
Abstract
Sulfur-protected enantiomerically pure 1-phosphanorbornane aldehyde 5 is obtained in good yield via Swern oxidation of the previously reported 1-phosphanorbornane alcohol. The aldehyde is further used to prepare a novel sulfur-protected 1-phosphanorbornane enamine.
GRAPHICAL ABSTRACT
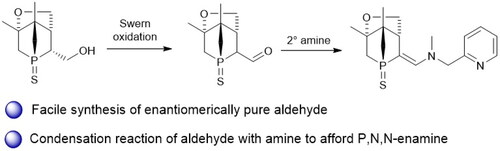
Introduction
Chiral compounds are especially important in life sciences and drug development, thus necessitating efficient and targeted syntheses.[Citation1] Accordingly, the fields of asymmetric synthesis and enantioselective catalysis are growing, requiring new tools and reagents.[Citation2] P-chiral phosphines find their applications in various fields.[Citation3] For example, these compounds are viewed as an efficient class of ligands employed in asymmetric homogeneous catalysis[Citation4] pioneered by the groundbreaking discovery of (ethane-1,2-diyl)bis[(2-methoxy-phenyl)(phenyl)phosphine] (R,R-DIPAMP) in the 1970s.[Citation5] Phosphole-based P-chiral ligands[Citation6] are a promising class of compounds readily available through functionalization of phospholes. Especially the ability of the tautomeric 2H-phos-pholes to undergo hetero-Diels–Alder cycloaddition reactions gives access to a wide range of phosphacycles.[Citation7] Recently, we reported the first phospha-aza-Diels–Alder cycloaddition between 2H-phospholes and the dienophile N-sulfonylimino ester to give racemic 1-phospha-2-azanorbornenes (PANs).[Citation8] Moreover, we could show that the reactive P–N bond of PANs can be cleaved with nucleophiles to afford both racemic[Citation8] and enantiopure[Citation9] 2,3-dihydrophosphole derivatives. Additionally, the ring-opening and ring-closing reactions of PANs with ethanol were studied, both experimentally and theoretically.[Citation10] We have also reported the unprecedented asymmetric phospha-Diels–Alder cycloaddition between the diene 2H-phosphole and the enantiopure dienophile (5R)-(L-menthyloxy)-2(5H)-furanone (MOxF) to yield sulfur-protected 1-phosphanorbornene 2 (Scheme 1).[Citation11] Furthermore, the chiral auxiliary could be cleaved to afford 1-phosphanorbornene diol 3, followed by a base-catalyzed selective Michael addition to give the enantiopure 1-phosphanorbornane alcohol 4.[Citation12] The latter compound is a highly resourceful starting material for further functionalization, such as the synthesis of 1-phosphanorbornane silyl ethers[Citation13] or 1-phosphanorbornane phosphine-phosphites[Citation14] as well as bisphosphines,[Citation12] which were studied as ligands in Ir,[Citation13] Pd[Citation14] or Rh[Citation14] complexes.
Scheme 1. Preparation of sulfur-protected 1-phosphanorbornane alcohol 4.[Citation12]
![Scheme 1. Preparation of sulfur-protected 1-phosphanorbornane alcohol 4.[Citation12]](/cms/asset/1c2a3637-1360-48a8-b1a6-8c227653efb2/gpss_a_2338393_sch0001_b.jpg)
Here we report the conversion of enantiomerically pure alcohol 4 to the corresponding aldehyde, which opens up a wealth of new possibilities for further modifications.
Results and discussion
Enantiomerically pure sulfur-protected 1-phosphanorborane alcohol 4 is readily prepared according to the literature (Scheme 1).[Citation12]
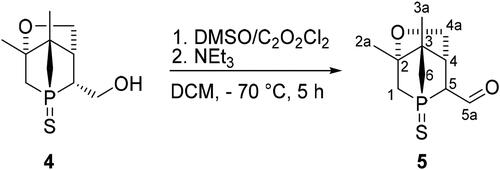
The aldehyde was obtained by Swern oxidation using the well-established procedure including oxalyl chloride, DMSO and triethylamine as base (Scheme 2). The critical point is to keep the temperature of the reaction mixture around −70 °C. In the case of higher temperatures, the formation of more by-products was observed. The reaction time was limited to 5 h, even though the reaction was not yet completed and a small amount of alcohol 4 was still present, as longer reaction times led to the formation of more by-products. The 31P{1H} NMR spectrum revealed small amounts of 4 and a singlet at 46 ppm for 5 as the major product. Moreover, in the 1H NMR spectrum the characteristic resonance for the aldehyde proton at 9.8 ppm was observed. However, the purification and isolation of 5 proved challenging.
Scheme 2. Preparation of 1-phosphanorbornane aldehyde 5 via Swern oxidation of 4.
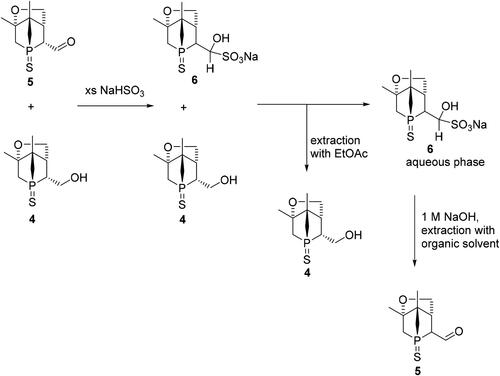
Followed by the reaction work-up described in the experimental part below, the concentrated mixture containing aldehyde 5, alcohol 4 and unknown by-products was dissolved in MeOH. Then the mixture was further allowed to react with an aqueous solution of NaHSO3, which is one of the commonly used liquid-liquid extraction methods for isolating aldehydes selectively (Scheme 3).[Citation15] The organic layer was treated with ethyl acetate to extract most of the alcohol and other by-products. The salt 6 is insoluble in organic solvents and was contained in the aqueous layer. The aqueous layer was separated, basified with NaOH and then extracted with ethyl acetate to afford sulfur-protected aldehyde 5, which still contained small amounts of 4. Compound 5 could be isolated and purified by column chromatography. Recrystallization in hot isopropanol resulted in the formation of the hemiacetal 7 (Scheme 4), which was obtained as single crystals suitable for X-ray crystallography. However, once the crystalline hemiacetal is dissolved in organic solvents, the aldehyde 5 is formed again and iPrOH is eliminated, thus making it impossible to characterize compound 7 by NMR or HRMS techniques. The molecular structure of the hemiacetal 7 was confirmed by XRD. Thus, the absolute configuration (P1-(R), C1-(S), C2-(R), C4-(S), C5-(R)) could be determined ().
Scheme 3. Isolation of sulfur-protected aldehyde 5 using NaHSO3.
Scheme 4. Formation of hemiacetal 7 via addition reaction of iPrOH with aldehyde 5.
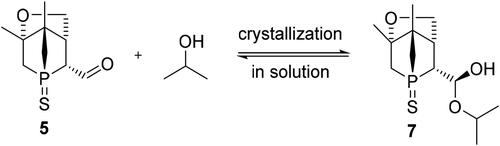
Figure 1. Molecular structure of hemiacetal 7. Ellipsoid displacements are at 50% probability. Selected bond lengths [pm] and angles [°]: S(1)–P(1) 194.35(5), P(1)–C(4) 182.3(2), C(4)–C(10) 152.1(2), O(2)–C(10) 140.5(2), O(3)–C(10) 141.2(2), O(3)–C(11) 143.3(2); C(7)–P(1)–C(3) 92.57(8), C(7)–P(1)–S(1) 123.94(6), C(3)–P(1)–C(4) 98.82(8), C(6)–O(1)–C(2) 110.5(1), C(10)–O(3)–C(11) 115.0(1).
![Figure 1. Molecular structure of hemiacetal 7. Ellipsoid displacements are at 50% probability. Selected bond lengths [pm] and angles [°]: S(1)–P(1) 194.35(5), P(1)–C(4) 182.3(2), C(4)–C(10) 152.1(2), O(2)–C(10) 140.5(2), O(3)–C(10) 141.2(2), O(3)–C(11) 143.3(2); C(7)–P(1)–C(3) 92.57(8), C(7)–P(1)–S(1) 123.94(6), C(3)–P(1)–C(4) 98.82(8), C(6)–O(1)–C(2) 110.5(1), C(10)–O(3)–C(11) 115.0(1).](/cms/asset/562636b1-dd77-4771-bf13-18d50ffa69fe/gpss_a_2338393_f0001_c.jpg)
Like other aldehydes,[Citation16] 5 is a highly versatile starting material, e.g. for introducing additional donor atoms to generate potentially multidentate or pincer ligands. This is readily achieved by a condensation reaction with amines. As a proof of concept, a slight excess of 2-[(methylamino)methyl]pyridine was allowed to react with aldehyde 5 for 1 h at 80 °C (Scheme 5). The 31P{1H} NMR spectrum of the reaction mixture revealed full consumption of 5 and formation of enamine 8 (singlet at 43 ppm). Single crystals of 8 were obtained by recrystallization from hot isopropanol. The structure was confirmed by single crystal XRD (), 2D NMR spectroscopy and high-resolution mass spectrometry.
Scheme 5. Synthesis of enamine 8.
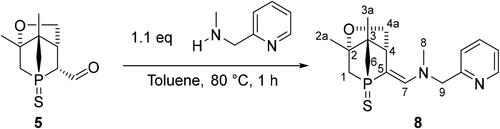
Figure 2. Molecular structure of 8 (P1-(R), C1-(S), C2-(R), C5-(R)). Ellipsoid displacements are at 50% probability. Selected bond lengths [pm] and angles [°]: S(1)–P(1) 194.96(7), P(1)–C(4) 178.4(2), N(1)–C(10) 135.9(3), O(1)–C(6) 143.0(3), N(1)–C(11) 144.9(3), N(2)–C(12) 133.6(3); C(4)–P(1)–C(3) 102.8(1), C(4)–P(1)–C(7) 92.8(1), C(4)–P(1)–S(1) 120.4(8), C(7)–P(1)–S(1) 124.22(8), C(6)–O(1)–C(2) 110.6(2), C(10)–N(1)–C(11) 121.0(2), C(10)–N(1)–C(17) 123.8(2), C(11)–N(1)–C(17) 115.2(2), C(16)–N(2)–C(12) 117.4(2).
![Figure 2. Molecular structure of 8 (P1-(R), C1-(S), C2-(R), C5-(R)). Ellipsoid displacements are at 50% probability. Selected bond lengths [pm] and angles [°]: S(1)–P(1) 194.96(7), P(1)–C(4) 178.4(2), N(1)–C(10) 135.9(3), O(1)–C(6) 143.0(3), N(1)–C(11) 144.9(3), N(2)–C(12) 133.6(3); C(4)–P(1)–C(3) 102.8(1), C(4)–P(1)–C(7) 92.8(1), C(4)–P(1)–S(1) 120.4(8), C(7)–P(1)–S(1) 124.22(8), C(6)–O(1)–C(2) 110.6(2), C(10)–N(1)–C(11) 121.0(2), C(10)–N(1)–C(17) 123.8(2), C(11)–N(1)–C(17) 115.2(2), C(16)–N(2)–C(12) 117.4(2).](/cms/asset/23d05719-636e-4c1f-ba1b-e14d36b20d7b/gpss_a_2338393_f0002_c.jpg)
Conclusions
xPreviously reported 1-phosphanorbornane alcohol 4 was successfully oxidized via Swern reaction to give 1-phosphanorbornane aldehyde 5. Compound 5 is a useful starting material for the production of chiral multidentate ligands, as confirmed by the condensation reaction with 2-[(methylamino)methyl]pyridine to afford enantiomerically pure enamine 8.
Experimental section
Reactions with air-sensitive compounds were carried out under dry high purity nitrogen using standard Schlenk techniques. THF was degassed and distilled from potassium. DMSO and DCM were degassed and stored over 4 Å activated molecular sieves. Compound 4 was prepared according to the literature.[Citation12] All other reagents are commercially available and were used as purchased. The NMR spectra were recorded with a Bruker Avance DRX 400 spectrometer (1H NMR 400.13 MHz, 13C NMR 100.63 MHz, 31P NMR 161.98 MHz). 13C{1H} NMR spectra were recorded as APT spectra. If not mentioned otherwise, CDCl3 was used as deuterated solvent. TMS was used as the internal standard in the 1H NMR spectra, and all other nuclei spectra were referenced to TMS using the Ξ-scale.[Citation17] The numbering scheme for compounds 5 and 8 are given in Schemes 2 and 5, respectively. High-resolution mass spectra (HRMS; ESI) were measured using a Bruker Daltonics APEX II FT-ICR spectrometer. IR spectra were obtained with an FTIR spectrometer (Nicolet iS5 FTIR by Thermo Scientific, Waltham, MA, USA) in the range of 400–4000 cm−1 in KBr. Iodine (saturated atmosphere) was used as staining reagent in thin-layer chromatography.
Synthesis of 5
Oxalyl chloride (0.184 mL, 2.15 mmol, 1.0 eq.) was added at −78 °C to a solution of DMSO (0.38 mL, 5.37 mmol, 2.5 eq.) in DCM (3 mL) and stirred for 45 min at this temperature. A solution of 4 (500 mg, 2.15 mmol, 1.0 eq.) in DCM (7 mL) was then added and the temperature was maintained between −60 and −78 °C for 3 h. The reaction was quenched by the addition of triethylamine (0.75 mL, 5.37 mmol, 2.5 eq.) (at −78 °C) and water (7 mL) was added. The cold bath was then removed and the reaction mixture was allowed to warm to rt for 30–45 min. The organic phase was separated, washed first with an HCl solution (1%, 7 mL) and then with a saturated Na2CO3 solution (10 mL). After drying over MgSO4 and removing the solvent, compound 5 was isolated via column chromatography as a white powder (318 mg, 64%). 1H NMR (400 MHz, CDCl3): δ = 10.02 (s, 1H, H-5a), 4.12 (dd, J = 9.4, 5.6 Hz, 1H, H-4a), 3.66 (d, J = 9.4 Hz, 1H, H-4a), 3.30 (dd, J = 17.4, 2.4 Hz, 1H, H-5), 3.05 (m, 1H, H-4), 2.36 (m, 1H, H-1/6), 2.10 − 1.94 (m, 2H, H-1 and H-6), 1.86 (dd, J = 13.6, 5.8 Hz, 1H, H-1/6), 1.30 (s, 3H, H-2a/3a), 1.23 (s, 3H, H-2a/3a) ppm. 13C{1H} NMR (101 MHz, CDCl3): δ = 198.5 (s, C-5a), 86.5 (s, C-quart.), 71.8 (d, J = 2.6 Hz, C-4a), 60 (d, J = 35.4 Hz, C-5), 50.7 (d, J = 19.0 Hz, C-quart.), 45.1 (d, J = 47.3 Hz, C-1 or C-6), 45.2 (d, J = 2 Hz, C-4), 38.5 (d, J = 50.5 Hz, C-1 or C-6), 23.7 (d, J = 7.4 Hz, C-2a or C-3a), 17.2 (d, J = 17.5 Hz, C-2a or C-3a) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ = 46.2 (s) ppm. 31P NMR (162 MHz, CDCl3): δ = 46.2 (m) ppm. ATR-IR (KBr): ṽ = 2963 (m), 2874 (w), 2853 (m), 2731 (w), 1705 (s), 1450 (m), 1416 (w), 1402 (m), 1383 (m), 1354 (m), 1318 (w), 1288 (m), 1267 (w), 1245 (m), 1222 (w), 1199 (w), 1169 (s), 1132 (m), 1096 (m), 1082 (m), 1047 (s), 1023 (s), 1008 (s), 977 (m), 929 (m), 908 (m), 884 (m), 862 (m), 802 (m), 781 (s), 737 (s), 667 (s), 580 (m), 558 (m), 461 (s) cm−1. HRMS (ESI(+), MeCN), m/z: found: 231.0608, calculated for [M + H]+: 231.0603; found: 237.0661, calculated for [M + Li]+: 237.0618; found: 253.0419, calculated for [M + Na]+: 253.0423.
Synthesis of 8
2-[(Methylamino)methyl]pyridine (61 μL, 59.5 mg, 0.49 mmol, 1.1 eq.) together with a small amount of MgSO4 (ca. 20 mg) were added to a solution of 5 (102 mg, 0.44 mmol, 1 eq.) in toluene (2 mL). The suspension was heated to 80 °C and stirred for 1 h. Then the solution was decanted and the solvent evaporated. The remaining solid was dissolved in a hot iPrOH/n-hexane (5 mL/8 mL) mixture and left overnight at −25 °C to give compound 8 as a yellow powder (121 mg, 82%). 1H NMR (400 MHz, CDCl3): δ = 8.63–8.48 (m, 1H), 7.71 (td, J = 7.7, 1.8 Hz, 1H), 7.23 (dd, J = 11.7, 7.5 Hz, 2H), 6.56 (dd, J = 19.5, 1.5 Hz, 1H, H-7), 4.44 (bs, 2H, H-9), 4.13 (dd, J = 8.0, 5.6 Hz, 1H, H-4a), 3.85 (d, J = 8.0 Hz, 1H, H-4a), 3.17 (dd, J = 11.9, 5.6 Hz, 1H, H-4), 2.97 (s, 3H, H-8), 2.10 − 2.00 (m, 2H, H-1 or H-6), 1.87 (dd, J = 12.4, 7.1 Hz, 1H, H-1 or H-6), 1.76 (ddd, J = 12.4, 9.7, 2.7 Hz, 1H, H-1 or H-6), 1.28 (s, 3H, H-2a or 3a), 1.17 (s, 3H, H-2a or 3a). 13C{1H} NMR (101 MHz, CDCl3): δ = 157.6 (s, C-quart. pyridyl), 149.7 (s, C-aryl), 143.6 (d, J = 23.7 Hz, C-7), 137.0 (s, C-aryl), 123.4 (s, C-aryl), 122.5 (s, C-aryl), 121 (s, C-aryl), 97.9 (d, J = 88.4 Hz, C-quart. C-5), 86.8 (d, J = 2.3 Hz, C-3 or C-2), 73.3 (d, J = 2.2 Hz, C-4a), 60.9 (s, C-9), 51.1 (d, J = 17.2 Hz, C-quart. C-2 or C-3), 49.7 (d, C-4), 45.5 (d, J = 50.7 Hz, C-1 or C-6), 41.2 (d, J = 54.9 Hz, C-1 or C-6), 39.7 (s, C-8), 24.1 (d, J = 7.3 Hz, C-2a or 3a), 18.5 (d, J = 14.6 Hz, C-2a or 3a) ppm. 31P{1H} NMR (162 MHz, CDCl3): δ = 43.7 (s) ppm. 31P NMR (162 MHz, CDCl3): δ = 43.7 (m) ppm. HRMS (ESI(+), MeCN), m/z: found: 335.1348, calculated for [M + H]+: 335.1341.
X-ray crystallography data
The data were collected on a Gemini diffractometer (Rigaku Oxford Diffraction) using Mo-Kα or Cu-Kα radiation and ω-scan rotation. Data reduction was performed with CrysAlisPro[Citation18] including the program SCALE3 ABSPACK for empirical absorption correction. All structures were solved by dual space methods with SHELXT[Citation19] and the refinement was performed with SHELXL.[Citation20] Non-hydrogen atoms were refined with anisotropic displacement parameters and a difference-density Fourier map was used to locate hydrogen atoms. Structure figures were generated with DIAMOND-4.[Citation21] CCDC deposition numbers given in Table S1 (SI) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/ (or from the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44)1223-336-033; or [email protected]).
Supplemental Material
Download MS Word (268.2 KB)Acknowledgement
We thank the DFG (HE 1376/46-1) and the Graduate School BuildMoNa for financial support.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- (a) Brooks, W. H.; Guida, W. C.; Daniel, K. G. The Significance of Chirality in Drug Design and Development. Curr. Top. Med. Chem. 2011, 11, 760–770. DOI: 10.2174/156802611795165098. (b) Chen, Q.-S.; Li, J.-Q.; Zhang, Q.-W. Application of Chiral Piperidine Scaffolds in Drug Design. Pharmaceut. Fronts 2023, 05, e1–e14. DOI: 10.1055/s-0043-1764218.
- (a) Fanourakis, A.; Docherty, P. J.; Chuentragool, P.; Phipps, R. J. Recent Developments in Enantioselective Transition Metal Catalysis Featuring Attractive Noncovalent Interactions between Ligand and Substrate. ACS Catal. 2020, 10, 10672–10714. DOI: 10.1021/acscatal.0c02957. (b) García Mancheño, O.; Waser, M. Recent Developments and Trends in Asymmetric Organocatalysis. Eur. J. Org. Chem. 2023, 26, e202200950. DOI: 10.1002/ejoc.202200950.
- (a) Corbridge, D. E. C. Phosphorus. Chemistry, Biochemistry and Technology (CRC Press, Boca Raton, 2013). (b) Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. DOI: 10.1021/jm200587f. (c) Forlani, G.; Berlicki, L.; Duò, M.; Dziędzioła, G.; Giberti, S.; Bertazzini, M.; Kafarski, P. Synthesis and Evaluation of Effective Inhibitors of Plant δ1-Pyrroline-5-Carboxylate Reductase. J. Agric. Food Chem. 2013, 61, 6792–6798. DOI: 10.1021/jf401234s. (d) Long, N.; Cai, X.-J.; Song, B.-A.; Yang, S.; Chen, Z.; Bhadury, P. S.; Hu, D.-Y.; Jin, L.-H.; Xue, W. Synthesis and Antiviral Activities of Cyanoacrylate Derivatives Containing an α-Aminophosphonate Moiety. J. Agric. Food Chem. 2008, 56, 5242–5246. DOI: 10.1021/jf800405m.
- (a) Xu, G.; Senanayake, C. H.; Tang, W. P-Chiral Phosphorus Ligands Based on a 2,3-Dihydrobenzo[d][1,3]Oxaphosphole Motif for Asymmetric Catalysis. Acc. Chem. Res. 2019, 52, 1101–1112. DOI: 10.1021/acs.accounts.9b00029. (b) Börner A. (Ed.) Phosphorus ligands in asymmetric catalysis (Wiley-VCH-Verl., Weinheim, 2008). (c) Kamer, P. C. J. (Ed.) Phosphorus(III) Ligands in Homogeneous Catalysis. Design and Synthesis (Wiley, Chichester, 2012). (d) Imamoto, T. Synthesis and Applications of High-Performance P-Chiral Phosphine Ligands. Proc. Jpn. Acad, Ser. B 2021, 97, 520–542. DOI: 10.2183/pjab.97.026. (e) Dutartre, M.; Bayardon, J.; Jugé, S. Applications and Stereoselective Syntheses of P-Chirogenic Phosphorus Compounds. Chem. Soc. Rev. 2016, 45, 5771–5794. DOI: 10.1039/C6CS00031B. (f) Cabré, A.; Riera, A.; Verdaguer, X. P-Stereogenic Amino-Phosphines as Chiral Ligands: From Privileged Intermediates to Asymmetric Catalysis. Acc. Chem. Res. 2020, 53, 676–689. DOI: 10.1021/acs.accounts.9b00633. (g) Xie, X.; Li, S.; Chen, Q.; Guo, H.; Yang, J.; Zhang, J. Synthesis and Application of Novel P-Chiral Monophosphorus Ligands. Org. Chem. Front. 2022, 9, 1589–1592. DOI: 10.1039/D1QO01819A.
- Vineyard, B. D.; Knowles, W. S.; Sabacky, M. J.; Bachman, G. L.; Weinkauff, D. J. Asymmetric Hydrogenation. Rhodium Chiral Bisphosphine Catalyst. J. Am. Chem. Soc. 1977, 99, 5946–5952. DOI: 10.1021/ja00460a018.
- (a) Yavari, K.; Aillard, P.; Zhang, Y.; Nuter, F.; Retailleau, P.; Voituriez, A.; Marinetti, A. Helicenes with Embedded Phosphole Units in Enantioselective Gold Catalysis. Angew. Chem. Int. Ed. 2014, 53, 861–865. DOI: 10.1002/anie.201308377. (b) Tang, W.; Qu, B.; Capacci, A. G.; Rodriguez, S.; Wei, X.; Haddad, N.; Narayanan, B.; Ma, S.; Grinberg, N.; Yee, N. K.; et al. Novel, Tunable, and Efficient Chiral Bisdihydrobenzooxaphosphole Ligands for Asymmetric Hydrogenation. Org. Lett. 2010, 12, 176–179. DOI: 10.1021/ol9025815.
- (a) Mathey, F. The Organic Chemistry of Phospholes. Chem. Rev 1988, 88, 429–453. DOI: 10.1021/cr00084a005. (b) Maitra, K.; Catalano, V. J.; Nelson, J. H. Intramolecular [4 + 2] Diels − Alder Cycloaddition of a 2H-Phosphole to Coordinated Unsaturated Phosphines, Phospholes, and an Arsine. J. Am. Chem. Soc. 1997, 119, 12560–12567. DOI: 10.1021/ja9724654. (c) Zhang, K.; Zhang, Q.; Wei, D.; Tian, R.; Duan, Z. Hetero-Diels–Alder Reactions of 2H-Phospholes with Allenes: Synthesis and Functionalization of 6-Methylene-1-Phosphanorbornenes. Org. Chem. Front. 2021, 8, 3740–3745. DOI: 10.1039/D1QO00535A. (d) Mathey, F. Transient 2H-Phospholes as Powerful Synthetic Intermediates in Organophosphorus Chemistry. Acc. Chem. Res. 2004, 37, 954–960. DOI: 10.1021/ar030118v.
- Wonneberger, P.; König, N.; Kraft, F. B.; Sárosi, M. B.; Hey-Hawkins, E. Access to 1-Phospha-2-Azanorbornenes by Phospha-aza-Diels–Alder Reactions. Angew. Chem. Int. Ed. Engl. 2019, 58, 3208–3211. DOI: 10.1002/anie.201811673.
- Ramazanova, K.; Lönnecke, P.; Hey-Hawkins, E. Facile Synthesis of Enantiomerically Pure P-Chiral 1-Alkoxy-2,3-Dihydrophospholes via Nucleophilic P-N Bond Cleavage of a 1-Phospha-2-Azanorbornene. Chem. Eur. J. 2023, 29, e202300790. DOI: 10.1002/chem.202300790.
- Ramazanova, K.; Müller, A. K.; Lönnecke, P.; Hollóczki, O.; Kirchner, B.; Hey-Hawkins, E. Ring-Opening Reaction of 1-Phospha-2-Azanorbornenes via P-N Bond Cleavage and Reversibility Studies. Molecules 2023, 28, 7163. DOI: 10.3390/molecules28207163.
- Möller, T.; Sárosi, M. B.; Hey-Hawkins, E. Asymmetric Phospha-Diels–Alder Reaction: A Stereoselective Approach towards P-Chiral Phosphanes through Diastereotopic Face Differentiation. Chem. Eur. J. 2012, 18, 16604–16607. DOI: 10.1002/chem.201203671.
- Möller, T.; Wonneberger, P.; Sárosi, M. B.; Coburger, P.; Hey-Hawkins, E. P-Chiral 1-Phosphanorbornenes: From Asymmetric phospha-Diels–Alder Reactions towards Ligand Design and Functionalisation. Dalton Trans. 2016, 45, 1904–1917. DOI: 10.1039/C5DT02564H.
- Ramazanova, K.; Chakrabortty, S.; Kallmeier, F.; Kretzschmar, N.; Tin, S.; Lönnecke, P.; de Vries, J. G.; Hey-Hawkins, E. Access to Enantiomerically Pure P-Chiral 1-Phosphanorbornane Silyl Ethers. Molecules 2023, 28, 6210. DOI: 10.3390/molecules28176210.
- Ramazanova, K.; Chakrabortty, S.; Müller, B. H.; Lönnecke, P.; de Vries, J. G.; Hey-Hawkins, E. Synthesis of P-Stereogenic 1-Phosphanorbornane-Derived Phosphine–Phosphite Ligands and Application in Asymmetric Catalysis. RSC Adv. 2023, 13, 34439–34444. DOI: 10.1039/D3RA07630J.
- Boucher, M. M.; Furigay, M. H.; Quach, P. K.; Brindle, C. S. Liquid–Liquid Extraction Protocol for the Removal of Aldehydes and Highly Reactive Ketones from Mixtures. Org. Process Res. Dev. 2017, 21, 1394–1403. DOI: 10.1021/acs.oprd.7b00231.
- Moloney, M. G. (Ed.) Organic Reaction Mechanisms 2019: An annual survey covering the literature dated January to December 2019, (Wiley, 2023)
- Harris, R. K.; Becker, E. D.; Cabral de Menezes, S. M.; Goodfellow, R.; Granger, P. NMR Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts. IUPAC Recommendations 2001. International Union of Pure and Applied Chemistry. Physical Chemistry Division. Commission on Molecular Structure and Spectroscopy. Solid State Nucl. Magn. Reson. 2002, 22, 458–483. DOI: 10.1002/mrc.1042.
- Rigaku Corporation. CrysAlisPro Software System (Rigaku Oxford Diffraction, Wroclaw, Poland, 1995–2023).
- Sheldrick, G. M. SHELXT – Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. DOI: 10.1107/S2053273314026370.
- Sheldrick, G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. DOI: 10.1107/S2053229614024218.
- Crystal Impact GbR, version 4.6.8, DIAMOND 4, K. Brandenburg, Bonn, Germany, 2022.