 to form the apoptosome, a complex that activates caspase-9. Caspase-9 then cleaves and activates the executioner caspases (such as caspase-3), leading to cell death. Upon DNA damage, radiation, or other genotoxic stress, p53 upregulates a host of Bcl2 family members to induce apoptosis through this pathway. These members include BAX, PMAIP1 (NOXA), BBC3 (PUMA), and BID (BH3 interacting domain dead agonist). BAX was one of the first pro-apoptotic genes identified with a p53 response element in its promoter (Miyashita et al. Citation1994; Miyashita and Reed Citation1995). BAX forms a homodimer at the mitochondrial membrane to promote cytochrome c release in response to genotoxic stress (McCurrach et al. Citation1997). NOXA and PUMA are additional Blc2 family members that are direct targets of p53. NOXA and PUMA tend to work in independent systems, although PUMA requires the presence of BAX to induce apoptosis () (Oda et al. Citation2000; Nakano and Vousden Citation2001; Shibue et al. Citation2003). Finally, Apaf-1 is also directly regulated by p53 and involved in the apoptotic response (Robles et al. Citation2001).
p53 also regulates several genes involved in the extrinsic signaling cascade of apoptosis. This pathway is mediated through death receptors such as the tumor necrosis factor receptor, or TNFR, family, which activate Caspase 8 and Caspase 3 to induce apoptosis. p53 transcriptionally activates a host of these receptors, including Fas and KILLER/DR5 (death receptor 5) (Müller et al. Citation1998; Wu et al. Citation1999; Attardi et al. Citation2000). BID intersects the extrinsic pathway with the intrinsic pathway. BID, another transcriptional target of p53, is cleaved by Caspase 8 to generate its active form, and then translocates to the mitochondria to oligomerize BAX and BAK (Sax et al. Citation2002). Despite the obvious importance of p53 to the induction of apoptosis, it is apparent from several studies that apoptosis does not fully explain p53-mediated tumor suppression. For example, these apoptotic genes are not inactivated by mutation in cancers (Aubrey et al. Citation2018). Like CDKN1A, knockout mice for BAX, PUMA and NOXA are not tumor-prone (Knudson et al. Citation1995; Jeffers et al. Citation2003; Kiryu-Seo et al. Citation2005). Even triple knockout mice (PUMA -/-, NOXA -/-, Cdkn1a -/-), in which cells are defective for p53-mediated apoptosis and growth arrest, do not show increased spontaneous tumor formation compared to wild type p53 mice (Valente et al. Citation2013). Whereas these p53 target genes are clearly important for p53 function, these combined studies pointed to other pathways required for tumor suppression by p53.
Key target genes for p53-mediated tumor suppression
FBXW7 and protein degradation
In 2004 the Balmain group conducted a mammalian genetic screen for p53-regulated tumor suppressor genes in a radiation-induced mouse model of lymphoma, and identified Fbxw7 as a p53-induced gene that functioned as a haplo-insufficient tumor suppressor (Mao et al. Citation2004). FBXW7 protein functions as the substrate recognition component of an SCF E3 ubiquitin ligase, and it controls the stability of a number of key oncogenic proteins including C-MYC, cyclin E, MCL1, mTOR, c-JUN, PLK1, NOTCH and AURKA; regulation of the latter is believed to control genomic stability (Yeh et al. Citation2018; Galindo-Moreno et al. Citation2020). One analysis indicated that Fbxw7 mutations exist in a minimum of 2.5% of all cancers, suggesting that this is a broadly influential tumor suppressor (Yeh et al. Citation2018). Interestingly, three groups recently showed that endogenous FBXW7 interacts with p53, and targets p53 for polyubiquitylation and proteasomal degradation (Galindo-Moreno et al. Citation2019; Tripathi et al. Citation2019; Cui et al. Citation2020); this finding of feedback regulation holds true for the p53 target gene PADI4 as well.
PADI4 and citrullination
The Murphy group focuses on African-centric germline hypomorphic variants of TP53. Their analysis of the Y107H germline variant of p53 revealed that the p53 target gene PADI4 is one of only three dozen p53 target genes with impaired transactivation in cells containing this cancer-associated variant (Indeglia et al. Citation2023). Interestingly, transactivation of PADI4 by p53 was also impaired in multiple non-transformed lymphoblastoid cell lines containing other heterozygous p53 hypomorphic mutations (P47S, Y107H, R175C, Y220H), suggesting that PADI4 transactivation is exquisitely sensitive to p53 function. PADI4 possesses potent tumor suppressive activity in cultured cells and in multiple syngeneic mouse models, and this gene is subject to mutation in tumors (Tanikawa et al. Citation2009; Tanikawa et al. Citation2012; Indeglia et al. Citation2023). PADI4 mutations in cancer were shown to create enzymatically inactive versions of the enzyme, thus solidifying that this gene as a tumor suppressor (Tanikawa et al. Citation2012). PADI4 is a peptidyl arginine deiminase enzyme, which converts arginine to the non-natural amino acid citrulline, and this protein can citrullinate key proteins involved in anti-tumor pathways, including GSK3β and NPM1 (Tanikawa et al. Citation2009; Stadler et al. Citation2013). Moreover, PADI4 functions normally in immune function, where citrullination is associated with immune stimulation; along these lines, activating mutations in PADI4 are associated with autoimmune disorders such as rheumatoid arthritis (Suzuki et al. Citation2003). In a common theme where-in p53 regulated tumor suppressors often feedback and regulate this pathway, the Murphy group showed that PADI4 interacts with p53 and stimulates its ability to transactivate a subset of genes involved in immune recognition of cancer and the efficacy of immune checkpoint inhibitors (Indeglia et al. Citation2023).
PTEN, genomic stability, cell survival, and metabolism
The group of Tak Mak was the first to show that the PTEN tumor suppressor is a direct transcriptional target of p53 (Stambolic et al. Citation2001); the Levine group subsequently showed that the transactivation of PTEN by p53 occurs in tissue- and stress-specific manner (Feng et al. Citation2007). The PTEN gene is mutated in some hereditary cancer syndromes, and this gene is also one of the most frequently mutated genes in sporadic cancer (for review, see Lee and Chen Citation2018). Along these lines, PTEN heterozygous knock-out mice are predisposed to cancer (Di Cristofano et al. Citation1998). The best understood function of PTEN is as a lipid phosphatase that controls AKT activity, though this protein possesses protein phosphatase and scaffolding functions as well (Lee and Chen Citation2018). Interestingly, deletion of AKT is sufficient to suppress tumor development in PTEN mutant mice (Chen et al. Citation2006). Notably, and similar to the cases for PADI4 and FBXW7, PTEN feeds back to regulate p53 function; interestingly, PTEN can negatively regulates p53 function through phosphatase-dependent and independent manners (Freeman et al. Citation2003; Li et al. Citation2006).
GLS2 and ferroptosis
In 2011 the Gu group generated an artificial mutant p53 mouse model in which three lysine residues in the DNA binding domain of p53 that were normally acetylated were mutated to arginine (the 3KR mouse) (Li et al. Citation2012). The 3KR mouse was unable to induce apoptosis, senescence, or growth arrest upon p53 activation in the thymus, and in mouse embryonic fibroblasts. Surprisingly, 3KR mice did not develop spontaneous tumors. This group went on to show that the 3KR mutant of p53 could confer the sensitivity of cells to ferroptosis. Ferroptosis in an iron- and lipid-peroxide mediated form of cell death. The Gu group showed that p53-null cells were resistant to ferroptosis, while p53 wild-type and p533KR remained sensitive. Several direct p53 target genes play roles in ferroptosis: these include SAT1, ALOX15 and PLA2G6 (the role of p53 in ferroptosis has recently been expertly reviewed; Liu and Gu Citation2022). One p53 target gene that plays a role in ferroptosis and deserves particular mention is GLS2. GLS2 is a direct p53 target gene that is subject to mutation in a small percentage of sporadic cancers, and GLS2 suppresses tumor development when overexpressed (Hu et al. Citation2010; Suzuki et al. Citation2010; Liu et al. Citation2014). Additionally, GLS2 knockout mice develop hepatocellular carcinomas and lymphoma, thus solidifying the status of GLS2 as a tumor suppressor (Suzuki et al. Citation2022). Interestingly, GLS2 was one of the first genes identified as a key regulator of ferroptosis (Gao et al. Citation2015). Data solidifying a role for ferroptosis and GLS2 in tumor suppression by p53 were generated in a mouse model for the p53 hypomorph P47S. The P47S mouse is prone to spontaneous hepatocellular cancer, and is defective in the regulation of ferroptosis; notably, this variant of p53 is capable of transactivating the majority of p53 target genes, but is defective for the transactivation of GLS2 (Jennis et al. Citation2016).
ZMAT3 and RNA splicing
Dysregulation of alternative splicing can lead to the production of aberrant mRNA and protein isoforms that contribute to cancer hallmarks such as uncontrolled cell proliferation, invasion, and metastasis. As one example, aberrant splicing of the CD44 gene has been shown to promote tumor invasion and metastasis in various cancers (Bernard et al. Citation2022). Similarly, abnormal splicing of the BCL2L1 gene has been linked to chemotherapy resistance in some cancers (Wahid et al. Citation2023). Wig-1, or Zmat3, was first described as a gene induced by a temperature sensitive p53 mutant (Varmeh-Ziaie et al. Citation1997). Zmat3 is induced by DNA damage in a wild type p53-dependent manner (Harms et al. Citation2004), and overexpression of Zmat3 promotes apoptosis in multiple human tumor-derived cell lines (Israeli et al. Citation1997). Attardi and colleagues identified Zmat3 as the top gene induced by the transcriptionally deficient p5325,26 mutant, which retains tumor suppressor function (Bieging-Rolett et al. Citation2020). This group found that Zmat3 exhibits potent tumor suppressor activity in Kras mutant lung and liver cancers, using multiple mouse models. The authors identified Zmat3 as an RNA binding protein uniquely positioned at the 3′ splice site of key regulators of tumor suppression, including Mdm2 and Mdm4, which feedback and regulate p53. Surprisingly, they found Zmat3 was a master regulator of alternative splicing, and that it targeted transcripts involved in diverse cellular processes. Although the tumor suppressor function of Zmat3 appears to be context dependent, the presence of mutations in Zmat3 in tumors identifies it as a classical tumor suppressor (Janic et al. Citation2018).
RFX7, BTG2 and transcriptional regulation
RFX7 was identified as a p53 target gene by the Fischer group (Coronel et al. Citation2021). This gene is subject to inactivating mutations in lymphoid malignancies (Fischer et al. Citation2020), and knockout of this gene enhances lymphoma development in mouse models (Weber et al. Citation2019). RFX7 is a transcription factor, and the Fischer group identified four established tumor suppressor genes that are transcriptional targets of RFX7 (PDCD4, PIK3IP1, MXD4 and PNRC1), thus solidifying a role for this p53 target gene in tumor suppression. A second p53 target gene with a role in transcriptional regulation is BTG2. BTG2 is a p53 target gene that shows anti-proliferative function by arresting cells at the G1/S phase of the cell cycle (Rouault et al. Citation1996). Like RFX7, BTG2 is subject to mutational inactivation in lymphoid cancers. Additionally, knockout of BTG2 enhances medulloblastoma in heterozygous Patched mutant mice (Farioli-Vecchioli et al. Citation2012). A summary of the key p53 target genes with roles in tumor suppression are highlighted in .
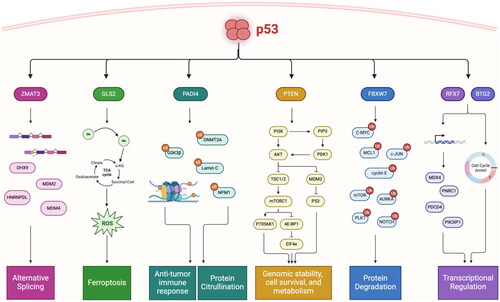
Figure 3. Chain of command: p53 transcriptionally activates “effectors” of tumor suppression that regulate distinct pathways. Critical target genes of p53 are mutated and inactivated in human cancers, have intrinsic tumor suppressive activity, and serve as master regulators of anti-tumor pathways. ZMAT3 regulates alternative splicing through binding to immature transcripts upstream of the 3’ splice site of critically important genes involved in tumor suppression and nonsense-mediated mRNA decay. GLS2 regulates ferroptosis by converting glutamine to glutamate to fuel the TCA cycle and generate reactive oxygen species (ROS) to trigger ferroptosis. PADI4 regulates immune activation through both transcriptional regulation and protein citrullination, and post translationally modifies key tumor suppressors to modulate their activity. PTEN collectively regulates genome stability, cell survival, and metabolism through the PI3K/AKT/mTOR pathway, as well as through inhibition of MDM2 to activate p53. FBXW7 is master regulator of protein degradation, and in particular controls the stability of key oncogenes in multiple cellular processes. RFX7 transcriptionally regulates downstream tumor suppressor genes, and BTG2 induces G1/S cell cycle arrest to protect against DNA-damage through its involvement in transcriptional regulation.
Concluding remarks
As the p53 field approaches 45 years of discovery, the importance of p53 transcriptional programs and target genes will be a major point of discussion moving forward. p53 has over 3000 transcriptional targets, and experimental findings suggest that the roles of these targets may be context and tissue-dependent. Indeed, recent work by Lozano’s group suggests that only 7 of these transcriptional targets are shared between multiple tissue groups, indicating that p53 plays a much larger tissue-specific role than previously appreciated (Pant et al. Citation2023). We propose that p53 is not just the “guardian of the genome”, but rather the “commander in chief” that follows a chain of command in response to stress or genotoxic injury. It follows that p53 communicates with the “generals” (ZMAT3, PADI4, GLS2, others) which in turn give orders to their battalion (alternative splicing, immune activation, ferroptosis) (). With this model, and further studies into the molecular functions of understudied p53 target genes, we can begin to uncover novel networks of tumor suppression that can be leveraged for putative therapies.
With many recent developments and the quest to develop novel therapeutic strategies for mutant p53 cancers, further studies are warranted to understand the role of p53 transcriptional targets in cell death, DNA repair, and immune activation. Owing to the complexity of the p53 pathway, some outstanding questions are proposed below:
To what extent is p53 transcriptional activity contributing to tumor suppression? Does the direct, mitochondrial pathway of apoptosis contribute to tumor suppression?
Since the discovery of p53, evidence has indicated that transcriptional activity is the prominent driver of p53-mediated tumor suppression (Kern et al. Citation1991). But even early on it was clear that the entire story is much more complicated. Early work by Karin’s group showed that p53 could induce apoptosis in the absence of transactivation of canonical p53 targets (Caelles et al. Citation1994). It was later found that p53 could induce transcription-independent cell death through direct interaction with BAX, BCL2 or BAK, leading to mitochondrial membrane permeabilization and subsequent release of cytochrome c from the mitochondrial intermembrane space (Schuler et al. Citation2000; Schuler et al. Citation2003; Mihara et al. Citation2003; Leu et al. Citation2004). Moreover, a version of p53 with a mitochondrial leader peptide was potently tumor suppressive in mouse models (Palacios et al. Citation2008). These findings supported a direct, transcription-independent role for p53 in cell death; notably, the direct mitochondrial pathway may explain why the A347D Li Fraumeni mutant of p53, which is transcriptionally dead but retains tumor suppressor function, is tumor suppressive (Choe et al. Citation2023).
How does p53 transactivate different target genes in different tissues, or under different stress conditions?
We are just beginning to understand the tissue-specific and context-specific transcriptional targets of p53. Recent work by Tanikawa and colleagues surveyed the p53 transcriptional network in vivo, using WT p53 mice and p53 -/- mice treated with radiation to induce the p53 response. Analysis of 24 tissues showed distinct transcriptional outputs with very little overlap of direct p53 targets (Tanikawa et al. Citation2017). Similarly, a recent study by the Lozano group used an MDM2 knockout mouse to probe the p53 response. Notably, overlay of induced gene expression in the pancreas, small intestine, ovary, kidney, and heart transcriptomes revealed only 7 p53 transcriptional targets that were common between all tissues: Ccng1, Mdm2, Eda2r, Psrc1, Gtse1, Polk, and Zfp365 (Moyer et al. Citation2020). Work by Isbel and colleagues showed that histone modifying factors can contextualize the p53 transcriptional landscape. This group used embryonic stem cells to show p53 is localized to open and closed chromatin in untreated cells, but can open chromatin and activate transcription only upon recruitment of the cofactor Trim24 (Isbel et al. Citation2023). There may also be specificity dictated by the p53 response element: for example the Lozano group showed dimerization mutants of p53 could transactivate genes containing half site response elements at promoter regions of PPARα target genes (Gencel-Augusto et al. Citation2023). It is interesting to note, that of the many tumor suppressive p53 target genes described above, at least two (PTEN and PADI4) are also tissue specific with regard to their transactivation by p53, suggesting that tumor suppression by p53 may have a tissue-specific regulatory component.
Can we leverage p53 target genes as drug targets to induce tumor suppression?
Compounds that target mutant p53, potentially to refold this protein into wild type conformation and activity, have long been sought, but many attempts to re-activate p53 through small molecule interaction have proven unsuccessful, and many of these compounds appear to kill cells in a manner independent of p53 (Hassin and Oren Citation2023). As such, recent efforts have turned to drugging p53 targets that either inhibit p53, bind to mutant p53, or act downstream of p53 to carry out tumor suppression. Perhaps the most well-known example is MDM2, where inhibitors of MDM2 began with the invention of nutlin-3a to bind covalently at the p53 binding pocket of MDM2 (Vassilev et al. Citation2004). However, nutlin-3a was unsuccessful in clinical trials due to hematological toxicity and the failure to induce apoptosis in most cancer types. More recent results for targeting MDM2 have been promising, in particular with the invention of proteolysis targeting chimeras (PROTACs) against MDM2. The Eischen laboratory showed that targeted degradation of MDM2 could significantly reduce the tumor burden in triple negative breast cancer, with no apparent toxicity (Adams et al. Citation2023). Further studies may benefit from shifting the focus from re-activating p53 directly to targeting downstream effectors of p53 function.
While canonical targets of p53, such as p21 and MDM2, are often upregulated in any p53-dependent response, many other targets appear to be context specific. The mechanisms underlying target gene specificity are still not well understood, and more studies are warranted. Unraveling the p53 transcriptional network will undoubtedly pave the way for novel therapies and synergy with existing therapies, such as immunotherapy. We have only just begun to identify non-canonical p53 targets that will streamline this process, with the goal of reactivating the p53 pathway in mutant tumors to reduce the burden of human cancer.
Final considerations
This review is unique is that it gathers information about less well-known p53 targets and highlights their roles in tumor suppression. Common themes emerge from this study: the first is the number of these targets that then feedback to regulate p53. Of these PADI4 stands out because it positively regulates p53, while the other proteins predominantly feedback to negatively regulate this protein. Additionally, a number of these key targets are regulated by p53 in tissue- or stress-specific manner (for example, PTEN and PADI4), and the significance of this regulation to tumor suppression remains unclear. Finally, it should be noted that whereas p53 is known to transactivate tumor suppressors involved in DNA repair like MLH1, this gene does not suppress tumors when overexpressed, so it is not discussed here.
Authors’ contributions
Conception and design of the review: M.E.M. Generation of the figures: A.I. Drafting of manuscript: A.I, M.E.M. Proofing and revision of manuscript: A.I., M.E.M.
Acknowledgements
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to apologize for all the investigators whose references could not be included in this article due to space constraints.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Funding
References
- Adams CM, Mitra R, Xiao Y, Michener P, Palazzo J, Chao A, Gour J, Cassel J, Salvino JM, Eischen CM, et al. 2023. Targeted MDM2 degradation reveals a new vulnerability for p53-inactivated triple-negative breast cancer. Cancer Discov. 13(5):1210–1229. doi: 10.1158/2159-8290.CD-22-1131.
- Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, Jacks T. 2000. PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev. 14(6):704–718. doi: 10.1101/gad.14.6.704.
- Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. 2018. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 25(1):104–113. doi: 10.1038/cdd.2017.169.
- Bernard A, Boidot R, Végran F. 2022. Alternative splicing in cancer and immune cells. Cancers (Basel). 14(7):1726. doi: 10.3390/cancers14071726.
- Bieging-Rolett KT, Kaiser AM, Morgens DW, Boutelle AM, Seoane JA, Van Nostrand EL, Zhu C, Houlihan SL, Mello SS, Yee BA, et al. 2020. Zmat3 is a key splicing regulator in the p53 tumor suppression program. Mol Cell. 80(3):452–469.e9. doi: 10.1016/j.molcel.2020.10.022.
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. 2011. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 145(4):571–583. doi: 10.1016/j.cell.2011.03.035.
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. 1995. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 377(6549):552–557. doi: 10.1038/377552a0.
- Caelles C, Helmberg A, Karin M. 1994. p53-Dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature. 370(6486):220–223. doi: 10.1038/370220a0.
- Chen J. 2016. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 6(3):a026104. doi: 10.1101/cshperspect.a026104.
- Chen M-L, Xu P-Z, Peng X-d, Chen WS, Guzman G, Yang X, Di Cristofano A, Pandolfi PP, Hay N. 2006. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/- mice. Genes Dev. 20(12):1569–1574. doi: 10.1101/gad.1395006.
- Choe JH, Kawase T, Xu A, Guzman A, Obradovic AZ, Low-Calle AM, Alaghebandan B, Raghavan A, Long K, Hwang PM, et al. 2023. Li–Fraumeni syndrome–associated dimer-forming mutant p53 promotes transactivation-independent mitochondrial cell death. Cancer Discov. 13(5):1250–1273. doi: 10.1158/2159-8290.CD-22-0882.
- Coronel L, Riege K, Schwab K, Förste S, Häckes D, Semerau L, Bernhart SH, Siebert R, Hoffmann S, Fischer M, et al. 2021. Transcription factor RFX7 governs a tumor suppressor network in response to p53 and stress. Nucleic Acids Res. 49(13):7437–7456. doi: 10.1093/nar/gkab575.
- Cui D, Xiong X, Shu J, Dai X, Sun Y, Zhao Y. 2020. FBXW7 confers radiation survival by targeting p53 for degradation. Cell Rep. 30(2):497–509.e4. doi: 10.1016/j.celrep.2019.12.032.
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. 1995. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 82(4):675–684. doi: 10.1016/0092-8674(95)90039-x.
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. 1998. Pten is essential for embryonic development and tumour suppression. Nat Genet. 19(4):348–355. doi: 10.1038/1235.
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B, et al. 1993. WAF1, a potential mediator of p53 tumor suppression. Cell. 75(4):817–825. doi: 10.1016/0092-8674(93)90500-p.
- El-Deiry WS. 2016. p21(WAF1) mediates cell cycle inhibition, relevant to cancer suppression and therapy. Cancer Res. 76(18):5189–5191. doi: 10.1158/0008-5472.CAN-16-2055.
- Engeland K. 2018. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 25(1):114–132. doi: 10.1038/cdd.2017.172.
- Farioli-Vecchioli S, Cinà I, Ceccarelli M, Micheli L, Leonardi L, Ciotti MT, De Bardi M, Di Rocco C, Pallini R, Cavallaro S, et al. 2012. Tis21 knock-out enhances the frequency of medulloblastoma in Patched1 heterozygous mice by inhibiting the Cxcl3-dependent migration of cerebellar neurons. J Neurosci. 32(44):15547–15564. doi: 10.1523/JNEUROSCI.0412-12.2012.
- Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. 2007. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 67(7):3043–3053. doi: 10.1158/0008-5472.CAN-06-4149.
- Fischer BA, Chelbi ST, Guarda G. 2020. Regulatory factor X 7 and its potential link to lymphoid cancers. Trends Cancer. 6(1):6–9. doi: 10.1016/j.trecan.2019.11.001.
- Fischer M. 2016. p21 governs p53’s repressive side. Cell Cycle. 15(21):2852–2853. doi: 10.1080/15384101.2016.1205393.
- Fischer M. 2017. Census and evaluation of p53 target genes. Oncogene. 36(28):3943–3956. doi: 10.1038/onc.2016.502.
- Freeman DJ, Li AG, Wei G, Li H-H, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, et al. 2003. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 3(2):117–130. doi: 10.1016/s1535-6108(03)00021-7.
- Galindo-Moreno M, Giráldez S, Limón-Mortés MC, Belmonte-Fernández A, Sáez C, Japón MÁ, Tortolero M, Romero F. 2020. p53 and FBXW7: sometimes two guardians are worse than one. Cancers (Basel). 12(4):985. doi: 10.3390/cancers12040985.
- Galindo-Moreno M, Giráldez S, Limón-Mortés MC, Belmonte-Fernández A, Reed SI, Sáez C, Japón MÁ, Tortolero M, Romero F. 2019. SCF(FBXW7)-mediated degradation of p53 promotes cell recovery after UV-induced DNA damage. Faseb J. 33(10):11420–11430. doi: 10.1096/fj.201900885R.
- Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. 2015. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 59(2):298–308. doi: 10.1016/j.molcel.2015.06.011.
- Gencel-Augusto J, Su X, Qi Y, Whitley EM, Pant V, Xiong S, Shah V, Lin J, Perez E, Fiorotto ML, et al. 2023. Dimeric p53 mutant elicits unique tumor-suppressive activities through an altered metabolic program. Cancer Discov. 13(5):1230–1249. doi: 10.1158/2159-8290.CD-22-0872.
- Harms K, Nozell S, Chen X. 2004. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci. 61(7–8):822–842. doi: 10.1007/s00018-003-3304-4.
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 75(4):805–816.
- Hassin O, Oren M. 2023. Drugging p53 in cancer: one protein, many targets. Nat Rev Drug Discov. 22(2):127–144. doi: 10.1038/s41573-022-00571-8.
- Ho WC, Fitzgerald MX, Marmorstein R. 2006. Structure of the p53 core domain dimer bound to DNA. J Biol Chem. 281(29):20494–20502. doi: 10.1074/jbc.M603634200.
- Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. 2010. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 107(16):7455–7460. doi: 10.1073/pnas.1001006107.
- Indeglia A, Leung JC, Miller SA, Leu JI-J, Dougherty JF, Clarke NL, Kirven NA, Shao C, Ke L, Lovell S, et al. 2023. An African-specific variant of TP53 reveals PADI4 as a regulator of p53-mediated tumor suppression. Cancer Discov. 13(7):1696–1719. doi: 10.1158/2159-8290.CD-22-1315.
- Isbel L, Iskar M, Durdu S, Weiss J, Grand RS, Hietter-Pfeiffer E, Kozicka Z, Michael AK, Burger L, Thomä NH, et al. 2023. Readout of histone methylation by Trim24 locally restricts chromatin opening by p53. Nat Struct Mol Biol. 30(7):948–957. doi: 10.1038/s41594-023-01021-8.
- Israeli D, Tessler E, Haupt Y, Elkeles A, Wilder S, Amson R, Telerman A, Oren M. 1997. A novel p53-inducible gene, PAG608, encodes a nuclear zinc finger protein whose overexpression promotes apoptosis. Embo J. 16(14):4384–4392. doi: 10.1093/emboj/16.14.4384.
- Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, Yang H, Tai L, Vandenberg CJ, Kueh AJ, et al. 2018. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med. 24(7):947–953. doi: 10.1038/s41591-018-0043-5.
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 4(4):321–328. doi: 10.1016/s1535-6108(03)00244-7.
- Jeffrey PD, Gorina S, Pavletich NP. 1995. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science. 267(5203):1498–1502. doi: 10.1126/science.7878469.
- Jennis M, Kung C-P, Basu S, Budina-Kolomets A, Leu JI-J, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, et al. 2016. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 30(8):918–930. doi: 10.1101/gad.275891.115.
- Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. 1991. Identification of p53 as a sequence-specific DNA-binding protein. Science. 252(5013):1708–1711. doi: 10.1126/science.2047879.
- Kiryu-Seo S, Hirayama T, Kato R, Kiyama H. 2005. Noxa is a critical mediator of p53-dependent motor neuron death after nerve injury in adult mouse. J Neurosci. 25(6):1442–1447. doi: 10.1523/JNEUROSCI.4041-04.2005.
- Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. 1995. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 270(5233):96–99. doi: 10.1126/science.270.5233.96.
- Laptenko O, Prives C. 2006. Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 13(6):951–961. doi: 10.1038/sj.cdd.4401916.
- Laptenko O, Shiff I, Freed-Pastor W, Zupnick A, Mattia M, Freulich E, Shamir I, Kadouri N, Kahan T, Manfredi J, et al. 2015. The p53 C-terminus controls site-specific DNA binding and promotes structural changes within the central DNA binding domain. Mol Cell. 57(6):1034–1046. doi: 10.1016/j.molcel.2015.02.015.
- Lee YR, Chen M. 2018. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol. 19(9):547–562. doi: 10.1038/s41580-018-0015-0.
- Leu JIJ, Dumont P, Hafey M, Murphy ME, George DL. 2004. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 6(5):443–450. doi: 10.1038/ncb1123.
- Levine AJ, Oren M. 2009. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 9(10):749–758. doi: 10.1038/nrc2723.
- Li AG, Piluso LG, Cai X, Wei G, Sellers WR, Liu X. 2006. Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol Cell. 23(4):575–587. doi: 10.1016/j.molcel.2006.06.028.
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. 2012. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 149(6):1269–1283. doi: 10.1016/j.cell.2012.04.026.
- Liu J, Zhang C, Lin M, Zhu W, Liang Y, Hong X, Zhao Y, Young KH, Hu W, Feng Z, et al. 2014. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget. 5(9):2635–2647. doi: 10.18632/oncotarget.1862.
- Liu Y, Gu W. 2022. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 29(5):895–910. doi: 10.1038/s41418-022-00943-y.
- Luo Y, Hurwitz J, Massagué J. 1995. Cell-cycle inhibition by independent CDK and PCNA binding domains in p21Cip1. Nature. 375(6527):159–161. doi: 10.1038/375159a0.
- Mao J-H, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. 2004. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 432(7018):775–779. doi: 10.1038/nature03155.
- Martín-Caballero J, Flores JM, García-Palencia P, Serrano M. 2001. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 61(16):6234–6238.
- McCurrach ME, Connor TM, Knudson CM, Korsmeyer SJ, Lowe SW. 1997. BAX-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc Natl Acad Sci USA. 94(6):2345–2349. doi: 10.1073/pnas.94.6.2345.
- Meek DW, Anderson CW. 2009. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 1(6):a000950–a000950. doi: 10.1101/cshperspect.a000950.
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. 2003. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 11(3):577–590. doi: 10.1016/s1097-2765(03)00050-9.
- Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. 1994. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 9(6):1799–1805.
- Miyashita T, Reed JC. 1995. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 80(2):293–299. doi: 10.1016/0092-8674(95)90412-3.
- Moyer SM, Wasylishen AR, Qi Y, Fowlkes N, Su X, Lozano G. 2020. p53 drives a transcriptional program that elicits a non-cell-autonomous response and alters cell state in vivo. Proc Natl Acad Sci USA. 117(38):23663–23673. doi: 10.1073/pnas.2008474117.
- Müller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, et al. 1998. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 188(11):2033–2045. doi: 10.1084/jem.188.11.2033.
- Nakano K, Vousden KH. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 7(3):683–694. doi: 10.1016/s1097-2765(01)00214-3.
- Natan E, Baloglu C, Pagel K, Freund SMV, Morgner N, Robinson CV, Fersht AR, Joerger AC. 2011. Interaction of the p53 DNA-binding domain with its n-terminal extension modulates the stability of the p53 tetramer. J Mol Biol. 409(3):358–368. doi: 10.1016/j.jmb.2011.03.047.
- Nguyen TA, Menendez D, Resnick MA, Anderson CW. 2014. Mutant TP53 posttranslational modifications: challenges and opportunities. Hum Mutat. 35(6):738–755. doi: 10.1002/humu.22506.
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 288(5468):1053–1058. doi: 10.1126/science.288.5468.1053.
- Palacios G, Crawford HC, Vaseva A, Moll UM. 2008. Mitochondrially targeted wild-type p53 induces apoptosis in a solid human tumor xenograft model. Cell Cycle. 7(16):2584–2590. doi: 10.4161/cc.7.16.6070.
- Pant V, Sun C, Lozano G. 2023. Tissue specificity and spatio-temporal dynamics of the p53 transcriptional program. Cell Death Differ. 30(4):897–905. doi: 10.1038/s41418-023-01123-2.
- Pfister NT, Prives C. 2017. Transcriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harb Perspect Med. 7(2):a026054. doi: 10.1101/cshperspect.a026054.
- Robles AI, Bemmels NA, Foraker AB, Harris CC. 2001. APAF-1 is a transcriptional target of p53 in DNA damage-induced apoptosis. Cancer Res. 61(18):6660–6664.
- Rouault JP, Falette N, Guéhenneux F, Guillot C, Rimokh R, Wang Q, Berthet C, Moyret-Lalle C, Savatier P, Pain B, et al. 1996. Identification of BTG2, an antiproliferative p53-dependent component of the DNA damage cellular response pathway. Nat Genet. 14(4):482–486. doi: 10.1038/ng1296-482.
- Rubin SM, Sage J, Skotheim JM. 2020. Integrating old and new paradigms of G1/S control. Mol Cell. 80(2):183–192. doi: 10.1016/j.molcel.2020.08.020.
- Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. 2002. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 4(11):842–849. doi: 10.1038/ncb866.
- Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P, Green DR. 2000. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J Biol Chem. 275(10):7337–7342. doi: 10.1074/jbc.275.10.7337.
- Schuler M, Maurer U, Goldstein JC, Breitenbücher F, Hoffarth S, Waterhouse NJ, Green DR. 2003. p53 triggers apoptosis in oncogene-expressing fibroblasts by the induction of Noxa and mitochondrial Bax translocation. Cell Death Differ. 10(4):451–460. doi: 10.1038/sj.cdd.4401180.
- Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T, Tanaka N, et al. 2003. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17(18):2233–2238. doi: 10.1101/gad.1103603.
- Stadler SC, Vincent CT, Fedorov VD, Patsialou A, Cherrington BD, Wakshlag JJ, Mohanan S, Zee BM, Zhang X, Garcia BA, et al. 2013. Dysregulation of PAD4-mediated citrullination of nuclear GSK3β activates TGF-β signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc Natl Acad Sci USA. 110(29):11851–11856. doi: 10.1073/pnas.1308362110.
- Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. 2001. Regulation of PTEN transcription by p53. Mol Cell. 8(2):317–325. doi: 10.1016/s1097-2765(01)00323-9.
- Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. 2018. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 25(1):133–143. doi: 10.1038/cdd.2017.174.
- Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, et al. 2003. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 34(4):395–402. doi: 10.1038/ng1206.
- Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. 2010. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 107(16):7461–7466. doi: 10.1073/pnas.1002459107.
- Suzuki S, Venkatesh D, Kanda H, Nakayama A, Hosokawa H, Lee E, Miki T, Stockwell BR, Yokote K, Tanaka T, et al. 2022. GLS2 is a tumor suppressor and a regulator of ferroptosis in hepatocellular carcinoma. Cancer Res. 82(18):3209–3222. doi: 10.1158/0008-5472.CAN-21-3914.
- Tanikawa C, Espinosa M, Suzuki A, Masuda K, Yamamoto K, Tsuchiya E, Ueda K, Daigo Y, Nakamura Y, Matsuda K, et al. 2012. Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat Commun. 3(1):676. doi: 10.1038/ncomms1676.
- Tanikawa C, Ueda K, Nakagawa H, Yoshida N, Nakamura Y, Matsuda K. 2009. Regulation of protein citrullination through p53/PADI4 network in DNA damage response. Cancer Res. 69(22):8761–8769. doi: 10.1158/0008-5472.CAN-09-2280.
- Tanikawa C, Zhang Y-Z, Yamamoto R, Tsuda Y, Tanaka M, Funauchi Y, Mori J, Imoto S, Yamaguchi R, Nakamura Y, et al. 2017. The transcriptional landscape of p53 signalling pathway. EBioMedicine. 20:109–119. doi: 10.1016/j.ebiom.2017.05.017.
- Tripathi V, Kaur E, Kharat SS, Hussain M, Damodaran AP, Kulshrestha S, Sengupta S. 2019. Abrogation of FBW7α-dependent p53 degradation enhances p53’s function as a tumor suppressor. J Biol Chem. 294(36):13224–13232. doi: 10.1074/jbc.AC119.008483.
- Valente LJ, Gray DHD, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A. 2013. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 3(5):1339–1345. doi: 10.1016/j.celrep.2013.04.012.
- Varmeh-Ziaie S, Okan I, Wang Y, Magnusson KP, Warthoe P, Strauss M, Wiman KG. 1997. Wig-1, a new p53-induced gene encoding a zinc finger protein. Oncogene. 15(22):2699–2704. doi: 10.1038/sj.onc.1201454.
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. 2004. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 303(5659):844–848. doi: 10.1126/science.1092472.
- Vousden KH, Lu X. 2002. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2(8):594–604. doi: 10.1038/nrc864.
- Wahid M, Pratoomthai B, Egbuniwe IU, Evans HR, Babaei-Jadidi R, Amartey JO, Erdelyi V, Yacqub-Usman K, Jackson AM, Morris JC, et al. 2023. Targeting alternative splicing as a new cancer immunotherapy-phosphorylation of serine arginine-rich splicing factor (SRSF1) by SR protein kinase 1 (SRPK1) regulates alternative splicing of PD1 to generate a soluble antagonistic isoform that prevents T cell exhaustion. Cancer Immunol Immunother. 72(12):4001–4014. doi: 10.1007/s00262-023-03534-z.
- Walker KK, Levine AJ. 1996. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci USA. 93(26):15335–15340. doi: 10.1073/pnas.93.26.15335.
- Weber J, de la Rosa J, Grove CS, Schick M, Rad L, Baranov O, Strong A, Pfaus A, Friedrich MJ, Engleitner T, et al. 2019. PiggyBac transposon tools for recessive screening identify B-cell lymphoma drivers in mice. Nat Commun. 10(1):1415. doi: 10.1038/s41467-019-09180-3.
- Wu GS, Burns TF, McDonald ER, Meng RD, Kao G, Muschel R, Yen T, el-Deiry WS. 1999. Induction of the TRAIL receptor KILLER/DR5 in p53-dependent apoptosis but not growth arrest. Oncogene. 18(47):6411–6418. doi: 10.1038/sj.onc.1203025.
- Yeh CH, Bellon M, Nicot C. 2018. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 17(1):115. doi: 10.1186/s12943-018-0857-2.